
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA unified strategy to reverse-prenylated indole alkaloids: total syntheses of preparaherquamide, premalbrancheamide, and (+)-VM-55599†‡
Jose B.
Roque
 ,
Eduardo V.
Mercado-Marin
,
Sven C.
Richter
,
Danilo
Pereira de Sant'Ana
,
Ken
Mukai
,
Yingda
Ye
and
Richmond
Sarpong
*
,
Eduardo V.
Mercado-Marin
,
Sven C.
Richter
,
Danilo
Pereira de Sant'Ana
,
Ken
Mukai
,
Yingda
Ye
and
Richmond
Sarpong
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: rsarpong@berkeley.edu
First published on 28th May 2020
Abstract
A full account of our studies toward reverse-prenylated indole alkaloids that contain a bicyclo[2.2.2]core is described. A divergent route is reported which has resulted in the synthesis of preparaherquamide, (+)-VM-55599, and premalbrancheamide. An intramolecular Dieckmann cyclization between an enolate and isocyanate was used to forge the bicyclo[2.2.2]diazaoctane core that is characteristic of these molecules. The pentacyclic indole scaffold was constructed through a one-pot Hofmann rearrangement followed by Fischer indole synthesis. The utilization of our previously reported indole peripheral functionalization strategy also led to natural products including malbrancheamides B, C, stephacidin A, notoamides F, I and R, aspergamide B, and waikialoid A. Ultimately, the divergent route that we devised provided access to a wide range of prenylated indole alkaloids that are differently substituted on the cyclic amine core.
Introduction
Secondary metabolites isolated from fungi of the genera Penicillium and Aspergillus continue to attract the interest of the chemical synthesis community because of their structural complexity and diverse biological activity. Among these natural products are a series of reverse-prenylated indole alkaloids that exhibit a wide range of bioactivity including, but not limited to, insecticidal, cytotoxic, anthelmintic, and anti-bacterial properties.1 Representative of this group are the paraherquamides (e.g., 2, Fig. 1), stephacidins (e.g., 5), and marcfortines (e.g., 7), all of which possess a bicyclo[2.2.2]diazaoctane core ring system (highlighted in red in 1). Structurally, members of this group are comprised of two amino acids, tryptophan (highlighted in blue in 2) and proline or isoleucine (highlighted in maroon in 3 and 6, respectively). These core motifs are in turn reverse-prenylated (highlighted in green in 5).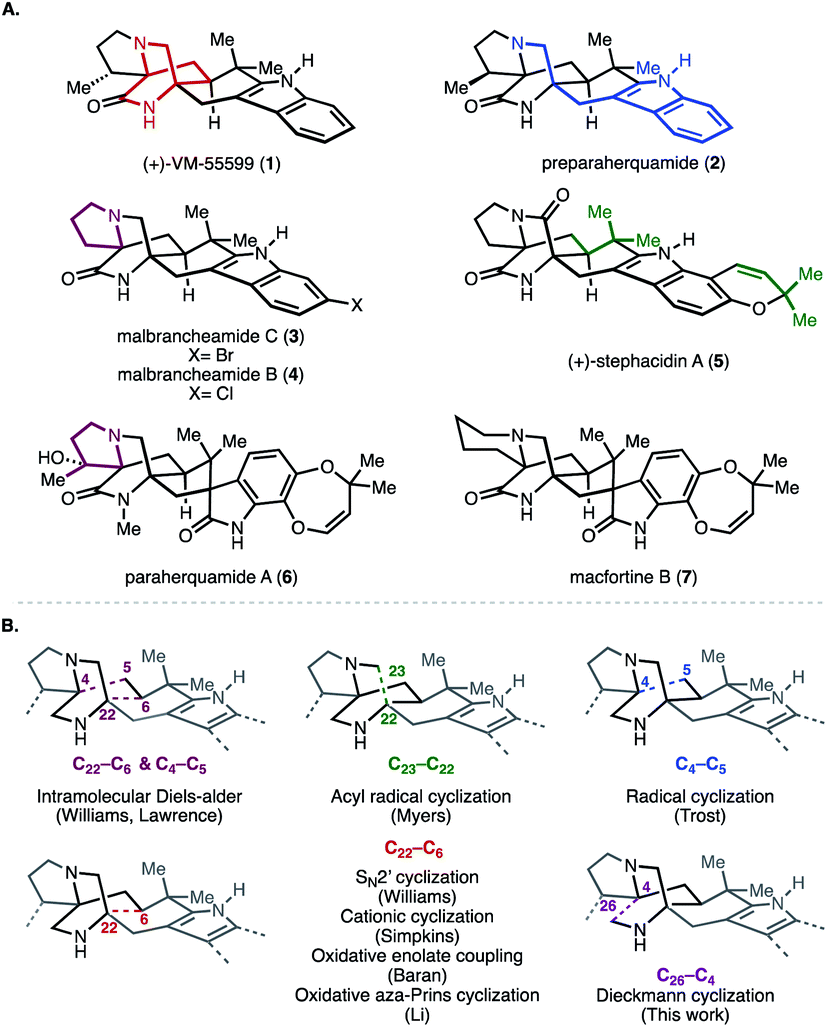 | ||
| Fig. 1 (A) Selected reverse-prenylated indole alkaloids. (B) Previous approaches to construct the bicyclo[2.2.2]diazaoctane ring system of reverse-prenylated indole alkaloids core. Numbering is based on stephacidin A. | ||
The different substitution and oxidation patterns present on the core framework of these indole alkaloids, along with their intriguing biogenesis, has spurred numerous synthetic campaigns to prepare them. Prior synthetic strategies can be organized around the construction of the bicyclo[2.2.2]diazaoctane core (Fig. 1B).2 For example, Williams and co-workers employed a biomimetic intramolecular Diels–Alder reaction to construct the bicyclo ring system en route to (−)-VM5599, rac-pre-paraherquamide, rac-marcfortine C, and (+) and (−)-versicolamide B (forming C22–C6 and C4–C5, Fig. 1B) and recently, Lawrence and co-workers leveraged a similar bioinspired approach to access (+)-brevianamide A.2 Notably, a complementary strategy employed by Williams and co-workers featured an intramolecular SN2′ cyclization in the syntheses of brevianamide B, (−)-paraherquamide A, (+)-paraherquamide B, stephacidins A and B, notoamide B, and avrainvillamide (forming C22–C6, Fig. 1B).3 Inspired by the seminal reports of the Mislow and Saegusa laboratories as well as previous work from their own laboratories, Baran and co-workers utilized an oxidative enolate coupling strategy to achieve the first total synthesis of stephacidin A (forming C22–C6, Fig. 1B).4 Further oxidative elaboration also resulted in syntheses of avrainvillamide and stephacidin B.4 Other strategies have relied on cationic, radical, acyl radical, and oxidative aza-Prins cyclizations (Fig. 1B).5–8
Despite the existing elegant strategies to access specific congeners within this family of natural products, a unified approach to access prenylated indole alkaloids that either possess or lack additional substituents on the cyclic amine ring (highlighted in maroon in Fig. 2A and labelled ring A in Fig. 2C for clarity) of the hexacyclic framework remained an outstanding challenge.9 Biosynthetically (Fig. 2A), natural products like paraherquamide A (6), which contain a methyl-proline residue (R′ = Me), are proposed to arise from a 4-electron oxidation of isoleucine to furnish an aldehyde intermediate which undergoes reductive amination.10,11 Alternatively, congeners like stephacidin A, which feature no substituents on the cyclic amine core, are derived from L-proline. Synthetically, it became evident that adhering to a bioinspired approach would require a series of challenging site selective late-stage oxidations in order to access each member of this reverse-prenylated indole alkaloid family (Fig. 2B).10,11 For example, issues of chemoselectivity were encountered in our own work with previously reported amide 7 and late-stage oxidation in the presence of the indole moiety proved difficult (8 in Fig. 2B).12–14 Given the aforementioned challenges and strategic value of divergent total synthesis for accessing related synthetic targets, a route was designed to leverage a versatile common intermediate (9, Fig. 2C) which contains a ketone as a synthetic handle on ring A for diversification. Because ketones engage in a plethora of organic reactions, we envisioned using the diverse reactivity of the carbonyl group to perform either late-stage nucleophilic additions to access members within the paraherquamide family, ring expansion to access macfortine natural products, or leverage enolate chemistry to access the mangrovamides. Another strategic design element embedded in the versatile common intermediate (9) is the unsubstituted indole motif, which was selected to maximize access to the diverse indole substitution patterns characteristic of the reverse-prenylated indole alkaloids through late-stage indole C–H functionalization.
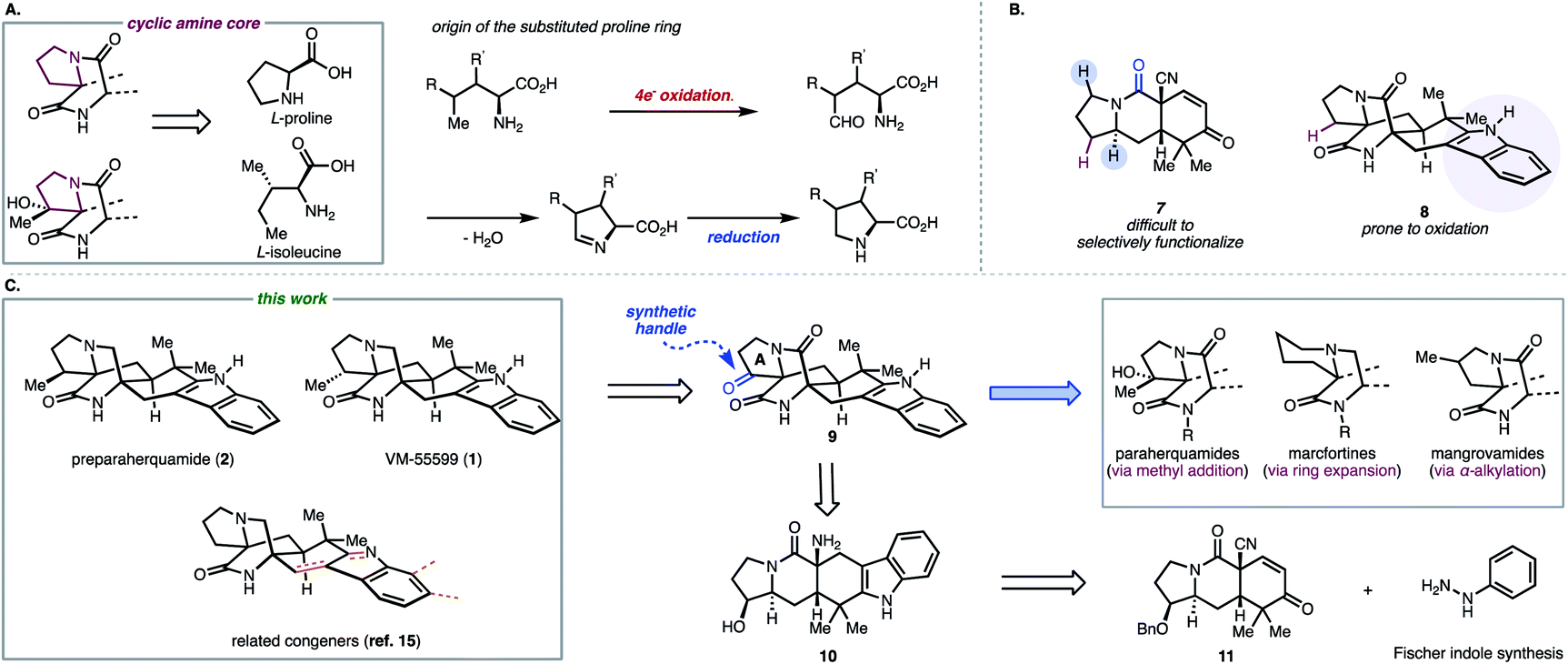 | ||
| Fig. 2 (A) Origin of cyclic amine core; (B) unsuccessful approaches; (C) retrosynthetic plan for a unified synthesis of reverse prenylated indole alkaloids. | ||
From 9, the bicyclo[2.2.2]diazaoctane structural motif could also be constructed using a similar strategy to our previously reported Dieckmann-type cyclization.13 Pentacyclic indole 10 was envisioned to arise from our previously reported tricyclic intermediate 11 and phenyl hydrazine by Fischer indole synthesis.13
Results and discussion
Our initial approach centered around a one-pot protocol for the construction of the bicyclo[2.2.2]diazaoctane core. It was envisioned that a Hofmann rearrangement performed in the absence of any external nucleophiles would generate isocyanate 13in situ, which could be intercepted by an attendant enol or enolate, through an intramolecular cyclization, to provide the bicyclo[2.2.2]diazaoctane ring (see 9) in a single-pot transformation.16 In order to explore the feasibility of this one-pot Hofmann/cyclization event, access to pentacyclic indole 12 was required. We commenced our synthetic studies with enone 11, which is available in gram-scale quantities (9 steps, 37% overall yield) from commercially available 1-tert-butyl 2-ethyl 3-oxopyrrolidine-1,2-dicarboxylate (Fig. 3B; see S1 in the ESI‡).13 Following hydrogenation of enone 11 using Pd/C, treatment with BBr3 effected cleavage of the benzyl group to provide ketone 14.13 Treatment of ketone 14 with phenyl hydrazine in aqueous sulfuric acid at 100 °C afforded pentacyclic indole 15 in 83% yield. Oxidation of the secondary alcohol group with Dess–Martin periodinane (DMP) gave the desired ketone (not shown) as an inseparable mixture of diastereomers in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (α:β epimers) that was taken directly to the next reaction because of stability issues. The use of the Ghaffar–Parkins platinum complex failed to provide any desired hydrated product (12) and led to a complex product distribution.17 An alternative procedure for nitrile hydration reported by Lee and co-workers using Wilkinson's catalyst and acetaldoxime in toluene at reflux led only to decomposition of starting material.18 Presumably, the enol (not shown) can compete for the binding site of the metal center in these cases leading to decomposition pathways. Given the difficulty in performing the nitrile hydration after oxidation of 15, an alternative sequence was explored. Nitrile 15 was hydrated with the Ghaffar–Parkins complex (16) to provide primary amide 17 in 96% yield. At this stage, traditional oxidation methods were explored, which led to either decomposition, low yields, or recovered starting material (Fig. 3B; see the ESI‡ for further details). Presumably, the presence of the nucleophilic primary amide in 17 adversely affected the oxidation of the secondary alcohol group. Because of the issues with functional group incompatibility, Hofmann rearrangement prior to oxidation of the secondary alcohol was explored. With alcohol carboxamide 17 in hand, conditions to effect the Hofmann rearrangement were investigated (see Fig. 3D). Treating 17 with phenyliodoso-trifluoromethyl acetate (PIFA) resulted in decomposition of the starting material. Interestingly, treating carboxamide 17 with Pb(OAc)4 in a mixture of DMF/MeOH at room temperature, resulted in the formation of [3.2.1] bicycle 21 as the sole product of the reaction in 89% yield instead of the expected methyl carbamate (18). Presumably, this [3.2.1] bicyclic system arises from an initial oxidation of the primary carboxamide to generate the N-acyl nitrene (19), which then interacts with the indole C2–C3 double bond forming aziridine 20. The indole nitrogen then facilitates opening of the aziridine at the C2 position, driven by release of ring strain (as shown by the red arrows). A proton transfer then delivers 21. Interestingly, while reminiscent of aspeverin,19 this unique ring system does not translate to any natural product scaffolds that have been reported to date. On the basis of these results, the Hofmann rearrangement would have to be accomplished prior to the installation of the indole moiety in order to avoid the formation of [3.2.1] bicycle 21.
:2 ratio (α:β epimers) that was taken directly to the next reaction because of stability issues. The use of the Ghaffar–Parkins platinum complex failed to provide any desired hydrated product (12) and led to a complex product distribution.17 An alternative procedure for nitrile hydration reported by Lee and co-workers using Wilkinson's catalyst and acetaldoxime in toluene at reflux led only to decomposition of starting material.18 Presumably, the enol (not shown) can compete for the binding site of the metal center in these cases leading to decomposition pathways. Given the difficulty in performing the nitrile hydration after oxidation of 15, an alternative sequence was explored. Nitrile 15 was hydrated with the Ghaffar–Parkins complex (16) to provide primary amide 17 in 96% yield. At this stage, traditional oxidation methods were explored, which led to either decomposition, low yields, or recovered starting material (Fig. 3B; see the ESI‡ for further details). Presumably, the presence of the nucleophilic primary amide in 17 adversely affected the oxidation of the secondary alcohol group. Because of the issues with functional group incompatibility, Hofmann rearrangement prior to oxidation of the secondary alcohol was explored. With alcohol carboxamide 17 in hand, conditions to effect the Hofmann rearrangement were investigated (see Fig. 3D). Treating 17 with phenyliodoso-trifluoromethyl acetate (PIFA) resulted in decomposition of the starting material. Interestingly, treating carboxamide 17 with Pb(OAc)4 in a mixture of DMF/MeOH at room temperature, resulted in the formation of [3.2.1] bicycle 21 as the sole product of the reaction in 89% yield instead of the expected methyl carbamate (18). Presumably, this [3.2.1] bicyclic system arises from an initial oxidation of the primary carboxamide to generate the N-acyl nitrene (19), which then interacts with the indole C2–C3 double bond forming aziridine 20. The indole nitrogen then facilitates opening of the aziridine at the C2 position, driven by release of ring strain (as shown by the red arrows). A proton transfer then delivers 21. Interestingly, while reminiscent of aspeverin,19 this unique ring system does not translate to any natural product scaffolds that have been reported to date. On the basis of these results, the Hofmann rearrangement would have to be accomplished prior to the installation of the indole moiety in order to avoid the formation of [3.2.1] bicycle 21.
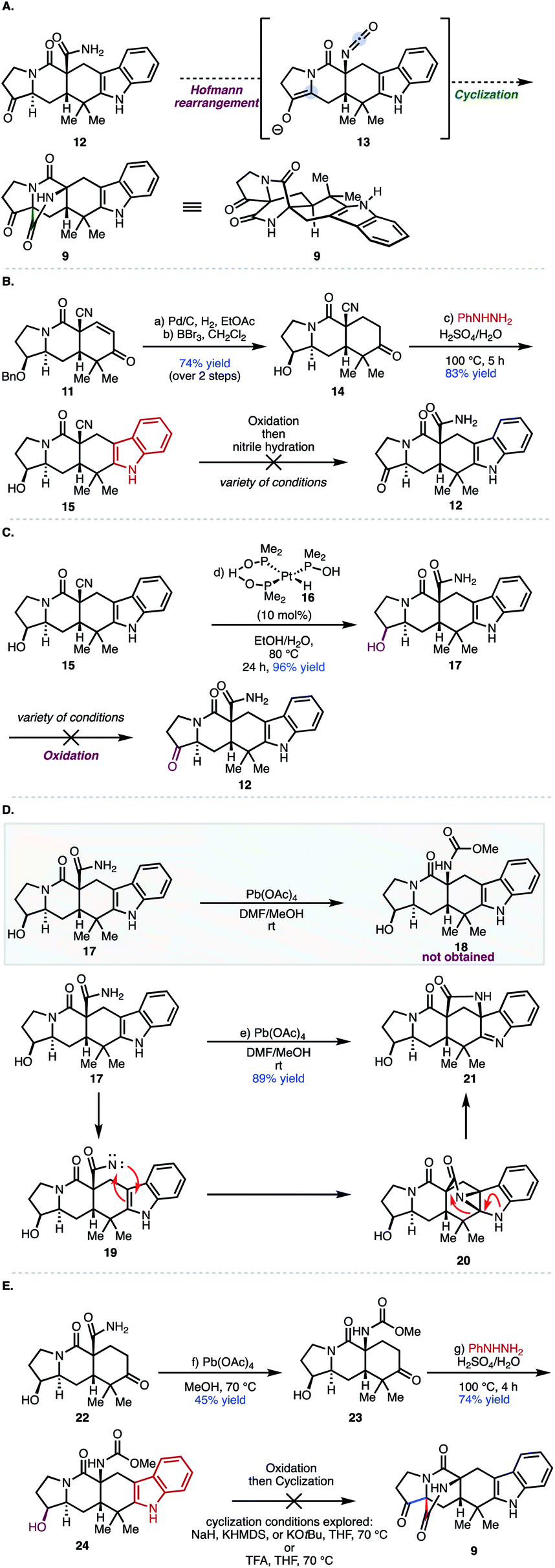 | ||
| Fig. 3 (A) Proposed one-pot construction of [2.2.2]-diazaoctane core; (B) attempts at synthesizing amide 12; (C) failed oxidation of 17; (D) formation of [3.2.1] bicycle 21. (E) attempted oxidation and cyclization of methylcarbamate 24; conditions: (a) Pd/C (10 wt%), H2, EtOAc, rt, 16 h; (b) BBr3, (4.6 equiv.), CH2Cl2, −78 °C, 15 min, 74% yield over 2 steps; (c) PhNHNH2 (4 equiv.), H2SO4/H2O (5% v/v), 100 °C, 5 h, 83% yield (d) 16 (10 mol%), EtOH/H2O, 24 h, 96% yield (e) Pb(OAc)4 (2 equiv.), DMF/MeOH, 2 h, rt, 89% yield (f) Pb(OAc)4 (5 equiv.), MeOH, 3.5 h, 70 °C, 45% yield (g) PhNHNH2 (4 equiv.), H2SO4/H2O (5% v/v), 100 °C, 4 h, 74% yield. | ||
Following from our prior studies, it was determined that Pb(OAc)4 in the presence of MeOH at room temperature was optimal for mediating the desired transformation to provide the methyl carbamate (23, Fig. 3E) accompanied by varying amounts of recovered starting material. After some optimization, it was established that elevated temperatures (70 °C) were required to attain full conversion of the starting material. However, reaction yields dropped dramatically upon scale-up and over 6 equivalents of Pb(OAc)4 were required to achieve complete consumption of the starting material. Nevertheless, methyl carbamate 23 was advanced through the Fischer indole synthesis to provide indole 24 in 74% yield. Subsequent oxidation with Dess–Martin periodinane (DMP) provided the desired ketone precursor (after oxidation of secondary alcohol highlighted in maroon), which was predisposed for the late-stage Dieckmann cyclization to afford the bicyclo[2.2.2]diazaoctane core. Unfortunately, treatment with a variety of bases (NaH, KHMDS, or KOtBu) or acid (e.g., TFA), were unsuccessful in providing the desired bicyclo[2.2.2] product (9). Presumably, the methyl carbamate is not sufficiently electrophilic for the cyclization step and/or the methoxide nucleofuge may subsequently serve as a better nucleophile in an undesired irreversible direction (cleaving the bond highlighted in blue in 9; Fig. 3E), leading to decomposition. Therefore, the installation of different carbamates was explored; carbamates bearing better leaving groups, such as phenols and polyfluoroalcohols, and thus less nucleophilic nucleofuges, emerged as an attractive option to minimize decomposition pathways.
Ultimately, a one-pot protocol was developed to access pentacyclic indole 25 (Scheme 1). It is worth noting that a modified work up procedure yielded two-step access to primary amide 22 from enone 11 and obviated the need for transition metal-catalyzed nitrile hydration. From 22, Hofmann rearrangement of the carboxamide was effected under mild conditions using (tosylimino)phenyl-λ3-iodane (PhINTs).20 Upon treatment with aqueous acid, the resulting isocyanate (23) was converted to the corresponding ammonium intermediate (24), which was directly subjected to phenylhydrazine to effect Fischer indolization, providing pentacyclic indole 25 in a single-pot operation from 22. Chemoselective amine carbamoylation of 25 in the presence of a secondary hydroxy group was achieved in high yield with phenyl chloroformate to afford phenyl carbamate 26. Oxidation of secondary alcohol 26 provided desired cyclization precursor 27. Treatment of 27 with K2CO3 in acetone yielded the bicyclo[2.2.2]diazaoctane core, presumably through Dieckmann cyclization of an intermediate enolate (generated from the ketone group) and the isocyanate group (generated in situ from the phenyl carbamate under basic conditions).13,21 Notably, the bicyclo[2.2.2]diazaoctane core was constructed in 5 steps from 11, which is an improvement over our prior work (10 steps from 11 in that case) and provides access to a wider range of congeners by leveraging the unsubstituted indole motif (vide infra).13
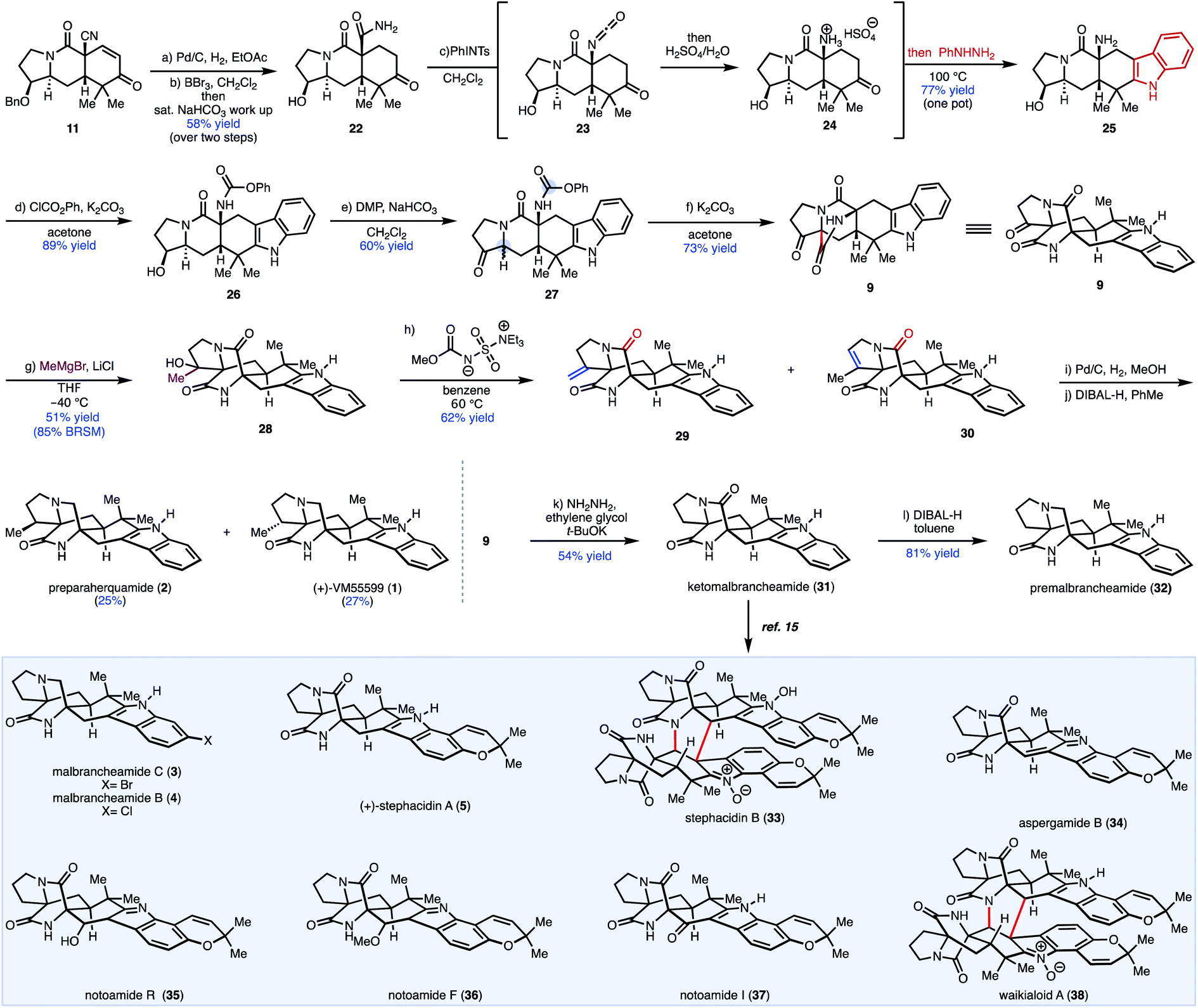 | ||
| Scheme 1 Synthesis of preparaherquamide (2), (+)-VM-55599 (1), ketomalbrancheamide (31), premalbrancheamide (32) and related congeners. (a) Pd/C (10 wt%), H2, EtOAc, rt, 16 h; (b) BBr3, (7.1 equiv.), CH2Cl2, −78 °C, 15 min then NaHCO3 (saturated aqueous), 24 h, 58% yield over 2 steps; (d) PhINTs (1.2 equiv.), CH2Cl2, rt, 2 h then H2SO4/H2O (5% v/v), 50 °C, 1 h; then PhNHNH2 (4 equiv.), 100 °C, 16 h, 77% yield (over 3 steps; one-pot) (e) ClCO2Ph (2.4 equiv.), K2CO3 (2 equiv.), acetone, rt, 8h, 89% yield. (e) DMP (1.5 equiv.), CH2Cl2, rt, 20 min, 60% yield; (f) K2CO3 (2 equiv.), acetone, 50 °C, 2 h, 74% yield; (g) MeMgBr (20 equiv.), LiCl (3 equiv.), THF, −40 °C, 2 h, 51% yield (85% BRSM) (h) Burgess reagent (10 equiv.), benzene, 60 °C, 2h, 63% yield (i) Pd/C (10 wt%), H2 (450 Psi), MeOH, rt, 16 h (j) DIBAL-H (19 equiv.), toluene, 0 °C to rt, 25% yield of 2 and 27% yield of 1. (k) NH2NH2 (1.1 equiv.), ethylene glycol, 70 °C, 17 h, then t-BuOK (5.0 equiv.), 170 °C, 2 h, 57% yield. (l) DIBAL-H (20 equiv.), toluene, 0 °C to rt, 81% yield. DMP = Dess–Martin periodinane. BRSM = Based on recovered starting material. | ||
The syntheses of preparaherquamide and (+)-VM55599 were accomplished through a four-step sequence that installed the requisite functionality on the five-membered ring (Scheme 1). Initial efforts toward olefination of ketone 9 were unsuccessful as Wittig olefinations lead to recovered starting material. A two-step nucleophilic addition followed by alcohol elimination was explored. For example, MeMgBr addition afforded tertiary alcohol 28 in low yields with significant recovered starting material. Presumably, side reactions like alpha deprotonation (enolization) result in low conversion. Re-subjecting the crude mixture to another cycle led to 48% yield of the desired tertiary alcohol (28). Efforts to attenuate the basicity of the Grignard reagent were unsuccessful as addition of cerium(III) chloride did not improve conversion and ultimately, it was found that the addition of LiCl led to a 51% yield (85% BRSM) of the desired tertiary alcohol 28.22 Treatment of 28 with the Burgess reagent gave a mixture of exocyclic (29) and endocyclic (30) alkenes in 71% yield (1:2 mixture). Hydrogenation of the mixture of alkenes (i.e., 29 and 30) using Pd/C, followed by chemoselective tertiary amide reduction with DIBAL-H, afforded epimeric natural products preparaherquamide (2) and (+)-VM-55599 (1) in 25% and 27% yield, respectively.
Having successfully accessed natural products bearing substituents on the pyrrolidine ring (ring A), we turned our efforts toward congeners derived from L-proline to showcase the utility of our unified approach (Scheme 1). Notably, from 9, indole alkaloids lacking substituents on ring A can be accessed through sequential reduction processes. For example, Wolff–Kishner reduction of the pyrrolidone ketone group in 9 yielded ketopremalbrancheamide (31, Scheme 1). From ketopremalbrancheamide, premalbrancheamide (32) was synthesized according to the precedent of Williams and co-workers.23,24 Thus, treating 31 with an excess of DIBAL-H effected chemoselective reduction of the tertiary amide. On the basis of our prior work on indole functionalization, ketomalbrancheamide (31) can be elaborated to malbrancheamides B and C, as well as stephacidins A and B, waikialoid A, aspergamide B, and finally notoamides F, I, and R.15
Conclusions
In conclusion, a unified approach was developed to access hexacyclic indole alkaloids that either possess or lack substituents on the cyclic amine. Specifically, studies that culminated in the total syntheses of (+)-VM55599 (1), preparaherquamide (2), and premalbrancheamide (31) as well access to ketomalbrancheamide (32) which can be elaborated to malbrancheamides B, C, stephacidin A, notoamides F, I and R, aspergamide B, and waikialoid A through our previously reported peripheral indole functionalization strategy is reported. This work serves as a blueprint for a unified approach to the synthesis of reverse-prenylated indole alkaloids possessing a bicyclo[2.2.2]diazaoctane core by addressing a key challenge posed by the diverse range of substitution found on the cyclic amine ring of the core framework. Our modular strategy hinged on leveraging a late-stage intermediate possessing a synthetic handle as well as rapid construction of the pentacyclic indole skeleton through a one-pot Hofmann rearrangement and Fischer indole synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. is grateful to the NIGMS for support of this work (R35 GM130345) J. B. R. thanks Bristol-Myers Squibb for a graduate fellowship. We are grateful for a Japan Society for the Promotion of Science Postdoctoral Fellowship to K. M., a Science without Borders Postdoctoral Fellowship to D. P. S. (Brazil; CSF/CNPq 200205/2014-5), the US National Science Foundation (NSF) Graduate Research Fellowship Program, the American Chemical Society Division of Organic Chemistry and the Hellman Graduate Awards Program (University of California (UC) Berkeley) for awards to E. V. M.-M. We thank College of Chemistry's NMR facility for resources provided and Dr Hasan Celik for NMR assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998.Notes and references
- K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Nat. Prod. Rep., 2018, 35, 532–558 RSC
.
-
(a) K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160–3174 RSC
-
(a) R. M. Williams, T. Glinka and E. Kwast, J. Am. Chem. Soc., 1988, 110, 5927–5929 CrossRef CAS
-
(a) C. A. Maryanoff, B. E. Maryanoff, K. Tang and K. Mislow, J. Am. Chem. Soc., 1973, 95, 5839–5840 CrossRef CAS
-
(a) F. C. Frebault and N. S. Simpkins, Tetrahedron, 2010, 66, 6585–6596 CrossRef CAS
- B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086–3087 CrossRef CAS PubMed
- S. B. Herzon and A. . G. Myers, J. Am. Chem. Soc., 2005, 127, 5342–5344 CrossRef CAS PubMed
- B. Zhang, W. Zheng, Z. Wang, D. Sun and C. Li, Angew. Chem., Int. Ed., 2016, 55, 10435–10438 CrossRef CAS PubMed
- K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160–3174 RSC
- E. M. Stocking, J. F. Sanz-Cervera, R. M. Williams and C. J. Unkefer, J. Am. Chem. Soc., 1996, 118, 7008–7009 CrossRef CAS
- A. E. Fraley and D. H. Sherman, FEBS J., 2020, 287, 1381–1402 CrossRef CAS PubMed
- C–H oxidations were unselective in the presence of tertiary amide (highlighted in blue). Efforts to reduce the tertiary amide in order to leverage remote amine functionalizations were unsuccessful due to competitive reduction of the nitrile and/or low conversions making this strategy not suitable for scale up.
- E. V. Mercado-Marin and R. Sarpong, Chem. Sci., 2015, 6, 5048–5052 RSC
- E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318–324 CrossRef CAS PubMed
- K. Mukai, D. P. de Sant'Ana, Y. Hirooka, E. V. Mercado-Marin, D. E. Stephens, K. G. M. Kou, S. C. Richter, N. Kelley and R. Sarpong, Nat. Chem., 2018, 10, 38–44 CrossRef CAS PubMed
-
Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., Hoboken, N. J., 2009 Search PubMed
- T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249 CrossRef CAS
- L. Lee, M. Kim, S. Chang and H.-Y. Lee, Org. Lett., 2009, 11, 5598–5601 CrossRef PubMed
- N.-Y. Ji, X.-H. Liu, F. P. Miao and M.-F. Qiao, Org. Lett., 2013, 15, 2327–2329 CrossRef CAS PubMed
- A. Yoshimura, M. W. Luedtke and V. V. Zhdankin, J. Org. Chem., 2012, 77, 2087–2091 CrossRef CAS PubMed
- For use of carbamates as precursors to isocyanates, see:
(a) D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 1999, 36, 148–172 CrossRef CAS
-
(a) T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS
- Y. Ding, T. J. Greshock, K. A. Miller, D. H. Sherman and R. M. Williams, Org. Lett., 2008, 10, 4863 CrossRef CAS PubMed
- For a formal synthesis of premalbrancheamide, see: J. G. Robins, K. Y. Kim, A. J. Chinn, J. S. Woo and J. R. Scheerer, J. Org. Chem., 2016, 81, 2293–2301 CrossRef CAS PubMed
Footnotes |
| † Dedicated to the memory of Prof. Robert M. Williams (1953–2020). |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02296a |
| This journal is © The Royal Society of Chemistry 2020 |