
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing metabolic alterations in breast cancer in response to molecular inhibitors with Raman spectroscopy and validated with mass spectrometry†
Xiaona
Wen‡
a,
Yu-Chuan
Ou‡
a,
Galina
Bogatcheva
b,
Giju
Thomas
c,
Anita
Mahadevan-Jansen
c,
Bhuminder
Singh
 b,
Eugene C.
Lin
d and
Rizia
Bardhan
*ef
b,
Eugene C.
Lin
d and
Rizia
Bardhan
*ef
aDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA
bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA
cVanderbilt Biophotonics Center, Vanderbilt University, Nashville, TN 37232, USA
dDepartment of Chemistry and Biochemistry, National Chung Cheng University, Chiayi 62106, Taiwan
eDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50012, USA. E-mail: rbardhan@iastate.edu
fNanovaccine Institute, Iowa State University, Ames, IA 50012, USA
First published on 20th August 2020
Abstract
Rapid and accurate response to targeted therapies is critical to differentiate tumors that are resistant to treatment early in the regimen. In this work, we demonstrate a rapid, noninvasive, and label-free approach to evaluate treatment response to molecular inhibitors in breast cancer (BC) cells with Raman spectroscopy (RS). Metabolic reprogramming in BC was probed with RS and multivariate analysis was applied to classify the cells into responsive or nonresponsive groups as a function of drug dosage, drug type, and cell type. Metabolites identified with RS were then validated with mass spectrometry (MS). We treated triple-negative BC cells with Trametinib, an inhibitor of the extracellular-signal-regulated kinase (ERK) pathway. Changes measured with both RS and MS corresponding to membrane phospholipids, amino acids, lipids and fatty acids indicated that these BC cells were responsive to treatment. Comparatively, minimal metabolic changes were observed post-treatment with Alpelisib, an inhibitor of the mammalian target of rapamycin (mTOR) pathway, indicating treatment resistance. These findings were corroborated with cell viability assay and immunoblotting. We also showed estrogen receptor-positive MCF-7 cells were nonresponsive to Trametinib with minimal metabolic and viability changes. Our findings support that oncometabolites identified with RS will ultimately enable rapid drug screening in patients ensuring patients receive the most effective treatment at the earliest time point.
Introduction
Dysregulation of the mitogen-activated protein kinases (MAPK) pathway plays a critical role in the proliferation and progression of breast cancer (BC). This dysfunction is achieved by activating the extracellular-signal-regulated kinase (ERK) (or Ras/Raf/MEK/ERK) signaling cascade,1 which has been associated with disease progression, metastasis, and treatment resistance in BC.2,3 Small molecule inhibitors (SMIs) that downregulate components of the ERK pathway have shown improved clinical outcomes in BC patients, and several clinical trials have shown successful outcomes (NCT03971409, NCT03395899).4 Current clinical measures rely on immunohistochemistry (IHC) analysis to guide initial treatments. But IHC is mostly qualitative, suffers from inter-patient variability, and often lacks accuracy when presented with heterogeneous tumors.5,6 Clinical decisions based on change in tumor size in response to therapy are inherently slow and low-throughput as a decrease in tumor burden can take several weeks. Therefore, an unmet clinical need exists for rapid, accurate, and cost-effective diagnostic tools that can guide the best treatment choices at the earliest time point and reduce mortality due to ineffective cancer therapies.Metabolic reprogramming is an emerging hallmark of cancer.7 Cancer cells reprogram their metabolism to maintain viability and proliferate to metastatic disease.8,9 Such metabolic rewiring goes beyond the well-known Warburg effect (glycolytic activity),10 and includes a cumulative change in phospholipids, amino acids, lipids and fatty acids.11,12 Emerging evidences now support that SMIs show an immediate inhibition of this altered metabolism in cancer cells before a reduction in tumor size is observed, presenting a more sensitive endpoint to treatment response.13,14 Metabolic response in clinical practice is traditionally measured with fluorodeoxyglucose-positron emission tomography (FDG-PET), which is both cost-prohibitive and has poor sensitivities to drug response.15 Further, FDG-PET is limited in evaluating treatment response of patients undergoing therapy with mitogen-activated protein kinase (MEK) inhibitors targeting the ERK pathway. This limitation of FDG-PET is primarily due to its ability to only visualize changes in glycolytic activity, whereas inhibition of MEK has minimal impact on glycolytic effect.16,17 It is also noteworthy that 18F-FDG uptake is often enhanced by endothelial proteins such as vascular endothelial growth factor, which is overexpressed in BC, resulting in false diagnosis.18
In this work, we address the limitations of current clinical measures by demonstrating the utility of Raman spectroscopy (RS) combined with multivariate statistical analysis for tracking alterations in multiple metabolites in response to SMIs of the ERK pathway in BC cells. RS is an established optical spectroscopy that measures inelastic scattering of photons induced by the vibrational bonds in samples.19–22 RS is a low-cost, rapid, label-free, and stain-free technique and has been utilized in breast cancer for diagnosis and surgical guidance.23–25 RS is also nondestructive allowing sample archival and retesting for accurate measure of therapeutic response.26–29 Here, we leveraged the advantages of Raman spectral mapping in its ability to first measure dynamic changes at the single-cell level with high sensitivity; second spatiotemporally resolve multiplexed metabolic changes; and third enable quantitative analysis. We treated triple-negative BC cell line MDA-MB-231 with Trametinib, a potent and specific MEK1/2 allosteric inhibitor,30 that downregulates MEK signaling in the ERK pathway. MDA-MB-231 has basal-like properties and is known to have BRAF and KRAS mutations, which represent two major oncogenic drivers in the ERK pathway (Scheme 1). Treatment response to Trateminib was assessed with RS and resulting data were analyzed with principal component analysis (PCA). PCA allowed us to reduce the data dimensions of the multiple peaks in RS to a smaller number of principal components or loadings that carry all of the relevant spectral information necessary for classification.31,32 We also treated MDA-MB-231 cells with phosphatidylinositol-3-kinase (PI3K) inhibitor (Alpelisib) and showed that these cells were resistant to this treatment. Finally, we studied an estrogen receptor (ER) positive BC cell line, MCF-7, which was resistant to Trametinib. Spectral trends from RS were then verified with mass spectrometry (MS), a gold standard in metabolomics. Note that MS by itself is not conducive to early and rapid drug screening since it is both time- and labor-intensive, and expensive. But RS trends validated with MS presents a complementary platform19,33 for rapid, high-throughput, and single-cell level drug screening (with RS) combined with ensemble analysis of large volume of cells (with MS) to simultaneously confirm the changes in multiple metabolites post-treatment (Scheme 1). By correlating the metabolic changes observed with RS and MS, we demonstrate that our approach can distinguish responders from nonresponders as a function of drug dosage, drug type targeting different signaling pathways, and cell type examining different BC lines. Our study goes beyond the traditional assays of cell viability and immunoblotting measurements, and highlights early and improved drug response selectivity with tremendous translational potential.
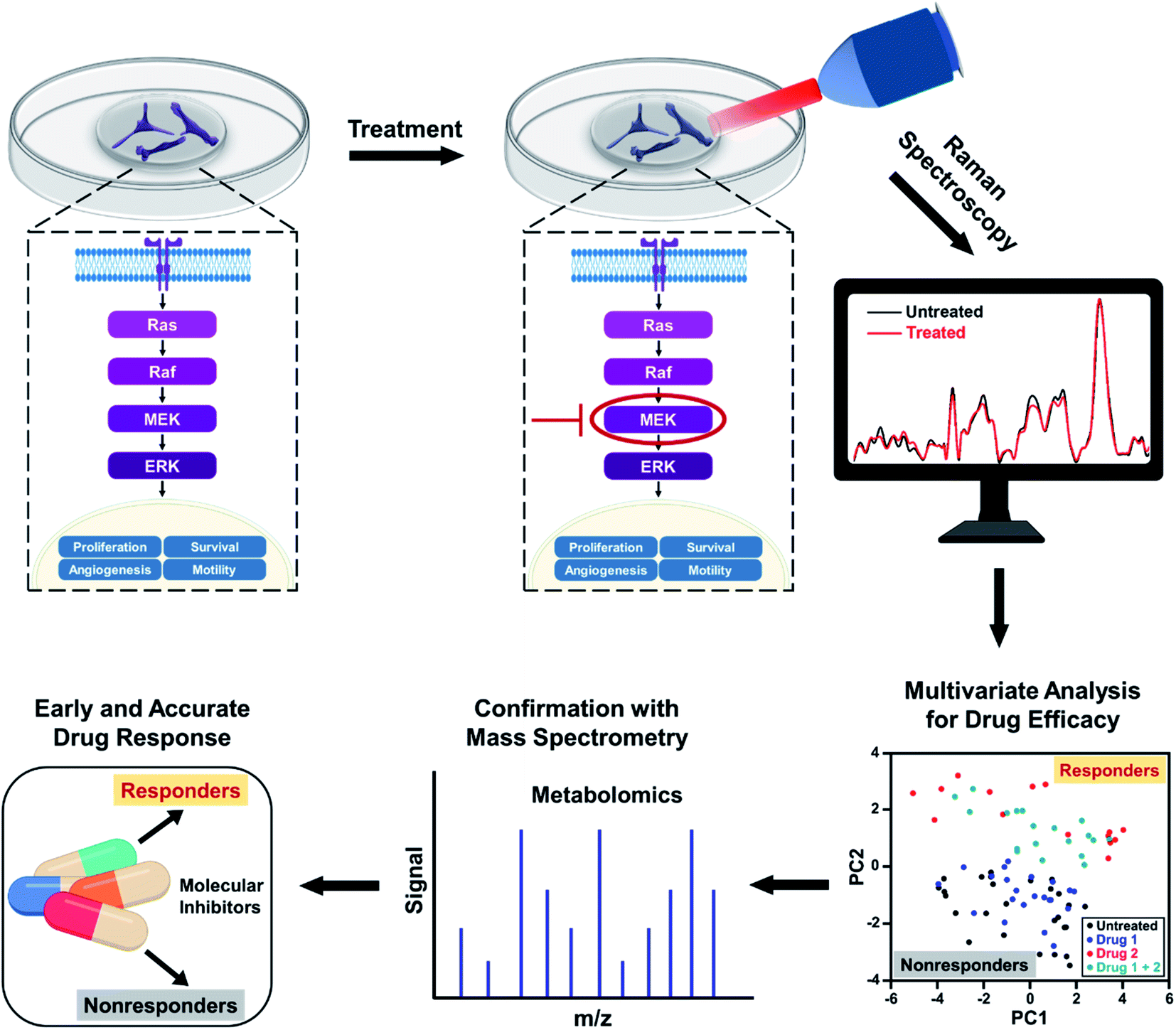 | ||
| Scheme 1 Schematic representation of metabolic changes probed in breast cancer cells with RS after treatment with small molecule inhibitors downregulating the ERK pathway. RS data combined with multivariate analysis and confirmed with mass spectrometry provide an early response to treatment distinguishing responders from nonresponders. | ||
Results and discussion
We recorded spatial Raman maps and measured the corresponding spectra of MDA-MB-231 cells in response to treatment with MEK inhibitor (MEKi), Trametinib. We measured ∼20–25 individual cells per treatment group (Fig. S1†) with a near-infrared 785 nm laser and 100× objective. Cells were seeded on poly-L-lysine coated calcium fluoride (CaF2) disks to minimize substrate-induced background, and fixed by 4% formaldehyde after 72 h of treatment. High resolution maps of a few cells were acquired in a single rectangular requisition at 2 μm by 2 μm resulting in 50–500 pixels per cell dictated by the size of the cell. Acquired spectra were then smoothed,34–37 background subtracted following literature methods38,39 and finally normalized for further analysis (details provided in the Experimental section). The representative original Raman spectra acquired from each cell and step-by-step spectral processing were shown in Fig. S2.† The mean normalized Raman spectra of the MDA-MB-231 cells treated with Trametinib at its working concentration (50 nM)40,41 showed significant changes in multiple peaks relative to untreated cells (Fig. 1a). To understand how the Raman footprint changed post-treatment, the difference spectrum was obtained by subtracting the mean normalized Raman spectrum of treated cells from the untreated cells (Fig. 1b). Here, positive differences indicate Raman peak intensities decreased after treatment, and negative differences indicate an increase in Raman peak intensities post-treatment. The key Raman peaks identified are shown with arrows, and the corresponding metabolites/proteins are listed in Table 1. These peak assignments were determined based on literature findings.42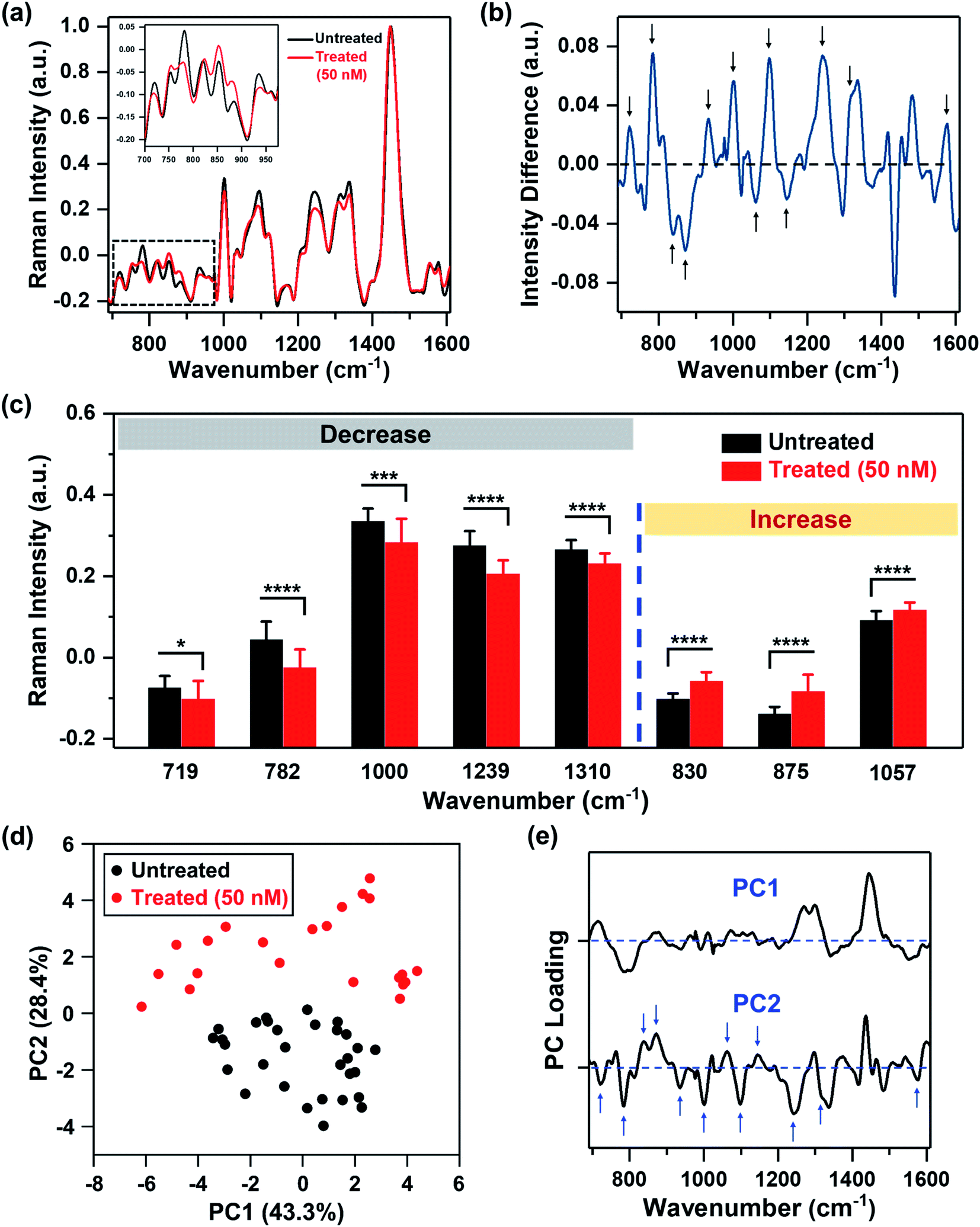 | ||
| Fig. 1 MDA-MB-231 cells treated with Trametinib and probed with RS. (a) Mean normalized Raman spectra of untreated cells (black) and cells treated with Trametinib at its working concentration 50 nM (red). Spectra were normalized to 1440 cm−1 biological peak. The inset shows a magnified view of the smaller wavenumber region. (b) Difference spectrum obtained from (a) by subtracting the mean normalized Raman spectrum of cells treated with Trametinib (50 nM) from the untreated cells to highlight changes in Raman footprint. Relevant peaks that either increase or decrease with treatment are shown with an arrow. (c) Selective Raman peaks that decreased with treatment including phosphatidylcholine (719 cm−1), DNA (782 cm−1), phenylalanine (1000 cm−1), amide III (1239 cm−1) and lipids & fatty acids (1310 cm−1). Selective Raman peaks that increased after treatment including tyrosine (830 cm−1), sphingomyelin (875 cm−1), and lipids (1057 cm−1). Here, * indicates p < 0.05, *** indicates p < 0.001, and **** indicates p < 0.0001 determined by Student's t-test. (d) Principal component analysis showing clustering of untreated cells relative to those treated with Trametinib represented in a scatter plot. (e) Corresponding PC loading showing both PC1 and PC2. Relevant peaks in PC2 that distinguish the treated from untreated group are shown with arrows. | ||
| Peak (cm−1) | Assignment |
|---|---|
| 719 | Phosphocholine and phosphatidylcholine |
| 782 | DNA |
| 787 | Phosphatidylserine |
| 830 | Tyrosine |
| 875 | Sphingomyelin |
| 935 | Proteins and amino acids |
| 1000 | Phenylalanine |
| 1057 | Lipids |
| 1094 | DNA |
| 1163 | Tyrosine |
| 1239 | Amide III |
| 1310 | Lipids and fatty acids |
| 1575 | DNA |
| 1582 | Phenylalanine |
We then quantified the changes in selective Raman peaks (Fig. 1c and S3†), and correlated the observed trends to metabolic response to SMIs of the ERK pathway in breast cancer. A decrease in the Raman peak at 719 cm−1 was observed post-treatment corresponding to phosphocholine and phosphatidylcholine (PC). Cancer cells are known to upregulate PC which is the most abundant phospholipid in cell membrane. Aberrant PC metabolism may result from enhanced choline kinase expression or activity.43 Due to the relevance of PC in tumor progression, it is considered as a predictive biomarker for monitoring tumor response.44,45 Emerging evidences suggest a strong correlation between choline metabolism and the ERK signaling cascade, where MEK inhibition decreases PC and its precursors,46,47 and this trend is well supported by our RS results. The difference spectrum also showed a decrease in the Raman footprint of DNA (782, 1094 and 1575 cm−1), indicating that the cytotoxic effect of Trametinib induced apoptosis and decrease in DNA replication.48 A decrease in cell viability with treatment response also reduced the ability of cells to synthesize proteins, which corresponded well with a decrease in Raman peaks of amide III (1239 cm−1), and phenylalanine (1000 and 1582 cm−1) that is an essential amino acid necessary for protein synthesis. A decrease in Raman footprint of lipids/fatty acids (1310 cm−1) was also observed, which is not surprising as cancer cells are known to rewire their metabolic circuit by dysregulating levels of lipids. Alterations in lipids often converge on the activation of the ERK pathway.49 Lipids and fatty acids metabolism is also mediated by crosstalk between receptor tyrosine kinases (RTKs) and downstream signaling via ERK.50 RTKs are overexpressed in most tumor types and activate cancer cells proliferation and survival. The epidermal growth factor receptor (EGFR) RTK is overexpressed in MDA-MB-231 cells,51 and EGFR aberrations are known to stimulate the ERK pathway.52 These correlations indicate that MEK inhibition with Trametinib is likely to reduce some lipids/fatty acids as observed with RS (and also validated with MS).
Our quantitative analysis also showed that several Raman peaks increased after treatment (Fig. 1c and S3†), including tyrosine (830 and 1163 cm−1), sphingomyelin (875 cm−1), and a subset of lipids (1057 cm−1). Tyrosine residues play a critical role in RTKs such as EGFR. EGFR overexpression results in phosphorylation of the protein tyrosine residues, which then activates downstream signaling via ERK.53 Therefore, we expect that MEK inhibition with Trametinib would result in dephosphorylation of tyrosine kinase. This dephosphorylation would manifest as an increase of tyrosine in Raman signal as the phosphorylated form of tyrosine decreased post-treatment as observed in our results. Sphingomyelin, a sphingosine-based phospholipid that exists in cell membranes, presents an anticancer role through ceramide release leading to apoptosis, and is known to augment the cytotoxicity of SMIs by activating sphingomyelinase enzyme activity.54,55 Therefore, an increase in Raman intensity of sphingomyelin post-treatment is expected given its potent biological role in cancer cell metabolism. Finally, a subset of lipids (1057 cm−1) increased post-treatment (confirmed with MS as discussed later), demonstrating that lipid metabolism in cancer is a highly complex phenomenon.
To identify major patterns in Raman spectra that distinguish the untreated cells from the treatment group (50 nM MEKi), we applied PCA to the data sets. The first and second principal component, PC1 and PC2, presented in a two-dimensional PC scatter plot (Fig. 1d) showed clear clustering between the groups with a variance level of 43.3% for PC1 and 28.4% for PC2. Each dot in the plot represents one cell/spectrum with dimension reduction. In the corresponding PC loading (Fig. 1e) spectral features in PC1 were likely due to the intrinsic variabilities between the cellular groups. Dominant peak positions in PC2 corresponded well to the peaks identified in the difference spectrum (Fig. 1b) supporting the metabolic alterations we discussed above.
Next, we probed the ability of RS in distinguishing responders from nonresponders by examining cellular response to treatment as a function of Trametinib concentration (1, 50 and 300 nM) (Fig. 2a). PCA scatter plot showed distinguishable clustering between the responsive and nonresponsive groups with a variance level of 45.4% and 19.6% for PC1 and PC2, respectively (Fig. 2b). Here, the cells treated with Trametinib at, and well above the working concentration (50 and 300 nM) were grouped on the negative side of PC2, and categorized as responders. The untreated cells and those treated with significantly low concentration of Trametinib (1 nM) were clustered together on the positive side of PC2, and categorized as nonresponders. The features in both PC1 and PC2 (Fig. S4†) had good concordance to the trends observed in Fig. 1e, and were also supported by ratiometric analysis of individual Raman peaks (Fig. S5†) as a function of Trametinib concentration. Ratiometric analysis of the different metabolites is well aligned with our discussion above on metabolic changes in response to treatment with MEKi.
 | ||
| Fig. 2 RS distinguishing responders from nonresponders as a function of Trametinib dosage. (a) Mean normalized Raman spectra of untreated MDA-MB-231 cells and those treated with various concentrations (1, 50 and 300 nM) of MEKi (Trametinib). Spectra were normalized to 1440 cm−1 biological peak. (b) PC scatter plot showing clustering of cells based on Trametinib concentrations differentiating responders from nonresponders. | ||
In addition to the ERK pathway, the mammalian target of rapamycin (mTOR) pathway consisting of the PI3K/AKT/mTOR cascade is also upregulated in BC.56 Whereas multiple SMIs, such as PI3K inhibitor (Alpelisib), have shown favorable outcomes in clinical trials (NCT02155088, NCT02998476, NCT01241500, etc.), BCs that are estrogen receptor (ER), progesterone receptor (PR) and/or human epidermal growth factor receptor 2 (HER2) negative are poorly responsive to these treatments. MDA-MB-231 cells have ER, PR, and HER2 triple-negative status and are known to be resistant to Alpelisib and other SMIs of the mTOR pathway.57,58 We performed RS and investigated the cellular response of MDA-MB-231 cells upon treatment with different concentrations of Alpelisib (0.5, 1 and 10 μM), where the working concentration of Alpelisib is ∼1 μM. Besides a slight decrease in amino acids (935 and 1000 cm−1) upon treatment with Alpelisib, overall minimal changes were observed in Raman peaks corresponding to DNA, phospholipids, lipids and fatty acids (Fig. S6a†) identified earlier. The PC scatter plot of PC1 vs. PC2 (Fig. 3a) clearly showed that across all concentrations of PI3Ki, there was no clustering demonstrating that MDA-MB-231 cells were resistant to Alpelisib even beyond its working concentration. The corresponding PC loading is shown in Fig. S6b.†
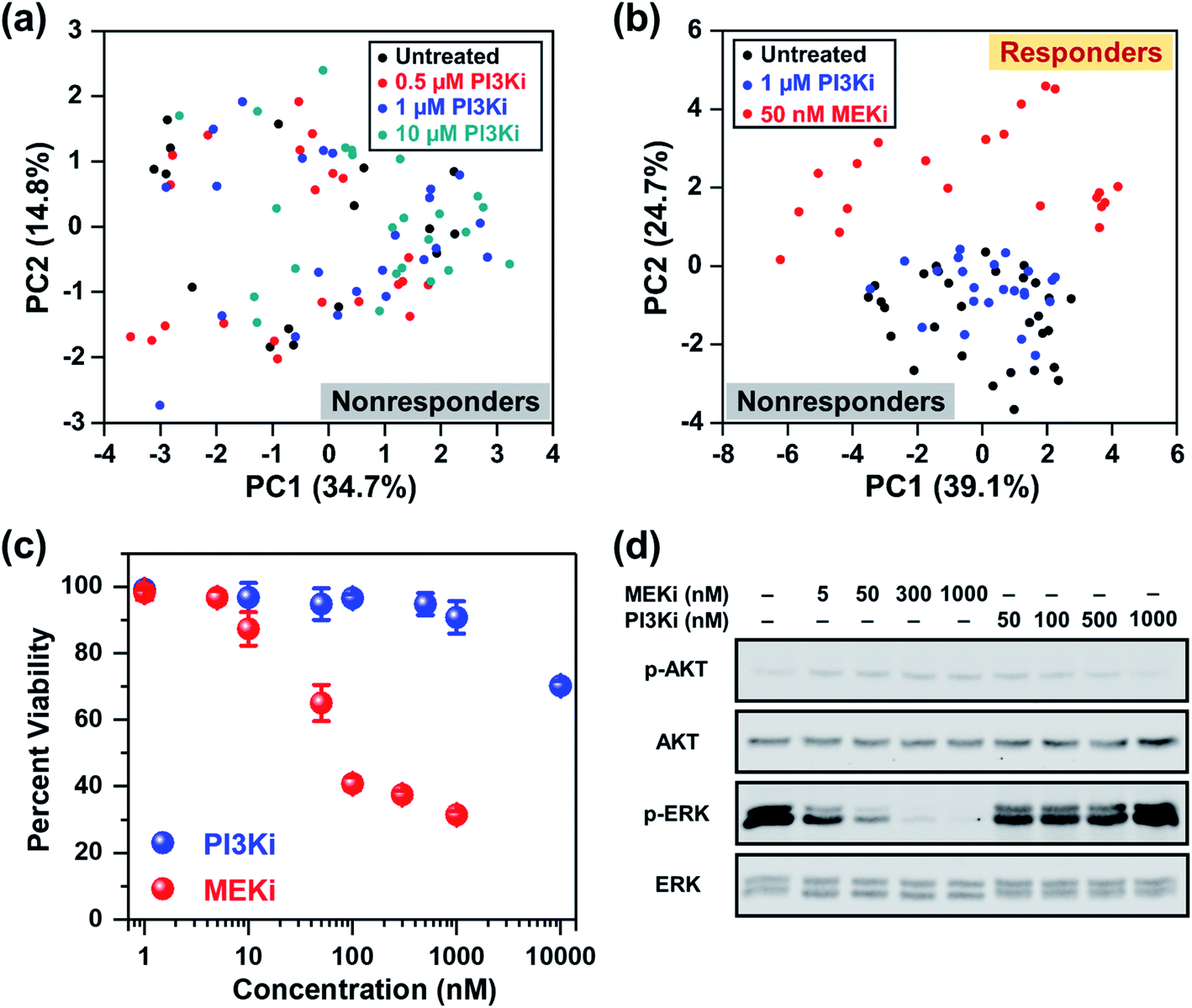 | ||
| Fig. 3 RS distinguishing responders from nonresponders as a function of drug type. (a) PCA scatter plot comparing MDA-MB-231 cells in response to different concentrations (0.5, 1 and 10 μM) of PI3Ki (Alpelisib) showing no distinct clustering among groups. (b) PCA scatter plot comparing untreated cells (black) to those treated with 1 μM of PI3Ki (Alpelisib, blue) and 50 nM of MEKi (Trametinib, red). Clear clustering of cells was observed for those responsive to treatment relative to nonresponders. (c) MTT viability assay of cells treated for 72 h with MEKi (Trametinib) or PI3Ki (Alpelisib) at 0–10 μM concentrations (n = 4 per concentration). Cell viability was measured at 540 nm. All data were presented as mean ± standard deviation. (d) Immunoblotting analysis of cells in response to treatment with MEKi (Trametinib) or PI3Ki (Alpelisib) at increasing concentrations showed reduced ERK1/2 phosphorylation for cells that responded to treatment. | ||
Next, we examined the ability of RS in distinguishing responders from nonresponders as a function of drug type. Here, cells were treated with 1 μM of PI3Ki or 50 nM of MEKi and compared to untreated cells. The mean normalized Raman spectra of the MDA-MB-231 cells treated with MEKi (50 nM) showed significant changes in multiple peaks compared to untreated cells or those treated with PI3Ki (1 μM) (Fig. S7a†). The spectral differences between untreated cells and those treated with PI3Ki (1 μM) were minimal. The PC scatter plot further confirmed the results and clearly differentiated cells that responded and those that were resistant to treatment (Fig. 3b). The untreated cells and those treated with PI3Ki (1 μM) were clustered together and defined as nonresponders, whereas the cells treated with MEKi (50 nM) were grouped and defined as responders. Additionally, PC1 and PC2 accounted for 39.1% and 24.7% of the total variance, respectively. The corresponding PC loading (Fig. S7b†) shared similar features as the PC loading shown in Fig. 1e. These results suggested that the metabolites summarized in Table 1 also played a critical role in differentiating BC cells response to PI3Ki (Alpelisib). Trends observed in RS were further verified with cell viability (MTT) assay (Fig. 3c). MDA-MB-231 cells were treated for up to 72 h and the percentage of viable cells was measured. MTT assay supported our findings with RS, and showed that cells were not responsive to PI3Ki (Alpelisib) where 90% of the cells were viable at the working concentration of the drug (1 μM) and 75% were viable at 10× higher concentration. The cells were highly responsive to MEKi (Trametinib) in a dose-dependent manner with ∼30% cells viable at the highest concentration of drug evaluated in our study (1 μM MEKi). We also performed immunoblotting assay (Fig. 3d) to further confirm the trends observed in RS and MTT assay, and determine if the observed trends among responders (MEKi treated) resulted from inhibition of the ERK pathway. Activation of the ERK pathway produces an abundance of phosphorylated ERK (p-ERK) which then stimulates downstream signaling. Therefore, inhibition of the ERK pathway with Trametinib reduced p-ERK, but minimal changes were observed in p-ERK for PI3K inhibition in BC cells. Immunoblotting results supported RS results discussed in Fig. 1c where upon treatment an increase in tyrosine was observed resulting from a decrease in phosphorylated tyrosine kinase. Western blot also showed neither inhibitors changed the levels of phosphorylated AKT, which is the downstream effector of PI3K in the mTOR cascade. We also examined the impact of combinatorial treatment of Trametinib (50 nM) + Alpelisib (1 μM) in MDA-MB-231 cells (Fig. S8†). PCA showed cells treated with combination therapy clustered with those treated with Trametinib alone. Additional details are provided in SI.
To further validate the metabolic changes observed in RS in response to molecular inhibitors, we performed LC-MS/MS on cell extracts (Fig. 4). Mass spectrometry (MS) is a gold standard in metabolomics sampling a large volume of cells and identifying specific metabolites with both high selectivity and sensitivity. MDA-MB-231 cells were treated with MEK (Trametinib, 50 nM) or P13K (Alpelisib, 1 μM) inhibitor at their working concentration. After 72 h of treatment, cells were resuspended in PBS, dried, weighted and then analyzed with MS. The metabolites assessed with MS were represented via a heat map where numbers shown are the ratio of integrated area of treated cells to those of untreated control cells. The ratio <1 indicates metabolites decreased with treatment, and ratio >1 indicates metabolites increased post-treatment. Representative metabolites were grouped into three main categories: amino acids (AAs), membrane phospholipids, and lipids and fatty acids. In response to treatment with Trametinib, the observed decrease in AAs was consistent with findings in the literature as cancer cells are known to have upregulated de novo synthesis of AAs and an increase in corresponding membrane transporters and metabolic enzymes.59,60 AAs are the building blocks of proteins and intricately participate in protein synthesis. Therefore, a decrease in AAs in BC cells after treatment was likely contributed by an inhibition of these catabolic enzymes. Further, decrease in cell viability post-treatment with Trametinib also reduced AAs and subsequent protein synthesis, a trend that was supported by RS (Fig. 1c, S3 and S5†). Additionally, a decrease in phosphatidylcholine and other choline precursors also supported the trends observed in RS and was consistent with literature findings noted earlier that demonstrate a decrease in choline metabolism with MEK inhibition. However, not all membrane phospholipids decreased with MEK inhibition in BC cells. MS measurement supported RS trends of increase in sphingomyelin after treatment with Trametinib, explained in the Fig. 1c discussion. We also observed a striking decrease in lipids and fatty acids metabolism with MEK inhibition supported by our RS findings as well (Fig. 1c and S5†). Note however, MS showed a subset of lipids increased post-treatment which was also observed in RS where the lipid peak at 1057 cm−1 (Fig. 1c) increased in intensity. These findings suggested that a future lipidomics study will be necessary to unravel the crosstalk between lipids/fatty acids metabolism and pro-oncogenic downstream signaling pathways. For cells treated with Alpelisib, a decrease in amino acids was observed in MS which likely resulted from a decrease in protein synthesis, also observable as a decrease in the RS footprint at 935 and 1000 cm−1 (Fig. S6a†). But overall, treatment with Alpelisib resulted in minimal changes in both membrane phospholipids, and lipids and fatty acids in MS. These results collectively show that MS validates the findings from RS, and these two techniques are complementary in providing rapid drug screening and corresponding metabolic rewiring at the cellular level.
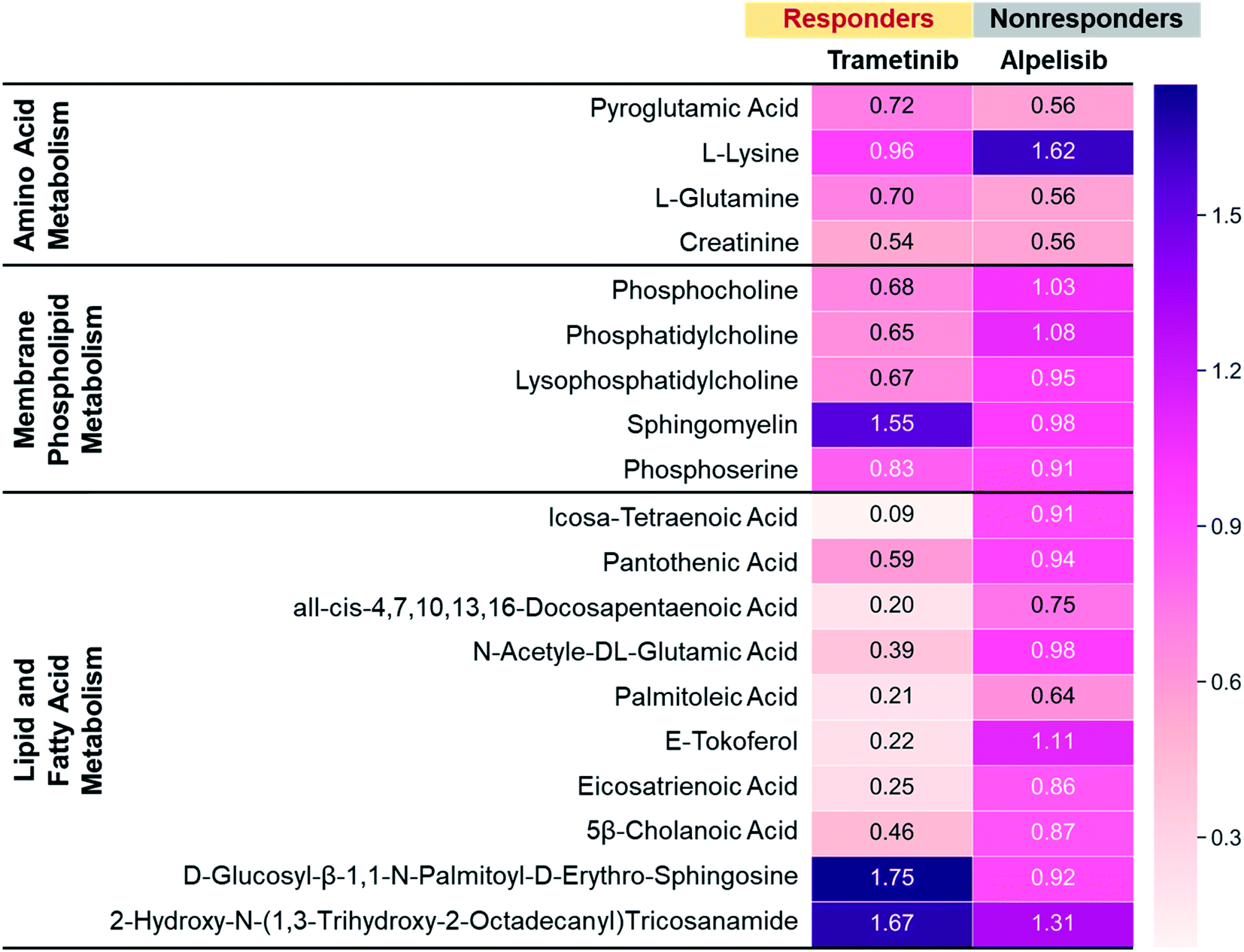 | ||
| Fig. 4 Mass spectroscopic analysis of MDA-MB-231 cells treated with Trametinib (MEKi) or Alpelisib (PI3Ki) at their working concentrations (MEKi: 50 nM; PI3Ki: 1 μM). The numbers were a ratio of treated cells to untreated cell control where closer to 1 indicated minimal changes in metabolites. All differential features (samples vs. controls) had a p value of <0.05. | ||
Thus far, our approach has focused on distinguishing responders from nonresponders within the same cell line as a function of dosage and drug type. Next, we evaluated if RS can determine treatment response in an ER-positive cell line, which represents a large subset of BC. We chose MCF-7 as a model of ER-positive cell line61 with no known KRAS mutations, and resistance to selective MEK inhibitors. MCF-7 cells were treated with Trametinib at various concentrations (1 nM, 50 nM and 1 μM), and cellular response was assessed with RS. Representative original Raman spectra of MCF-7 cells, and Raman spectra after smoothing and background subtraction were shown in Fig. S9.† Minimal changes were observed in most of the Raman peaks as a function of drug concentration (Fig. 5a) and PCA scatter plot corroborated that MCF-7 cells were resistant to Trametinib with no clear clustering of cells for any of the concentrations (Fig. 5b). These findings were further confirmed with cell viability assay (Fig. 5c) which showed MCF-7 cells were not responsive to MEK inhibition with Trametinib where 80% of the cells remained viable even at a significant high concentration of 1 μM. A comparison of the PC loading for MCF-7 cells treated with Trametinib (Fig. S10†) with those of MDA-MB-231 cells (Fig. 1e, S4, S7b and S8c†) showed significant differences in treatment response between the two BC cells supporting the cell viability results of treatment resistance. A few features in PC loading of MCF-7 (Fig. S10,† indicated by arrows) arose from DNA (782 cm−1), amino acids (856 cm−1), proteins (935 and 1123 cm−1), and lipids/fatty acids (1330 cm−1) were likely due to decrease in cell viability at the highest concentration of Trametinib as observed in RS (Fig. 5a) and viability assay (Fig. 5c).
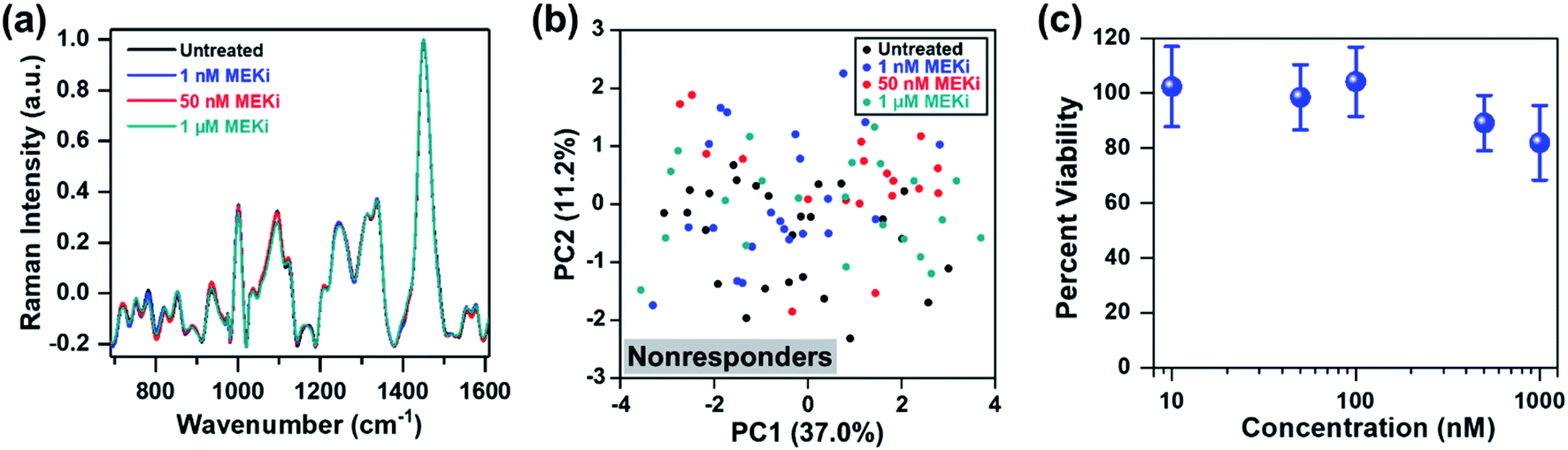 | ||
| Fig. 5 MCF-7 cells treated with Trametinib and interrogated with RS. (a) Mean normalized Raman spectra of untreated cells (black) relative to those treated with MEKi (Trametinib) at 1 nM (blue), 50 nM (red) and 1 μM (cyan). Spectra were normalized to 1440 cm−1 biological peak. Minimal changes in Raman footprint were observed. (b) PCA scatter plot comparing untreated cells with those treated with Trametinib showing no distinct clustering among groups. (c) MTT viability assay of cells treated for 72 h with Trametinib at various concentrations (n = 4 for each concentration). Cell viability was measured at 540 nm. All data were presented as mean ± standard deviation. | ||
Conclusions
In summary, this work addresses a critical need in early and accurate drug screening which goes beyond the traditional approaches (e.g., viability assay and immunoblotting) in distinguishing responders from nonresponders. Here, we performed RS to probe metabolic reprogramming in breast cancer cells, and evaluated treatment response to potent and clinically approved SMIs by examining alterations in metabolites. Our findings demonstrate that RS combined with multivariate analysis presents a powerful platform with tremendous clinical significance differentiating BC cells that are responsive to SMIs as a function of dosage, drug type, and cell type. Findings with RS were verified with MS, a workhorse in metabolomics, by examining various oncometabolites. Primarily we showed that triple-negative MDA-MB-231 BC cells were responsive to Trametinib, an inhibitor of the ERK pathway, and nonresponsive to Alpelisib, an inhibitor of the mTOR pathway, supported by a collective change in DNA, membrane phospholipids, amino acids, lipids and fatty acids. We also demonstrated that ER-positive MCF-7 BC cells were resistant to Trametinib with minimal changes in metabolites and no classification among treatment groups as seen in PCA. Our results suggest that these oncometabolites represent an important hallmark of cancer that can be targeted to both treat cancer and understand resistance mechanisms.62 Whereas this work demonstrated our capability in 2D cultures, our future work will focus on the utility of organotypic cultures to understand metabolic rewiring in a system that recapitulates the human tumor microenvironment. As our understanding expands in how cancer cells become addicted to specific metabolic pathways,63 we anticipate that RS validated with MS and combined with sophisticated machine learning algorithms64,65 will enable us to identify novel anticancer drugs that target these metabolic vulnerabilities. Ultimately, we expect this platform will have significant impact on cancer patients identifying those resistant to drug combinations even before therapy begins and guiding clinical decisions to an optimal treatment plan.Experimental
Cell culture
MDA-MB-231 cells were purchased from the American Type Culture Collection (ATCC, HTB-26). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Thomas Scientific). MCF-7 cells (ATCC, HTB-22) were cultured in Eagle's minimum essential medium (EMEM, ATCC) supplemented with 10% FBS (ATCC), 1% penicillin/streptomycin (Gibco), and 0.01 mg mL−1 human recombinant insulin (invitrogen). The cells were maintained at 37 °C and 5% CO2. Afterwards, MDA-MB-231 or MCF-7 cells were seeded on 6 well plates at approximately 60% confluency. Once the cells have attached to the plates, different concentrations of MEKi (Trametinib, Selleck Chemical), PI3Ki (Alpelisib, Selleck Chemical), or a combination of two in complete media were added. After 72 h of incubation, old media were removed and the cells were trypsinized. Detached cells were centrifuged at 125 × g for 7 min to remove dead cells and then re-seeded on poly-L-lysine (Sigma) coated calcium fluoride windows (CaF2, Crystran). Once the cells reattached on the CaF2 disks, media were removed. Next, the cells on CaF2 disks were washed three times with phosphate buffer saline (PBS), fixed by 4% formaldehyde (methanol-free) at room temperature for 10 min, washed with PBS three times again, and dried at room temperature prior to Raman mapping.In vitro Raman mapping and analysis
The cells on CaF2 disks were visualized using a Renishaw inVia Raman microscope system. Brightfield images of the cells were captured with a 100× objective. A rectangular area capturing 3–4 cells was selected for Raman mapping at 2 μm by 2 μm resolution. Raman spectra for the entire map were obtained with a 785 nm laser (10 mW) with 1200 lines per mm grating with 10 s acquisition time. Afterwards, cosmic ray removal was performed with nearest neighbor method using the Renishaw WiRE 3.4 software. A custom MATLAB (R2019a) code was used to perform smoothing and background correction. The spectra were first smoothed by using the Savitzky and Golay filter with fifth order and coefficient value of 47 (points). An automated and modified polyfit method was applied to remove fluorescent background by using a 11th order polynomial with a threshold of 0.0001. To eliminate non-cell pixels from the rectangular map, biological peak at 1440 cm−1 was selected to generate the cellular masks. The pixels were considered cell, or “mask” when the intensities were higher than the set threshold. A Gaussian function was utilized to smooth the edges of the cell mask. The brightfield optical images were used to ensure the accuracy of the final cell masks. Clusters of pixels in the Raman map were then averaged, normalized using standard normal variate method and defined as one cell for PCA analysis. PCA was performed by using the MATLAB built-in “pca” function where the analyzed data were mean centered by default. Therefore, the variations of PC score shown in Fig. 1d, 2b, 3a, b, 5b and S8b† represent the results of PCA. These variations were not contributed by any biases from the raw data. Two principal components were plotted to discriminate each treatment group.Viability assay
MDA-MB-231 or MCF-7 cells were passaged and seeded on 96 well plates. Once the cells were attached to the plates overnight, different concentrations of MEKi (Trametinib) or PI3Ki (Alpelisib) in complete media were added to the wells. After 72 h of treatment, old media in each well were removed and replaced with 100 μL of fresh media mixed with 10 μL of 12 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Thermo Fisher Scientific). After 3 h of incubation, 85 μL of the media solution in each well was removed and 50 μL of dimethyl sulfoxide (DMSO) was added to solubilize and dissolve the formazan. The plates were incubated at 37 °C for 10 min, and the absorbance of each well was read at 540 nm using a Biotek Synergy H1 plate reader.Western blot
MDA-MB-231 or MCF-7 cells were passaged, counted, and seeded on 6 well plates. The number of cells seeded on each well was counted by a scepter to ensure that each sample contained the same number of cells and proteins. Once the cells were attached to the plates overnight, different concentrations of MEKi (Trametinib) or PI3Ki (Alpelisib) in complete media were added to the wells. After 6 h of treatment, lysis buffer was added to the cells and incubated at 4 °C for 30 min. Lysates were then centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 15 min. Equal amounts of proteins were mixed with Laemmli buffer and boiled for 5 min. The samples were then loaded on 10% SDS-PAGE, and subsequently transferred onto nitrocellulose membranes through electrophoresis overnight in a cold room. The membranes were blocked before adding primary antibodies. Primary and secondary antibodies were prepared in the blocking buffer. All antibodies were purchased from Cell Signaling Technology. Primary antibodies were against AKT (9272S), phosphor-AKT (9271S), ERK 1/2 (9102S) and phosphor-ERK 1/2 (9101S).
000g at 4 °C for 15 min. Equal amounts of proteins were mixed with Laemmli buffer and boiled for 5 min. The samples were then loaded on 10% SDS-PAGE, and subsequently transferred onto nitrocellulose membranes through electrophoresis overnight in a cold room. The membranes were blocked before adding primary antibodies. Primary and secondary antibodies were prepared in the blocking buffer. All antibodies were purchased from Cell Signaling Technology. Primary antibodies were against AKT (9272S), phosphor-AKT (9271S), ERK 1/2 (9102S) and phosphor-ERK 1/2 (9101S).
Mass spectrometry
MDA-MB-231 cells were first seeded on 6 well plates and then treated with either MEKi (Trametinib) or PI3Ki (Alpelisib) for 72 h. After treatment, cells were washed with 1× PBS three times and detached by trypsin (Gibco). Trypsin was removed by centrifuge at 125 × g, and cells were resuspended in a small volume of 1× PBS. A lyophilizer (Labconco) was then utilized to remove any solvent. Dried cells were stored at −80 °C. The snap frozen cell pellets were weighed and reconstituted in a specified volume of methanol/water (3:1) to yield a cell density of 12.4 mg mL−1. Cells were then flash frozen on dry ice and thawed at 5 °C three times to facilitate complete lysis. An aliquot of cell lysate (700 L) was combined with chloroform (500 L) in a clean glass vial, vortexed vigorously, and centrifuged at 3000 × g for 5 min to achieve efficient phase separation. The aqueous (top) layer containing polar metabolites was transferred to a clean Eppendorf tube, evaporated to dryness under a gentle stream of nitrogen gas, and reconstituted in 200 L of acetonitrile/water (2:1) containing 250 M tyrosine (phenyl-3,5-d2) internal standard. Forty-two amino acid analogues were measured in the aqueous fraction by a targeted HILIC-MS/MS method. The chloroform (bottom) layer containing nonpolar metabolites was transferred to a clean glass vial, evaporated to dryness under a gentle stream of nitrogen gas, and reconstituted in 170 L of methanol containing 3 M carbamazepine and 30 L of a 10× solution of SPLASH LIPIDOMIX standard (Avanti Polar Lipids). The chloroform fraction was analyzed by reverse phase LC-high resolution MS for untargeted metabolomics.
For analysis of amino acids, LC-MS/MS analysis was performed using an Acquity UPLC system (Waters) interfaced with a TSQ Quantum Ultra™ triple-stage quadrupole mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was equipped with an IonMax source housing and a heated electrospray ionization (ESI) probe. Individual reference standards of all analytes were infused into the mass spectrometer for the optimization of ESI and selected reaction monitoring (SRM) parameters. Detection was based on SRM using the following optimized source parameters (positive ionization): spray voltage at 5 kV; capillary temperature at 300 °C; vaporizer temperature at 185 °C; tube lens of 52 V at m/z 184; N2 sheath gas pressure 50 (arbitrary units); and N2 auxiliary gas pressure 5 (arbitrary units). Data acquisition and quantitative spectral analysis were done using Thermo-Finnigan Xcalibur version 2.0.7 SP1 and Thermo-Finnigan LCQuan version 2.5.6, respectively. A Zic-cHILIC analytical column (3 μm, 2.1 × 150 mm, Merck SeQuant) was used for all chromatographic separations. Mobile phases were made up of 0.2% acetic acid and 15 mM ammonium acetate in (A) H2O/CH3CN (9:1) and in (B) CH3CN/CH3OH/H2O (90:5:5). Gradient conditions were as follows: 0–2 min, B = 85%; 2–5 min, B = 85–30%; 5–9 min, B = 30%; 9–11 min, B = 30–85%; and 11–20 min, B = 85%. The flow rate was maintained at 300 L min−1, and the total chromatographic run time was 20 min. The sample injection volume was 10 L. The autosampler injection valve and the sample injection needle were flushed and washed sequentially with mobile phase A (1 mL) and mobile phase B (1 mL) between each injection.
For analysis of untargeted lipidomics, discovery metabolomics data were acquired using a Vanquish ultrahigh performance liquid chromatography (UHPLC) system interfaced to a Q Exactive HF quadrupole/orbitrap mass spectrometer (Thermo Fisher Scientific). Samples were injected a total of four times. Two injections were made in positive ESI mode followed by two injections in negative mode. Chromatographic separation was performed with a reverse-phase Acquity BEH C18 column (1.7 m, 2.1 × 150 mm, Waters) at a flow rate of 300 L min−1. Mobile phases were made up of 10 mM ammonium acetate in (A) H2O/CH3CN (1:1) and in (B) CH3CN/iPrOH (1:1). Gradient conditions were as follows: 0–1 min, B = 20%; 1–8 min, B = 20–100%; 8–10 min, B = 100%; 10–10.5 min, B = 100–20%; and 10.5–15 min, B = 20%. The total chromatographic run time was 20 min, and the sample injection volume was 10 L. Mass spectra were acquired over a precursor ion scan range of m/z 100 to 1200 at a resolving power of 30000 using the following ESI source parameters: spray voltage at 5 kV (3 kV in negative mode); capillary temperature 300 °C; S-lens RF level at 60 V; N2 sheath gas pressure 40 (arbitrary units); N2 auxiliary gas pressure 10 (arbitrary units); and auxiliary gas temperature at 100 °C. MS/MS spectra were acquired for the top-five most abundant precursor ions with an MS/MS AGC target of 105, a maximum MS/MS injection time of 100 ms, and a normalized collision energy of 30 eV. Chromatographic alignment, peak picking, and statistical comparisons were performed using Compound Discoverer version 3.0 (Thermo Fisher Scientific). All differential features (samples vs. controls) having a p value of <0.05 and a fold change of >1.5 were processed for molecular matches in the Chemspider, mzCloud, HMDB, and KEGG databases based on precursor ion exact masses (±5 ppm) and MS/MS fragmentation patterns. Metabolite matches were then filtered to exclude biologically irrelevant drugs and environmental contaminants. The finalized list of putative identifications was mapped to relevant biological pathways using the Metabolika software module. Pooled QCs were injected to assess the performance of the LC and MS instruments at the beginning and at the end of each sequence. The results of mass spectrometry were visualized using the heatmap generated by Python.
Statistical analysis
All data were presented as mean ± standard deviation. Differences between two groups were assessed using GraphPad Prism 8 with unpaired two-sided Student's t-tests for the calculation of p values. Here, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001 and **** indicates p < 0.0001.Author contributions
XW, YO and RB conceived the idea of the project and directed the project. XW and YO performed the majority of experiments. XW, YO and RB wrote the manuscript. ECL, GT and AM-J helped with Raman data analysis. BS and GB assisted with the immunoblotting experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XW acknowledges support from the National Science Foundation (NSF) grant CMMI-1634856. YO was supported through the National Center for Advancing Translational Sciences CTSA award and CDMRP Peer Reviewed Cancer Research Program grant W81XWH1810139. BS acknowledges support from the VUMC Faculty Research Scholars and VICC SPORE P50CA236733 Career Enhancement Program. RB acknowledges support from the CDMRP Peer Reviewed Cancer Research Program grant W81XWH2010620.References
- K. K. Haagenson and G. S. Wu, Cancer Metastasis Rev., 2010, 29, 143–149 CrossRef CAS.
- C. Bartholomeusz, A. M. Gonzalez-Angulo, P. Liu, N. Hayashi, A. Lluch, J. Ferrer-Lozano and G. N. Hortobágyi, Oncologist, 2012, 17, 766–774 CrossRef CAS.
- I. C. Salaroglio, E. Mungo, E. Gazzano, J. Kopecka and C. Riganti, Int. J. Mol. Sci., 2019, 20, 2505 CrossRef CAS.
- J. M. Schafer, B. D. Lehmann, P. I. Gonzalez-Ericsson, C. B. Marshall, J. S. Beeler, L. N. Redman, H. Jin, V. Sanchez, M. C. Stubbs, P. Scherle, K. N. Johnson, Q. Sheng, J. T. Roland, J. A. Bauer, Y. Shyr, B. Chakravarthy, B. C. Mobley, S. W. Hiebert, J. M. Balko, M. E. Sanders, P. C. C. Liu and J. A. Pietenpol, Sci. Transl. Med., 2020, 12, eaaw8275 CrossRef CAS.
- S. J. Potts, J. S. Krueger, N. D. Landis, D. A. Eberhard, G. David Young, S. C. Schmechel and H. Lange, Lab. Invest., 2012, 92, 1342–1357 CrossRef.
- S. R. Sompuram, K. Vani, A. K. Schaedle, A. Balasubramanian and S. A. Bogen, Arch. Pathol. Lab. Med., 2018, 142, 851–862 CrossRef CAS.
- R. J. DeBerardinis and N. S. Chandel, Sci. Adv., 2016, 2, e1600200 CrossRef.
- G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472–482 CrossRef CAS.
- P. S. Ward and C. B. Thompson, Cancer Cell, 2012, 21, 297–308 CrossRef CAS.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS.
- S. Beloribi-Djefaflia, S. Vasseur and F. Guillaumond, Oncogenesis, 2016, 5, e189 CrossRef CAS.
- R. Munir, J. Lisec, J. V. Swinnen and N. Zaidi, Br. J. Cancer, 2019, 120, 1090–1098 CrossRef.
- N. J. Serkova and S. G. Eckhardt, Front. Oncol., 2016, 6, 152 Search PubMed.
- O. Folger, L. Jerby, C. Frezza, E. Gottlieb, E. Ruppin and T. Shlomi, Mol. Syst. Biol., 2011, 7, 501 CrossRef CAS.
- C. Caldarella, G. Treglia and A. Giordano, Clin. Breast Cancer, 2014, 14, 241–248 CrossRef.
- A. Lodi, S. Woods and S. Ronen, NMR Biomed., 2014, 27, 700–708 CrossRef CAS.
- M. Morelli, J. Tentler, G. Kulikowski, A. Tan, E. Bradshaw-Pierce, T. Pitts, A. Brown, S. Nallapareddy, J. Arcaroli, N. Serkova, M. Hidalgo, F. Ciardiello and S. Eckhardt, Clin. Cancer Res., 2012, 18, 1051–1062 CrossRef CAS.
- M. M. Abouzied, E. S. Crawford and H. A. Nabi, J. Nucl. Med. Technol., 2005, 33, 145–155 Search PubMed.
- L. Xiao, C. Wang, C. Dai, L. E. Littlepage, J. Li and Z. D. Schultz, Angew. Chem., Int. Ed., 2020, 59, 3439–3443 CrossRef CAS.
- Y.-C. Ou, J. A. Webb, C. M. O'Brien, I. J. Pence, E. C. Lin, E. P. Paul, D. Cole, S.-H. Ou, M. Lapierre-Landry, R. C. DeLapp, E. S. Lippmann, A. Mahadevan-Jansen and R. Bardhan, Nanoscale, 2018, 10, 13092–13105 RSC.
- Y.-C. Ou, X. Wen, C. A. Johnson, D. Shae, O. D. Ayala, J. A. Webb, E. C. Lin, R. C. DeLapp, K. L. Boyd, A. Richmond, A. Mahadevan-Jansen, M. Rafat, J. T. Wilson, J. M. Balko, M. N. Tantawy, A. E. Vilgelm and R. Bardhan, ACS Nano, 2020, 14, 651–663 CrossRef CAS.
- X. Wen, Y.-C. Ou, H. F. Zarick, X. Zhang, A. B. Hmelo, Q. J. Victor, E. P. Paul, J. M. Slocik, R. R. Naik, L. M. Bellan, E. C. Lin and R. Bardhan, Bioeng. Transl. Med., 2020, e10165 Search PubMed.
- G. Thomas, T. Q. Nguyen, I. J. Pence, B. Caldwell, M. E. O'Connor, J. Giltnane, M. E. Sanders, A. Grau, I. Meszoely, M. Hooks, M. C. Kelley and A. Mahadevan-Jansen, Sci. Rep., 2017, 7, 13548 CrossRef CAS.
- S. K. Paidi, A. Rizwan, C. Zheng, M. Cheng, K. Glunde and I. Barman, Cancer Res., 2017, 77, 247 CrossRef CAS.
- T. J. E. Hubbard, A. Shore and N. Stone, Analyst, 2019, 144, 6479–6496 RSC.
- S. Qiu, M. Li, J. Liu, X. Chen, T. Lin, Y. Xu, Y. Chen, Y. Weng, Y. Pan, S. Feng, X. Lin, L. Zhang and D. Lin, Biomed. Opt. Express, 2020, 11, 1819–1833 CrossRef.
- F. Draux, C. Gobinet, J. Sulé-Suso, M. Manfait, P. Jeannesson and G. D. Sockalingum, Analyst, 2011, 136, 2718–2725 RSC.
- Z. Farhane, F. Bonnier, O. Howe, A. Casey and H. J. Byrne, J. Biophotonics, 2018, 11, e201700060 CrossRef.
- A. S. Mondol, N. Töpfer, J. Rüger, U. Neugebauer, J. Popp and I. W. Schie, Sci. Rep., 2019, 9, 12653 CrossRef.
- S. Patra, V. Young, L. Llewellyn, J. N. Senapati and J. Mathew, Asian Pac. J. Cancer Prev., 2017, 18, 2209–2213 Search PubMed.
- F. Bonnier and H. J. Byrne, Analyst, 2012, 137, 322–332 RSC.
- I. Pence and A. Mahadevan-Jansen, Chem. Soc. Rev., 2016, 45, 1958–1979 RSC.
- A. K. Meher and Y.-C. Chen, Anal. Chem., 2016, 88, 9151–9157 CrossRef CAS.
- X. Audier, S. Heuke, P. Volz, I. Rimke and H. Rigneault, APL Photonics, 2020, 5, 011101 CrossRef CAS.
- Y. Hernández, L. K. Lagos and B. C. Galarreta, Sens. Biosensing Res., 2020, 28, 100331 CrossRef.
- S. Niebling, H. Y. Kuchelmeister, C. Schmuck and S. Schlücker, Chem. Sci., 2012, 3, 3371–3377 RSC.
- D. Bovenkamp, A. Micko, J. Püls, F. Placzek, R. Höftberger, G. Vila, R. Leitgeb, W. Drexler, M. Andreana, S. Wolfsberger and A. Unterhuber, Molecules, 2019, 24, 3577 CrossRef CAS.
- F. Sinjab, G. Sicilia, D. W. Shipp, M. Marlow and I. Notingher, Appl. Spectrosc., 2017, 71, 2595–2607 CrossRef CAS.
- S. Fore, J. Chan, D. Taylor and T. Huser, J. Opt., 2011, 13, 044021 CrossRef.
- X. Dai, H. Xia, S. Zhou, Q. Tang and F. Bi, Cancer Lett., 2019, 442, 202–212 CrossRef CAS.
- E. Y. Leung, J. E. Kim, M. Askarian-Amiri, G. W. Rewcastle, G. J. Finlay and B. C. Baguley, PLoS One, 2014, 9, e105792 CrossRef.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- I. Dobrzyńska, B. Szachowicz-Petelska, B. Darewicz and Z. A. Figaszewski, J. Membr. Biol., 2015, 248, 301–307 CrossRef.
- K. Glunde and N. J. Serkova, Pharmacogenomics, 2006, 7, 1109–1123 CrossRef CAS.
- N. M. S. Al-Saffar, H. Troy, A. R. de Molina, L. E. Jackson, B. Madhu, J. R. Griffiths, M. O. Leach, P. Workman, J. C. Lacal, I. R. Judson and Y.-L. Chung, Cancer Res., 2006, 66, 427 CrossRef CAS.
- M. P. Morelli, J. J. Tentler, G. N. Kulikowski, A.-C. Tan, E. L. Bradshaw-Pierce, T. M. Pitts, A. M. Brown, S. Nallapareddy, J. J. Arcaroli, N. J. Serkova, M. Hidalgo, F. Ciardiello and S. G. Eckhardt, Clin. Cancer Res., 2012, 18, 1051–1062 CrossRef CAS.
- S. Acciardo, L. Mignion, N. Joudiou, C. Bouzin, J.-F. Baurain, B. Gallez and B. F. Jordan, Oncotarget, 2018, 9, 16832–16846 CrossRef.
- B. Klotz, S. Kneitz, Y. Lu, W. Boswell, J. Postlethwait, W. Warren, R. B. Walter and M. Schartl, G3: Genes, Genomes, Genet., 2019, 9, 2267 CrossRef CAS.
- S. Beloribi-Djefaflia, S. Vasseur and F. Guillaumond, Oncogenesis, 2016, 5, e189 CrossRef CAS.
- C. Muñoz-Pinedo, N. El Mjiyad and J. E. Ricci, Cell Death Discovery, 2012, 3, e248 CrossRef.
- K. Subik, J.-F. Lee, L. Baxter, T. Strzepek, D. Costello, P. Crowley, L. Xing, M.-C. Hung, T. Bonfiglio, D. G. Hicks and P. Tang, Breast Cancer, 2010, 4, 35–41 Search PubMed.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef.
- A. E. Maennling, M. K. Tur, M. Niebert, T. Klockenbring, F. Zeppernick, S. Gattenlöhner, I. Meinhold-Heerlein and A. F. Hussain, Cancers, 2019, 11, 1826 CrossRef CAS.
- B. Ogretmen, Nat. Rev. Cancer, 2018, 18, 33–50 CrossRef CAS.
- M. Cheng, Z. M. Bhujwalla and K. Glunde, Front. Oncol., 2016, 6, 266 Search PubMed.
- K. S. Saini, S. Loi, E. de Azambuja, O. Metzger-Filho, M. L. Saini, M. Ignatiadis, J. E. Dancey and M. J. Piccart-Gebhart, Cancer Treat. Rev., 2013, 39, 935–946 CrossRef CAS.
- S.-H. Cheng, Y.-M. Tseng, S.-H. Wu, S.-M. Tsai and L.-Y. Tsai, Sci. Rep., 2017, 7, 15976 CrossRef.
- M. Elkabets, S. Vora, D. Juric, N. Morse, M. Mino-Kenudson, T. Muranen, J. Tao, A. B. Campos, J. Rodon, Y. H. Ibrahim, V. Serra, V. Rodrik-Outmezguine, S. Hazra, S. Singh, P. Kim, C. Quadt, M. Liu, A. Huang, N. Rosen, J. A. Engelman, M. Scaltriti and J. Baselga, Sci. Transl. Med., 2013, 5, 196ra199 Search PubMed.
- L. Vettore, R. L. Westbrook and D. A. Tennant, Br. J. Cancer, 2020, 122, 150–156 CrossRef CAS.
- B.-H. Choi and J. L. Coloff, Cancers, 2019, 11, 675 CrossRef CAS.
- D. N. Metsiou, K. E. Siatis, E. Giannopoulou, D. J. Papachristou, H. P. Kalofonos, A. Koutras and G. Athanassiou, Ann. Biomed. Eng., 2019, 47, 1711–1724 CrossRef.
- M. Yang, T. Soga and P. J. Pollard, J. Clin. Invest., 2013, 123, 3652–3658 CrossRef CAS.
- C. Vernieri, S. Casola, M. Foiani, F. Pietrantonio, F. de Braud and V. Longo, Cancer Discovery, 2016, 6, 1315 CrossRef CAS.
- C.-S. Ho, N. Jean, C. A. Hogan, L. Blackmon, S. S. Jeffrey, M. Holodniy, N. Banaei, A. A. E. Saleh, S. Ermon and J. Dionne, Nat. Commun., 2019, 10, 4927 CrossRef.
- S. L. Goldenberg, G. Nir and S. E. Salcudean, Nat. Rev. Urol., 2019, 16, 391–403 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02221g |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |