
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Hydroxybenzimidazole as a structurally modifiable platform for N-oxyl radicals for direct C–H functionalization reactions†
Tomomi
Yoshii
a,
Saori
Tsuzuki
 a,
Shunya
Sakurai
a,
Ryu
Sakamoto
a,
Julong
Jiang
b,
Miho
Hatanaka
bc,
Akira
Matsumoto
d and
Keiji
Maruoka
*ade
a,
Shunya
Sakurai
a,
Ryu
Sakamoto
a,
Julong
Jiang
b,
Miho
Hatanaka
bc,
Akira
Matsumoto
d and
Keiji
Maruoka
*ade
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: maruoka.keiji.4w@kyoto-u.ac.jp
bInstitute for Research Initiatives, Division for Research Strategy, Graduate School of Materials Science, Data Science Center, Nara Institute of Science and Technology, Ikoma, Nara 630-0192, Japan
cPRESTO, Japan Science and Technology (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo, Kyoto 606-8501, Japan
eSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China
First published on 18th May 2020
Abstract
Methods for direct functionalization of C–H bonds mediated by N-oxyl radicals constitute a powerful tool in modern organic synthesis. While several N-oxyl radicals have been developed to date, the lack of structural diversity for these species has hampered further progress in this field. Here we designed a novel class of N-oxyl radicals based on N-hydroxybenzimidazole, and applied them to the direct C–H functionalization reactions. The flexibly modifiable features of these structures enabled facile tuning of their catalytic performance. Moreover, with these organoradicals, we have developed a metal-free approach for the synthesis of acyl fluorides via direct C–H fluorination of aldehydes under mild conditions.
Introduction
The direct functionalization of C–H bonds has been recognized as an innovative approach to realize step- and atom-economical synthesis of various functionalized molecules. Considerable research efforts have been devoted to the development of a variety of C–H functionalization reactions based on transition-metal-catalyzed or photoredox-promoted approaches.1,2 On the other hand, transition-metal-free and non-photolytic direct C–H functionalization reactions still remain a significant challenge,3 although they will provide new synthetic strategies with complementary reactivity and selectivity.4 In this context, N-oxyl radicals have attracted much attention as promising organoradical catalysts for these transformations.5 As represented by N-hydroxyphthalimide (NHPI), several N-hydroxy compounds have been used to generate active N-oxyl radicals in situ, which selectively abstract hydrogen atoms from C–H bonds of organic molecules. The resulting alkyl radicals react with various radical acceptors to afford the corresponding functionalized products.6 In order to improve the catalytic efficiency and stability of the key N-oxyl radicals, several N-hydroxy compounds as organoradical precursors have been designed based on the structure of NHPI and utilized in the same type of transformations (Fig. 1a).7 Most of the reported N-hydroxy compounds, however, have similar structures featuring carbonyl groups adjacent to hydroxylamine moiety as a reactive site. While these carbonyl groups significantly contribute to enhancing the catalytic performance of N-oxyl radicals,8 only a few sites are available for structural modification due to their existence. As a result, such a lack of structural diversity for these N-hydroxy compounds renders it difficult to alter the specific properties of the corresponding N-oxyl radicals such as bond dissociation energies (BDEs), a fundamental parameter to estimate their reactivities in the hydrogen atom transfer process. Accordingly, further progress in this field demands a completely novel design of N-oxyl radicals with structural diversity. Herein, we report the design and synthesis of novel organoradical species based on N-hydroxybenzimidazoles (NHBIs), which contain multiple modification sites (Fig. 1b), and demonstrate their distinct reactivities as organoradical catalysts and efficient radical initiators in the direct C–H functionalization reactions. | ||
| Fig. 1 (a) Representative examples of N-hydroxy compounds. (b) Design of N-hydroxybenzimidazole as N-oxyl radical platform. | ||
N-Hydroxybenzimidazoles, which are planar and stable heterocycles bearing an N-hydroxy moiety, have recently attracted interest in the field of biological and pharmaceutical sciences as anti-virulence or anti-cancer agents.9,10 However, they have rarely been used in synthetic organic chemistry and, to the best of our knowledge, studies on the potential of NHBIs to generate the corresponding N-oxyl radicals have not yet been conducted.11 In this context, we became interested in the potential of NHBIs as novel organoradical precursors based on the following features: (1) NHBIs can be readily prepared from 2-nitroaniline derivatives in a few steps (see the ESI†); (2) substituents can be easily introduced at both the aromatic ring and the 2-position of the benzimidazole moiety; (3) NHBIs contain additional modification sites such as the nitrogen atom at the 3-position of the benzimidazole moiety and the counteranion of the resulting benzimidazolium species, which may potentially be exploited for further functionalization.
Results and discussion
To test our hypothesis, we initially carried out density functional theory (DFT) calculations in order to estimate the BDE values of the O–H bonds in our designed NHBI derivatives (1a–f) (Fig. 2).12 As expected, the results of the DFT calculations revealed that the BDE values for 1 can be tuned within a wide range (∼10 kcal mol−1) by facile modifications such as the introduction of substituents at the aromatic ring or the 2-position of the imidazole moiety, or by N-alkylation at the 3-position. Moreover, some of synthesized NHBI derivatives have the similar or even higher BDEs compared to that of NHPI, which has been widely used as an efficient catalyst for several hydrogen atom abstraction reactions.6 Thus, these results indicate that N-oxyl radicals derived from NHBI derivatives potentially work as organoradical catalysts for direct C–H functionalization via hydrogen atom abstraction.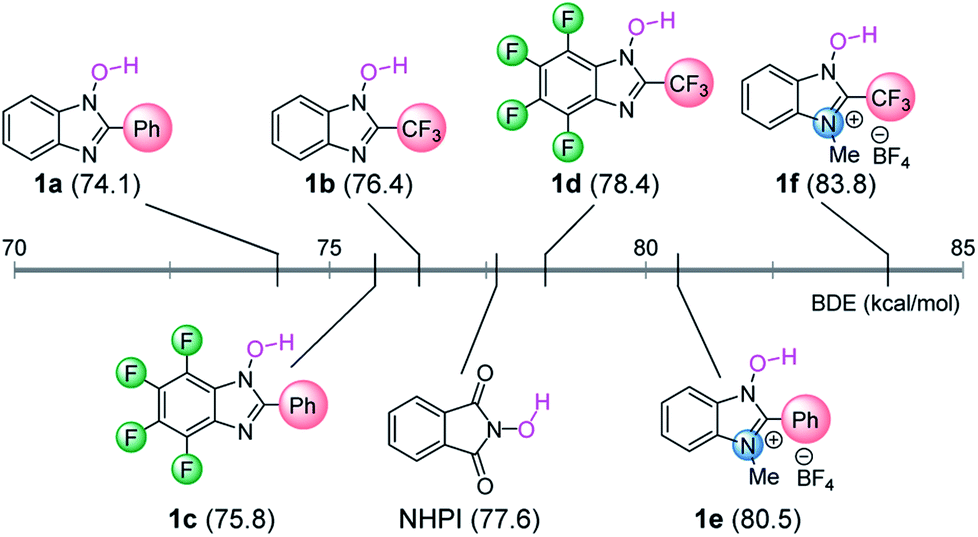 | ||
| Fig. 2 Calculated BDE values for O–H bond in 1 and NHPI. Calculations were performed at B3LYP-D3/6-311G(d,p) in SMD (MeCN) level of theory. For details, see the ESI.† | ||
In order to evaluate the catalytic activities of NHBI derivatives, we attempted to apply them to the reported C–H functionalization reactions. Among several transformations catalyzed by N-oxyl radicals, we selected the benzylic C–H amination of ethylbenzene (2) as a model reaction, which was originally reported as the NHPI-catalyzed reaction (Scheme 1).6d After the reaction was run using diethyl azodicarboxylate (3) in the presence of 10 mol% of NHPI at 80 °C for 24 h, the desired aminated product 4 was obtained in moderate yield. On the other hand, yields of 4 for reactions using NHBI derivatives varied significantly, depending on their substituents. Notably, when 1d was used under the same reaction conditions, the yield of 4 was much higher than the use of NHPI. On the contrary, cationic NHBI derivative 1e, which has a highest value of BDE among examined N-hydroxy compounds, did not show remarkable catalytic activity for this transformation, probably due to difficult regeneration of active N-oxy radicals. These experimental results clearly indicate that our designed NHBI derivatives can be applied to direct C–H functionalization reactions.
 | ||
| Scheme 1 Evaluation of 1 as organoradical catalyst for benzylic C–H amination. Yields were determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. | ||
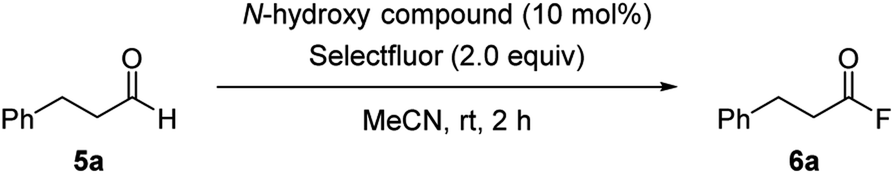
Having confirmed the potential of NHBI derivatives as active organoradical precursors, we set about exploring their distinct reactivities for the development of direct C–H functionalization reactions. Our continuing concern about methods for generation and application of acyl radical species13,14 led us to investigate the direct functionalization of aldehydes via C(sp2)–H bond activation by NHBI derivatives. To our delight, we found that in the presence of catalytic amount of NHBI, the aldehydic C–H bond was directly converted into C–F bond by using Selectfluor as a fluorine atom transfer reagent, affording the corresponding acyl fluorides (Scheme 2). Acyl fluorides have recently attracted much attention in synthetic and biological chemistry owing to their unique reactivity, which is not observable in commonly employed acyl chlorides or acid anhydrides.15,16 While several synthetic methods for acyl fluorides have been reported, they are typically prepared by using toxic fluorination reagents and/or precious metal catalysts.17,18 Therefore, the development of their different synthetic approach with high practicability is in great demand.
 | ||
| Scheme 2 Aldehydic C–H fluorination reaction using NHBI. | ||
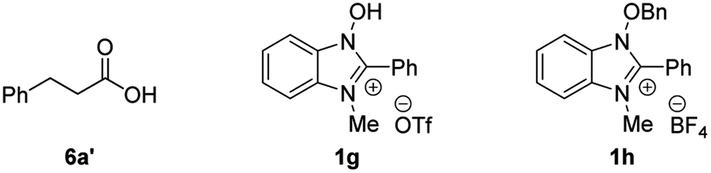
Initially, the effect of NHBI derivatives was assessed in the reaction of 3-phenylpropanal (5a) with Selectfluor (Table 1). Although the reaction using 10 mol% of 1a did not afford the desired acyl fluoride 6a (entry 1), fluorine-substituted NHBIs 1b–d afforded 6a in moderate to good yields (entries 2–4). It is noteworthy that cationic NHBI derivative 1e, which was not active for the aforementioned benzylic C–H amination reaction (Scheme 1), turned out to be effective for this C–H fluorination reaction (entry 5). During the optimization, we observed the formation of 3-phenylpropionic acid (6a′) as a side product, which could potentially be formed during the reaction of an acyl radical with oxygen.19 Thus, we carried out the reaction under an inert atmosphere of argon, which further improved the yield of 6a with the generation of 6a′ sufficiently suppressed (entry 6). On the other hand, the introduction of trifluoromethyl group at the 2-position of benzimidazolium structure gave only a small amount of the product (entry 7). While the effect of counteranion for cationic NHBI derivative is almost negligible (entry 8), the importance of free hydroxylamine moiety of NHBI was confirmed by the reaction with N-benzyloxybenzimidazole 1h (entry 9). Neither 6a or 6a′ was obtained in the absence of NHBI derivative (entry 10). As a comparative study, NHPI and its derivatives were used for this transformation (entries 11–13). Although the reactions proceeded moderately in the presence of these N-hydroxy compounds, the yields of 6a were much lower than the use of 1e.
|
|
||||
|---|---|---|---|---|
| Entry | N-Hydroxy compound | Yield of 6a (%) | Yield of 6a′ (%) | Recovery of 5a (%) |
| a Unless otherwise specified, reactions were carried out in MeCN for 2 h in the presence of 5a (0.20 mmol), Selectfluor (2.0 equiv.) and N-hydroxy compound (10 mol%). b Yields of 6a were determined by 1H NMR spectroscopy using benzotrifluoride as the internal standard. c The reaction was conducted under an atmosphere of argon. N.D.: not detected. | ||||
| 1 | 1a | N.D. | 10 | 81 |
| 2 | 1b | 28 | 21 | 45 |
| 3 | 1c | 33 | 28 | 39 |
| 4 | 1d | 61 | 16 | 8 |
| 5 | 1e | 64 | 12 | 11 |
| 6c | 1e | 77 | 5 | 18 |
| 7c | 1f | 4 | 17 | 83 |
| 8c | 1g | 72 | 2 | 9 |
| 9c | 1h | Trace | Trace | 97 |
| 10c | — | N.D. | Trace | 96 |
| 11c | NHPI | 47 | 18 | 26 |
| 12c | 4-Nitro-NHPI | 19 | 36 | 15 |
| 13c | THICA | 46 | 17 | 16 |
|
||||
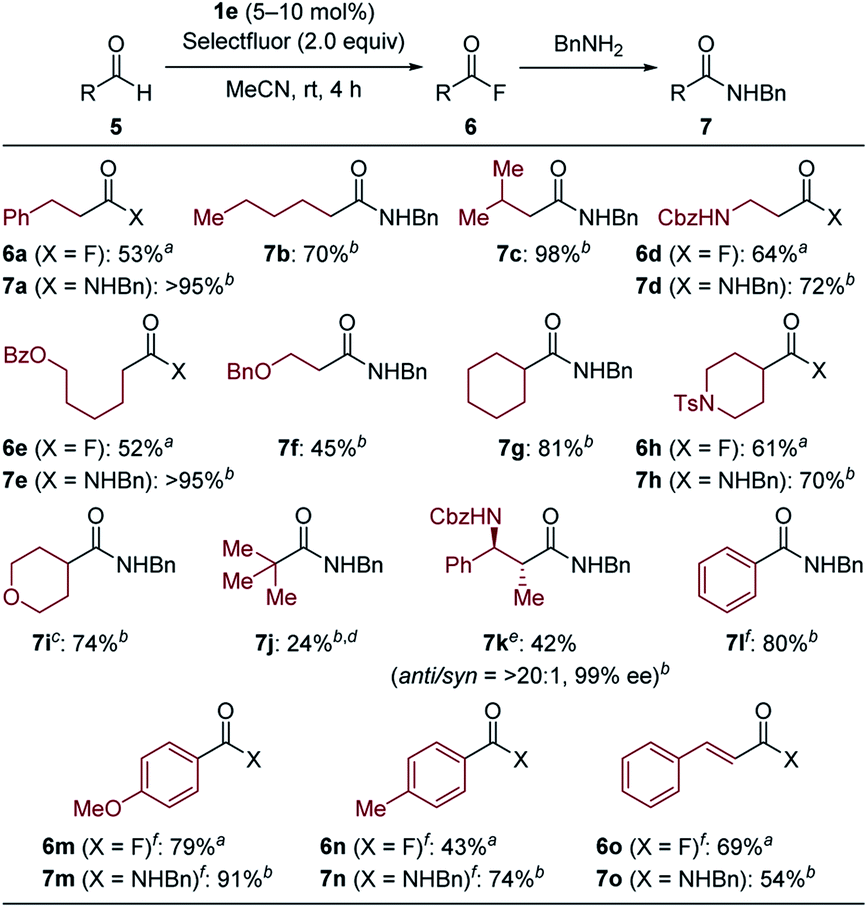
With the optimal NHBI derivative in hand, the scope of aldehyde was then investigated (Table 2). While isolated yields of products were determined after one-pot conversion into benzyl amides 7 to avoid loss of acyl fluorides 6 due to their volatility and/or instability on silica-gel, several reactions could be scaled up to 2.0 mmol and the obtained acyl fluorides were successfully isolated after flash column chromatography in acceptable yields. The reactions of aliphatic aldehydes proceeded even at room temperature, affording the desired products in moderate to high yields. Desired products bearing benzylic C–H bond (6a–7a), acyclic or cyclic ether moieties (7f and 7i) and nitrogen-containing functional groups (6d–7d, 6h–7h and 7k) were successfully obtained, highlighting the advantage of mild reaction conditions. Although pivalaldehyde gave the corresponding product 7j in low yield due to competitive decarbonylation,18d functionalized aldehyde with two stereogenic centers provided the desired benzylamide 7k without epimerization. Moreover, products from aryl aldehydes (7l, 6m–7m and 6n–7n) and trans-cinnamaldehyde (6o–7o) could also be obtained at increased reaction temperature.
| a The reaction was carried out in MeCN for 4 h in the presence of 5 (2.0 mmol), Selectfluor (2.0 equiv.) and 1e (10 mol%). Isolated as acyl fluoride 6. b The reaction was carried out in MeCN for 4 h in the presence of 5 (0.20 mmol), Selectfluor (1.0 equiv.) and 1e (5.0 mol%), followed by a treatment with benzylamine (2.0 equiv.) for 2 h. Isolated as benzylamide 7. c The reaction was conducted at 50 °C. d NMR yield. e The reaction was conducted in the presence of 1e (10 mol%). f The reaction was conducted at 80 °C. |
|---|
|
The direct conversion of aldehydes into acyl fluorides and the subsequent reaction with various nucleophiles allow for the divergent synthesis of carbonyl compounds (Scheme 3). Other than benzylamine, secondary amines (8a–b), amino acid derivatives (8c–d) and even less nucleophilic oxazolidinone (8e) were also applicable to the one-pot procedure. The use of other nucleophiles such as alcohol (8f–h) and thiol (8i) provided the corresponding esters and thioester in good to high yields. This protocol was also applied to the conversion of β-hydroxy aldehyde 9 into β-lactone 10 in one pot.
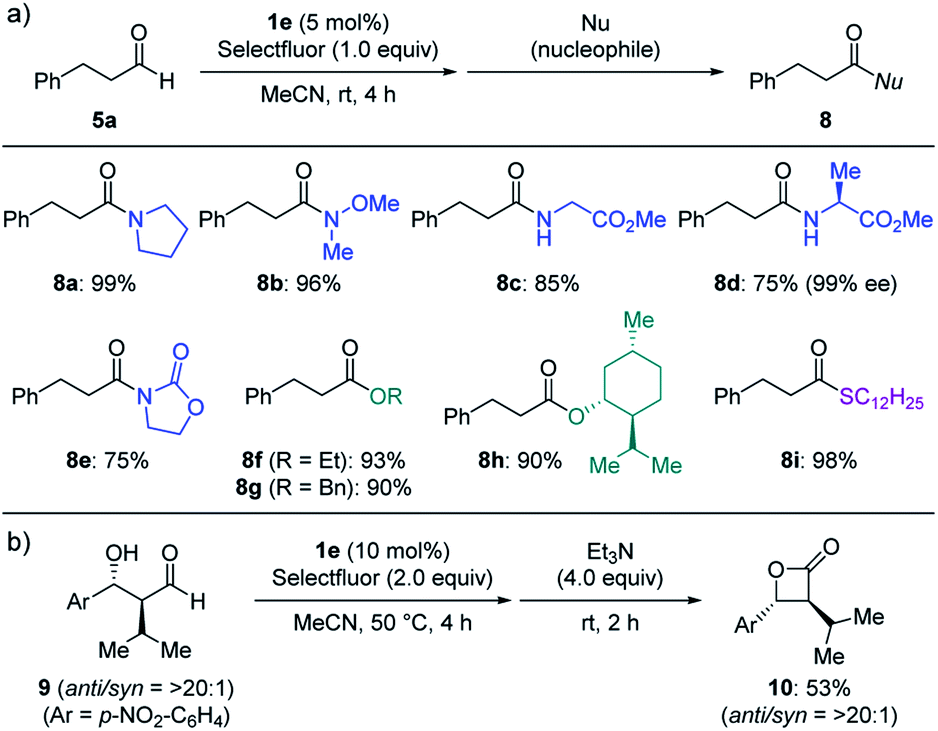 | ||
| Scheme 3 Subsequent transformations of acyl fluoride. (a) The reaction was carried out in MeCN for 4 h in the presence of 5a (0.20 mmol), Selectfluor (1.0 equiv.) and 1e (5 mol%), followed by a treatment with a nucleophile (2.0 equiv.) and triethylamine (0–2.0 equiv.) for 2 h. Isolated yields. (b) Synthesis of β-lactone 10. The reaction was carried out in MeCN for 4 h in the presence of 9 (0.20 mmol), Selectfluor (2.0 equiv.) and 1e (10 mol%), followed by a treatment with triethylamine (4.0 equiv.) for 2 h. Isolated yields. | ||
Moreover, starting from isolated acyl fluoride 6a, several unsymmetrical ketones (11a–e) could be synthesized via the reaction with various carbon nucleophiles (Scheme 4).20 Both the arylation of 6a by a palladium-catalyzed cross-coupling with phenylboronic acid and the Friedel–Crafts reaction with 1,3-dimethoxybenzene (A) afforded aryl ketones 11a and 11b, respectively, in good yields. Alternatively, the reaction with a Wittig reagent furnished β-keto ester 11c, while the use of allyltrimethylsilane afforded β,γ-unsaturated ketone 11d in high yield. Finally, the reaction with silyl enol ether B in the presence of tributyltin fluoride afforded β-keto ester 11e quantitatively. These transformations demonstrate the utility of acyl fluorides as versatile synthetic intermediates.
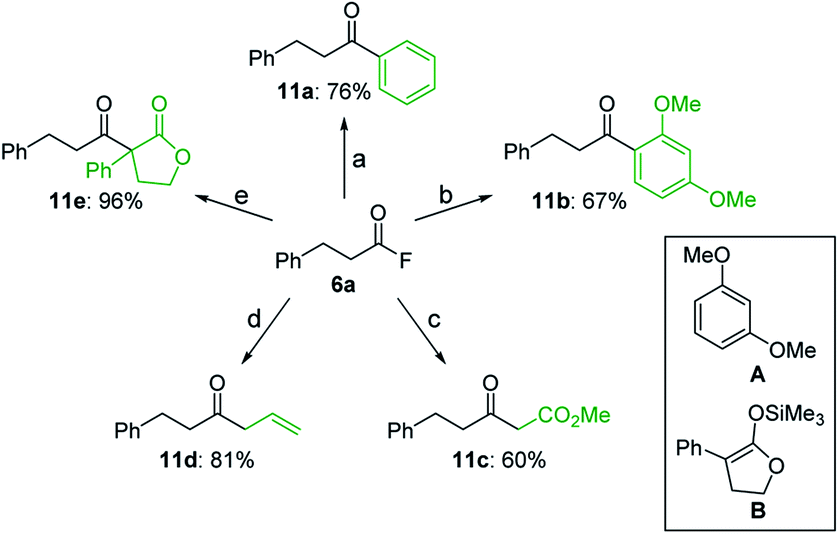 | ||
Scheme 4 Synthesis of unsymmetrical ketones from 6a. (a) PhB(OH)2 (1.5 equiv.), Pd(OAc)2 (1.0 mol%), P(p-MeO–C6H4)3 (4.0 mol%), KF (1.5 equiv.), toluene, 120 °C. (b) A (1.0 equiv.), TMSOTf (1.2 equiv.), MeCN, rt. (c) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me (1.0 equiv.), KF (6.0 equiv.), MeCN, 90 °C. (d) allyltrimethylsilane (1.2 equiv.), TiCl4 (1.0 equiv.), CH2Cl2, −78 °C. (e) B (2.0 equiv.), n-Bu3SnF (2.0 equiv.), toluene, 120 °C. CHCO2Me (1.0 equiv.), KF (6.0 equiv.), MeCN, 90 °C. (d) allyltrimethylsilane (1.2 equiv.), TiCl4 (1.0 equiv.), CH2Cl2, −78 °C. (e) B (2.0 equiv.), n-Bu3SnF (2.0 equiv.), toluene, 120 °C. | ||
To better understand the reaction mechanism, several experiments were carried out (Scheme 5). The addition of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as a radical scavenger under standard conditions completely inhibited the formation of acyl fluoride (Scheme 5a). In addition, when a stoichiometric reaction of 1e with styrene and TEMPO was run in the presence of Selectfluor, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 adduct of 1e, styrene and TEMPO was detected by high-resolution mass spectrometry (Scheme 5b). On the other hand, Selectfluor is known to be activated under photoirradiation condition, generating the corresponding radical cation 12.21 This species can work as a strong hydrogen atom abstraction reagent for C–H activation of various hydrocarbons.21a–c,22 Based on recent reports, we conducted the C–H oxidative arylation of cyclooctane with isoquinoline in the presence of a catalytic amount of 1e (Scheme 5c).21b,c As a result, arylated product 13 could be obtained in good yield even without photoirradiation. We also confirmed 13 was not detected in the absence of 1e and photoirradiation. Considering a high BDE value for C–H bond of cyclooctane (95.8 kcal mol−1, see the ESI†), a reaction pathway where the N-oxyl radical derived from 1e abstracts a hydrogen atom from C–H bond of cyclooctane would not be feasible. Therefore, these results indicate that our NHBI/Selectfluor system allows for the efficient generation of highly reactive radical cation 12, which is a key species for the activation of aldehydic C–H bond.
:1:1 adduct of 1e, styrene and TEMPO was detected by high-resolution mass spectrometry (Scheme 5b). On the other hand, Selectfluor is known to be activated under photoirradiation condition, generating the corresponding radical cation 12.21 This species can work as a strong hydrogen atom abstraction reagent for C–H activation of various hydrocarbons.21a–c,22 Based on recent reports, we conducted the C–H oxidative arylation of cyclooctane with isoquinoline in the presence of a catalytic amount of 1e (Scheme 5c).21b,c As a result, arylated product 13 could be obtained in good yield even without photoirradiation. We also confirmed 13 was not detected in the absence of 1e and photoirradiation. Considering a high BDE value for C–H bond of cyclooctane (95.8 kcal mol−1, see the ESI†), a reaction pathway where the N-oxyl radical derived from 1e abstracts a hydrogen atom from C–H bond of cyclooctane would not be feasible. Therefore, these results indicate that our NHBI/Selectfluor system allows for the efficient generation of highly reactive radical cation 12, which is a key species for the activation of aldehydic C–H bond.
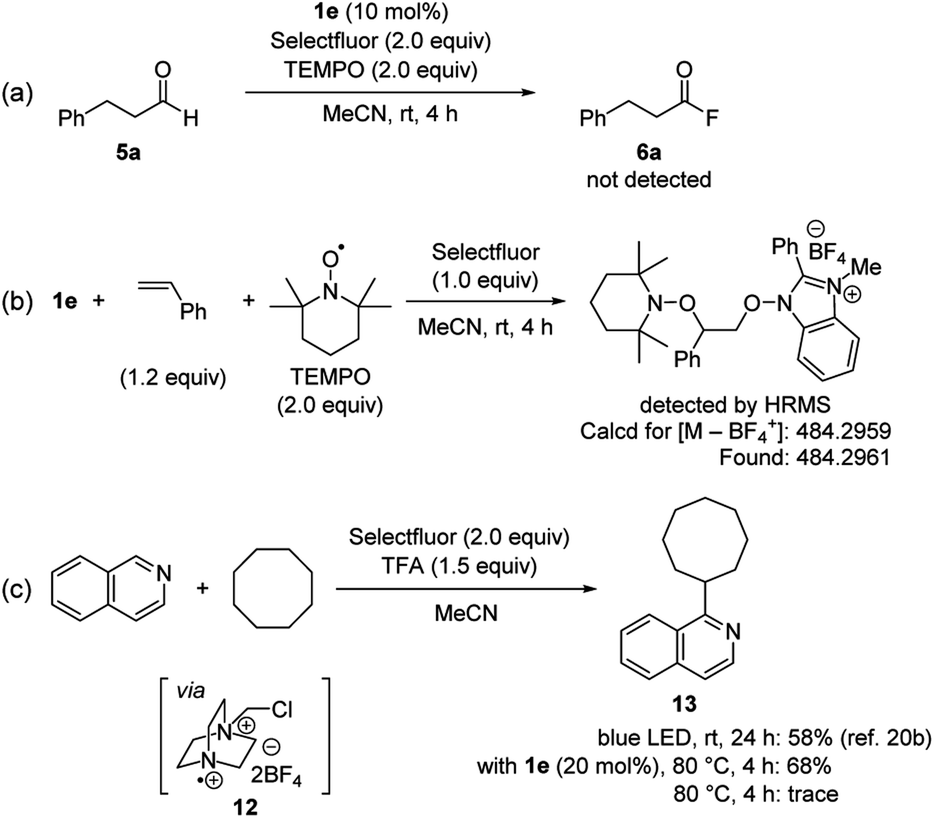 | ||
| Scheme 5 Mechanistic studies. | ||
Based on these studies, we propose a reaction mechanism as shown in Fig. 3. In the presence of NHBI 1, radical cation 12 is generated from Selectfluor with concomitant formation of N-oxyl radical 14. Then 12 abstracts a hydrogen atom from aldehyde 5 to form the corresponding acyl radical 15. Subsequently, a fluorine atom of another Selectfluor is trapped by 15 to give the desired acyl fluoride 6 and radical cation 12, and the reaction proceeds via a chain process until completion.
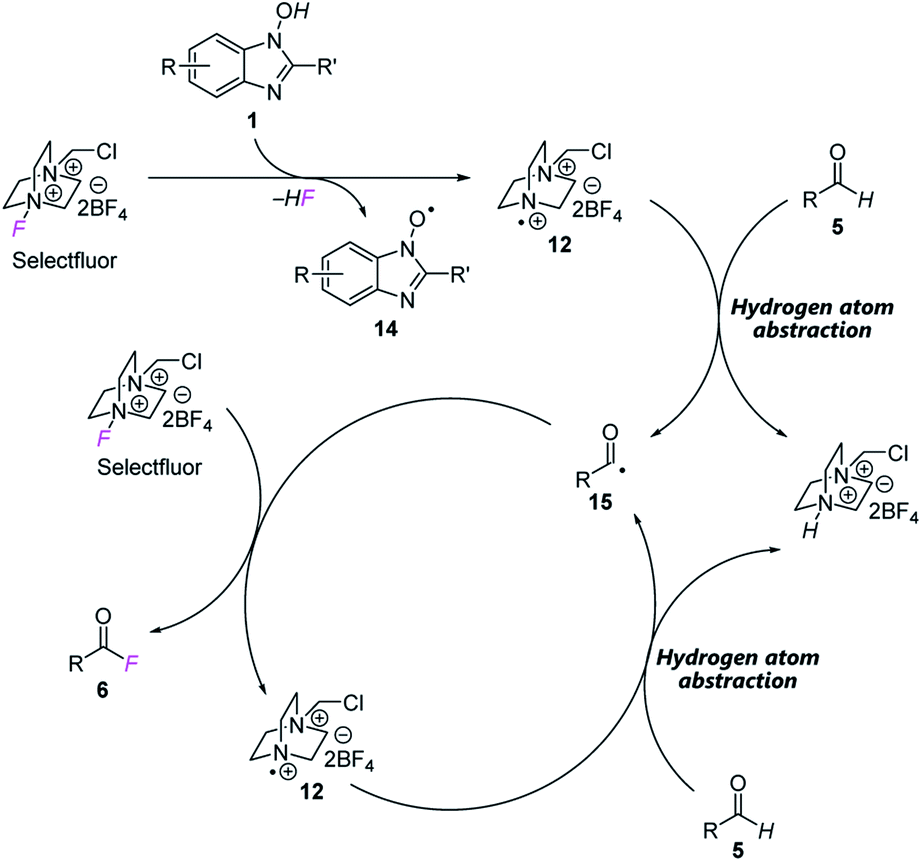 | ||
| Fig. 3 Proposed reaction mechanism. | ||
With respect to the initiation process generating two different radicals from 1 and Selectfluor, further theoretical studies by DFT calculation provided some insights into the remarkable reactivity difference among NHBI derivatives (Fig. 4). Namely, when this process is divided into two steps as shown in Fig. 4a, the energy gap between LUMO of putative oxoammonium 16 and HOMO of monocationic amine 17 is widely changed depending on the substitution pattern of NHBI structure. In addition, these values of HOMO–LUMO energy gap are in proportion to the Gibbs free energy difference (ΔG) for step II (Fig. 4b). It should be noted this correlation is not the only determinant for the efficiency of initiation process; the exceptionally lower reactivity of 1f (Table 1, entry 7) would be attributed to an unfavorable uphill in free energy for the step I, which makes it difficult to be oxidized by Selectfluor to form the corresponding oxoammonium species leading to step II (see the ESI†). Thus, these results demonstrated that the highly designable structures of 1 allow for the tuning of their electronic state, which can alter the energy profile of the process for generation of active radical species.
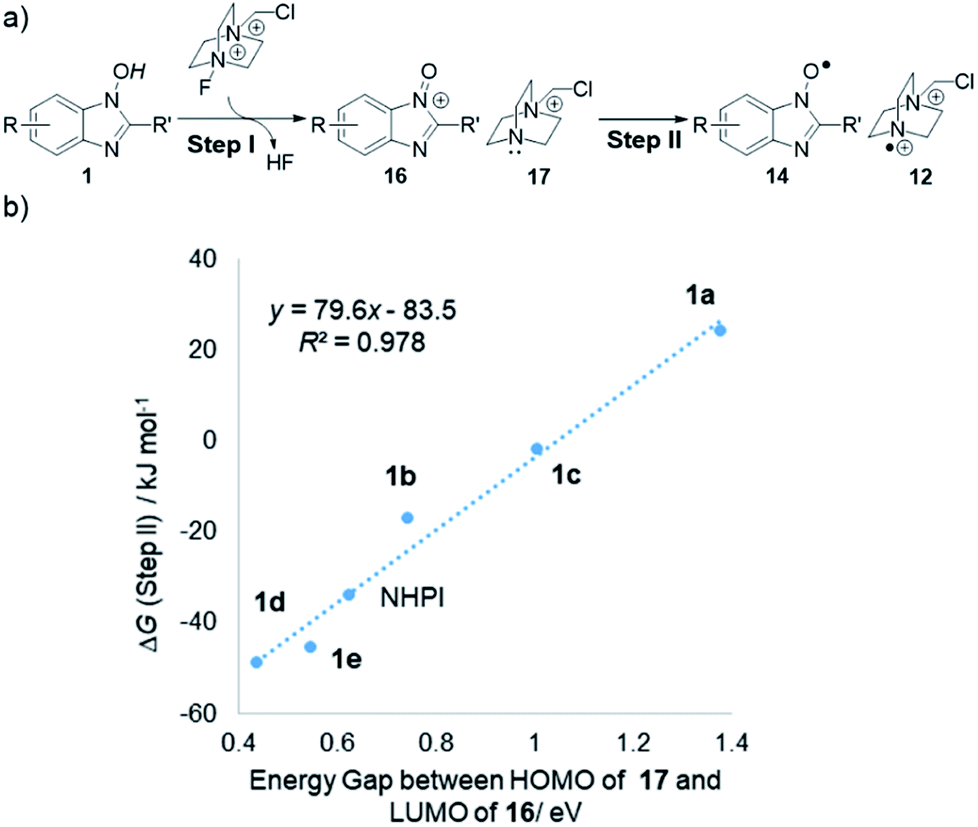 | ||
| Fig. 4 (a) Proposed mechanism for initiation process by 1 and Selectfluor. (b) Correlation of energy gap between HOMO of 17 and LUMO of 16 (in eV) and Gibbs free energy difference (ΔG) for step II (in kJ mol−1). | ||
Conclusions
In conclusion, we have synthesized a novel class of N-oxyl radicals based on NHBI, which is a flexibly modifiable structural platform. The evaluation of these NHBI derivatives as organoradical catalysts revealed that the substituents on these structures significantly alter their catalytic performance in benzylic C–H amination reaction. Moreover, we have developed a novel metal-free and non-photolytic method for the synthesis of acyl fluorides by using a catalytic amount of NHBI derivatives. Mechanistic studies indicated a distinct character of NHBI derivatives as an efficient radical initiator to generate a more active radical species for hydrogen atom abstraction. Further investigations into the applications of NHBI derivatives to other C–H activation strategies, as well as the development of novel NHBI catalysts, are currently underway in our laboratory. We believe that these studies indicate a new direction for the chemistry of N-oxyl radicals, which will spur further research on organoradical catalysis for direct C–H activation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant number JP26220803 and JP17H06450 (Hybrid Catalysis). R. S. acknowledges the Kyoto University Research Development Program 2017. S. S. is thankful for the Grant-in-Aid for JSPS Fellowship for Young Scientists.Notes and references
- For selected reviews on metal-catalyzed C–H functionalization reactions, see: (a) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (c) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (e) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- For selected reviews on photolytic C–H functionalization reactions, see: (a) M. Uygur and O. García Mancheño, Org. Biomol. Chem., 2019, 17, 5475–5489 RSC; (b) L. Revathi, L. Ravindar, W.-Y. Fang, K. P. Rakesh and H.-L. Qin, Adv. Synth. Catal., 2018, 360, 4652–4698 CrossRef CAS; (c) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (d) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS PubMed; (e) S. Protti, M. Fagnoni and D. Ravelli, ChemCatChem, 2015, 7, 1516–1523 CrossRef CAS.
- (a) V. Charushin and O. V. Chupakhin, Metal Free C–H Functionalization of Aromatics. Nucleophilic Displacement of Hydrogen, Springer International Publishing, Switzerland, 2014 Search PubMed; (b) R. Narayan, K. Matcha and A. P. Antonchick, Chem.–Eur. J., 2015, 21, 14678–14693 CrossRef CAS PubMed; (c) R. Narayan, S. Manna and A. P. Antonchick, Synlett, 2015, 26, 1785–1803 CrossRef CAS; (d) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769–5804 CrossRef CAS.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- For reviews of N-oxyl radicals as organoradical catalysts, see: (a) K. Chen, P. Zhang, Y. Wang and H. Li, Green Chem., 2014, 16, 2344–2374 RSC; (b) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC; (c) L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296–1310 CrossRef CAS PubMed; (d) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed; (e) Y. Ishii and S. Sakaguchi, Catal. Today, 2006, 117, 105–113 CrossRef CAS; (f) R. A. Sheldon and I. W. C. E. Arenas, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (g) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS.
- For selected examples of the use of NHPI and its derivatives as organoradical catalysts, see: (a) S. Sakaguchi, Y. Nishiwaki, T. Kitamura and Y. Ishii, Angew. Chem., Int. Ed., 2001, 40, 222–224 CrossRef CAS; (b) S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352–2353 RSC; (c) T. Hirabayashi, S. Sakaguchi and Y. Ishii, Angew. Chem., Int. Ed., 2004, 43, 1120–1123 CrossRef CAS PubMed; (d) Y. Amaoka, S. Kamijo, T. Hoshikawa and M. Inoue, J. Org. Chem., 2012, 77, 9959–9969 CrossRef CAS PubMed; (e) Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed; (f) K. Kiyokawa, K. Takemoto and S. Minakata, Chem. Commun., 2016, 52, 13082–13085 RSC; (g) J. Liu, K.-F. Hu, J.-P. Qu and Y.-B. Kang, Org. Lett., 2017, 19, 5593–5596 CrossRef CAS PubMed; (h) S. H. Combe, A. Hosseini, L. Song, H. Hausmann and P. R. Schreiner, Org. Lett., 2017, 19, 6156–6159 CrossRef CAS PubMed; (i) K. Kiyokawa, R. Ito, K. Takemoto and S. Minakata, Chem. Commun., 2018, 54, 7609–7612 RSC; (j) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef CAS PubMed; (k) L. Ye, Y. Tian, X. Meng, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 1129–1133 CrossRef CAS PubMed.
- (a) A. Shibamoto, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2002, 43, 8859–8861 CrossRef CAS; (b) N. Hirai, N. Sawatari, N. Nakamura, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2003, 68, 6587–6590 CrossRef CAS PubMed; (c) X. Baucherel, I. W. C. E. Arends, S. Ellwood and R. A. Sheldon, Org. Process Res. Dev., 2003, 7, 426–428 CrossRef CAS; (d) Y. Kadoh, K. Oisaki and M. Kanai, Chem. Pharm. Bull., 2016, 64, 737–753 CrossRef CAS PubMed.
- R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi and L. Greci, J. Org. Chem., 2003, 68, 1747–1754 CrossRef CAS PubMed.
- For the synthesis of N-hydroxybenzimidazoles, see: (a) D. B. Livingstone and G. Tennant, J. Chem. Soc., Chem. Commun., 1973, 96–97 RSC; (b) I. W. Harvey, M. D. McFarlane, D. J. Moody and D. M. Smith, J. Chem. Soc. Perkin Trans. I, 1988, 1939–1943 RSC; (c) J. M. Gardiner and J. Procter, Tetrahedron Lett., 2001, 42, 5109–5111 CrossRef CAS; (d) J. M. Gardiner, A. D. Goss, T. Majid, A. D. Morley, R. G. Pritchard and J. E. Warren, Tetrahedron Lett., 2002, 43, 7707–7710 CrossRef CAS; (e) N. H. Ansari and B. C. G. Söderberg, Tetrahedron Lett., 2017, 58, 4717–4720 CrossRef CAS PubMed; (f) N. H. Ansari, A. L. Jordan and B. C. G. Söderberg, Tetrahedron, 2017, 73, 4811–4821 CrossRef CAS.
- For biological activities of N-hydroxybenzimidazoles, see: (a) J. M. Gardiner, C. R. Loyns, A. Burke, A. Khan and N. Mahmood, Bioorg. Med. Chem. Lett., 1995, 5, 1251–1254 CrossRef CAS; (b) T. E. Bowser, V. J. Bartlett, M. C. Grier, A. K. Verma, T. Warchol, S. B. Levy and M. N. Alekshun, Bioorg. Med. Chem. Lett., 2007, 17, 5652–5655 CrossRef CAS PubMed; (c) O. K. Kim, L. K. Garrity-Ryan, V. J. Bartlett, M. C. Grier, A. K. Verma, G. Medjanis, J. E. Donatelli, A. B. Macone, S. Ken Tanaka, S. B. Levy and M. N. Alekshun, J. Med. Chem., 2009, 52, 5626–5634 CrossRef CAS PubMed; (d) M. C. Grier, L. K. Garrity-Ryan, V. J. Bartlett, K. A. Klausner, P. J. Donovan, C. Dudley, M. N. Alekshun, S. Ken Tanaka, M. P. Draper, S. B. Levy and O. K. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 3380–3383 CrossRef CAS PubMed; (e) W. Yue and H. Wang, Monatsh. Chem., 2015, 146, 2079–2086 CrossRef CAS; (f) A. E. Marsden, J. M. King, M. A. Spies, O. K. Kim and T. L. Yahr, Antimicrob. Agents Chemother., 2016, 60, 766–776 CrossRef CAS PubMed.
- The use of N-hydroxybenzimidazole for amide condensation reagents has been reported: (a) N. D. Kokare, R. R. Nagawade, V. P. Rane and D. B. Shinde, Synthesis, 2007, 5, 766–772 Search PubMed; (b) N. D. Kokare, R. R. Nagawade, V. P. Rane and D. B. Shinde, Tetrahedron Lett., 2007, 48, 4437–4440 CrossRef CAS; (c) N. D. Kokare and D. B. Shinde, J. Heterocycl. Chem., 2008, 45, 981–986 CrossRef CAS.
- (a) G. Da Silva and J. W. Bozzelli, J. Phys. Chem. C, 2007, 111, 5760–5765 CrossRef CAS; (b) K. Chen, L. Jia, C. Wang, J. Yao, Z. Chen and H. Li, ChemPhysChem, 2014, 15, 1673–1680 CrossRef CAS PubMed; (c) K. Chen, J. Yao, Z. Chen and H. Li, J. Catal., 2015, 331, 76–85 CrossRef CAS.
- For a review on acyl radical chemistry, see: C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2069 CrossRef CAS PubMed.
- (a) S. A. Moteki, A. Usui, S. Selvakumar, T. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060–11064 CrossRef CAS PubMed; (b) S. Selvakumar, R. Sakamoto and K. Maruoka, Chem.–Eur. J., 2016, 22, 6552–6555 CrossRef CAS PubMed; (c) S. Selvakumar, Q. K. Kang, N. Arumugam, A. I. Almansour, R. S. Kumar and K. Maruoka, Tetrahedron, 2017, 73, 5841–5846 CrossRef CAS; (d) R. Sakamoto, N. Hirama and K. Maruoka, Org. Biomol. Chem., 2018, 16, 5412–5415 RSC.
- (a) Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed; (b) N. Blanchard and V. Bizet, Angew. Chem., Int. Ed., 2019, 58, 6814–6817 CrossRef CAS PubMed; (c) G. Prabhu, N. Narendra, Basavaprabhu, V. Panduranga and V. V. Sureshbabu, RSC Adv., 2015, 5, 48331–48362 RSC.
- (a) N. Kielland, M. Vendrell, R. Lavilla and Y.-T. Chang, Chem. Commun., 2012, 48, 7401–7403 RSC; (b) I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjugate Chem., 2017, 28, 1452–1457 CrossRef CAS PubMed.
- (a) G. S. Lal, G. P. Fez, R. J. Pesaresi and F. M. Prozonic, J. Org. Chem., 1999, 64, 7048–7054 CrossRef CAS; (b) C. Chen, C.-T. Chien and C.-H. Su, J. Fluorine Chem., 2002, 115, 75–77 CrossRef CAS; (c) J. M. White, A. R. Tunoori, B. J. Turunen and G. I. Georg, J. Org. Chem., 2004, 69, 2573–2576 CrossRef CAS PubMed; (d) T. Gustafsson, R. Gilmour and P. H. Seeberger, Chem. Commun., 2008, 26, 3022–3024 RSC; (e) F. Beaulieu, L.-P. Beauregard, G. Courchesne, M. Couturier, F. LaFlamme and A. L'Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS PubMed; (f) J.-G. Kim and D. O. Jang, Synlett, 2010, 20, 3049–3052 Search PubMed; (g) G. M. Lee, R. Clément and R. T. Baker, Catal. Sci. Technol., 2017, 7, 4996–5003 RSC; (h) T. Scattolin, K. Deckers and F. Schoenebeck, Org. Lett., 2017, 19, 5740–5743 CrossRef CAS PubMed; (i) S. B. Munoz, H. Dang, X. Ispizua-Rodriguez, T. Mathew and G. K. S. Prakash, Org. Lett., 2019, 21, 1659–1663 CrossRef CAS PubMed; (j) Z. Yang, S. Chen, F. Yang, C. Zhang, Y. Dou, Q. Zhou, Y. Yan and L. Tang, Eur. J. Org. Chem., 2019, 34, 5998–6002 CrossRef.
- Several methods for direct C–H fluorination of aldehydes have been reported: (a) R. E. Banks, N. J. Lawrence and A. L. Popplewell, Synlett, 1994, 831–832 CrossRef CAS; (b) O. Roy, B. Marquet, J. P. Alric, A. Jourdan, B. Morel, B. R. Langlois and T. Billard, J. Fluorine Chem., 2014, 167, 74–78 CrossRef CAS; (c) M. Ochiai, A. Yoshimura, M. M. Hoque, T. Okubo, M. Saito and K. Miyamoto, Org. Lett., 2011, 13, 5568–5571 CrossRef CAS PubMed; (d) M. Meanwell, J. Lehmann, M. Eichenberger, R. E. Martin and R. Britton, Chem. Commun., 2018, 54, 9985–9988 RSC.
- To rule out the possibility of the generation of acyl fluorides from the in situ-generated acid 6a′, we attempted the reaction of 6a′ with Selectfluor in the presence of the NHBI catalyst. However, the formation of acyl fluoride 6a was not observed.
- (a) J. A. Birrell, J.-N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872–13875 CrossRef CAS PubMed; (b) Y. Ogiwara, Y. Maegawa, D. Sakino and N. Sakai, Chem. Lett., 2016, 45, 790–792 CrossRef CAS; (c) K. V. Raghavendra Rao and Y. Vallée, Tetrahedron, 2016, 72, 4442–4447 CrossRef CAS.
- (a) L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef PubMed; (b) X.-A. Liang, L. Niu, S. Wang, J. Liu and A. Lei, Org. Lett., 2019, 21, 2441–2444 CrossRef CAS PubMed; (c) H. Zhao and J. Jin, Org. Lett., 2019, 21, 6179–6184 CrossRef CAS PubMed; (d) F. J. Aguilar Troyano, F. Ballaschk, M. Jaschinski, Y. Özkaya and A. Gómez-Suárez, Chem.–Eur. J., 2019, 25, 14054–14058 CrossRef CAS PubMed.
- (a) C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed; (b) D. D. Bume, S. A. Harry, T. Lectka and C. R. Pitts, J. Org. Chem., 2018, 83, 8803–8814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational studies and characterization for all relevant compounds. See DOI: 10.1039/d0sc02134b |
| This journal is © The Royal Society of Chemistry 2020 |