
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-driven simultaneous water oxidation and quinone reduction by a nano-structured conjugated polymer without co-catalysts†‡
Jully
Patel
a,
Xiaojiao
Yuan
b,
Stéphanie Mendes
Marinho
a,
Winfried
Leibl
 *a,
Hynd
Remita
*b and
Ally
Aukauloo
*ac
*a,
Hynd
Remita
*b and
Ally
Aukauloo
*ac
aInstitut des Sciences du vivant Frédéric Joliot, SB2SM/Institut de Biologie Intégrative de la Cellule I2BC, UMR 9198, CEA, CNRS, Université Paris Sud, F-91191 Gif sur Yvette, France. E-mail: winfried.leibl@cea.fr
bInstitut de Chimie Physique (ICP), UMR 8000 CNRS, Université Paris Sud, Université Paris-Saclay, F-91405 Orsay Cedx, France. E-mail: hynd.remita@universite-paris-saclay.fr
cInstitut de Chimie Moléculaire et des Matériaux d'Orsay, UMR-CNRS 8182, Université Paris-Sud, Paris-Saclay, F-91405 Orsay, France. E-mail: ally.aukauloo@universite-paris-saclay.fr
First published on 12th June 2020
Abstract
In artificial photosynthesis, chemists are aiming to borrow principles from natural photosynthesis to develop photoelectrochemical cells (PEC) for water splitting. The water plastoquinone photo-oxidoreductase enzyme, also known as photosystem II, uses light to perform the four-electron, four-proton oxidation of water to dioxygen and stores reducing equivalents in reduced forms of quinones which are ultimately used in dark reactions for the synthesis of energy-rich molecules. We report a nano-structured semiconducting conjugated polymer based on poly(diphenylbutadiyne) (nano-PDPB) and its photocatalytic activities towards the water oxidation reaction under visible light irradiation when dispersed in water in the absence of any sacrificial agents or co-catalysts. Charge recovery at the nano-PDPB directly or delayed in time was exemplified by the reduction of quinone acting as a hydrogen reservoir. In the absence of quinones as electron acceptors H2O2 formation was detected, stemming from the partial reduction of O2.
Introduction
Solar energy could be harnessed and stored as a fuel using water as a renewable source of electrons and protons.1 Among the different materials under intense investigation towards this aim, organic semiconductors have captured immense attention for their synthetic readiness, robustness and handles to adjust the energy levels of their conduction and valence bands. Domen and Antonietti first reported the use of g-C3N4 as a photocatalyst for hydrogen production.2 These findings triggered a vibrant research effort in the design of organic semiconductors for photocatalytic water splitting. Unfortunately, progress towards functional materials for the overall water splitting reaction has been mitigated due to the challenging and limiting step of the four-electron water oxidation reaction.3 Two strategies are currently employed to bypass this hurdle, the adjunction of a known metal-based water oxidation co-catalyst or replacing water by sacrificial electron donors which provide the electrons necessary for the production of hydrogen. Henceforth, great effort is needed to engineer organic photocatalytic materials that can genuinely perform the water oxidation reaction coupled with usage of the liberated electrons and protons.4 Advancement in this prolific research theme has recently been reviewed based on the important discoveries by Mullins with a bird's eye view on the challenges ahead of chemists to reach cost-effective metal-free photocatalysts.5,6 Significant examples from Xu and co-workers demonstrated overall water splitting using only conjugated microporous polymers.7 Another original approach was to develop the Z-scheme scenario mimicking the serial operation of two charge separation systems found in oxygenic photosynthesis.8,9 Heterostructures interfacing two different polymer photocatalysts for O2 and H2 release, respectively, were successfully demonstrated.8,9 Liu et al. also reported a metal-free CDots/C3N4 system that leads to overall water splitting with an overall conversion efficiency of ∼2% under visible light irradiation.10 Beyond the photon absorption and charge separation process further challenges come from coupling the photo oxidation and reduction chemistry and separating O2 and H2 in the gas mixture. The inherent chemical complexity to build such systems together with the challenge of suppressing short-circuit reactions between the oxidation and reduction parts also hampers advancement in this field.10–19 Therefore, decoupling the oxidative and the reductive processes in time could be an alternative way to capture and store solar energy.Here we report that visible light irradiation of a suspension of nano-structured conjugated polymers based on polydiphenylbutadiyne (nano-PDPB herein) in water leads to O2 evolution. Intriguingly, no H2 was detected. Instead we showed that the electrons and protons from water oxidation are stored on the nanostructured semiconductor and could be recovered as a reduced form of quinone paralleling the functions of photosystem II (Scheme 1).
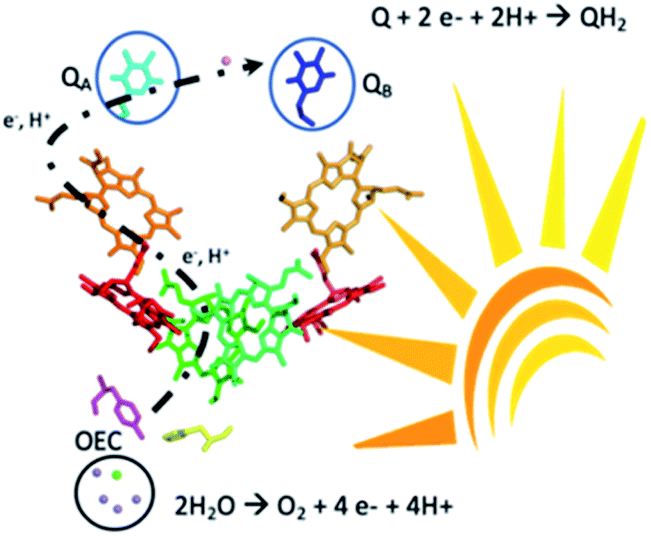 | ||
| Scheme 1 Map of the main cofactors of water plastoquinone photo-oxidoreductase (PSII) and reactions for water oxidation at the oxygen evolving complex (OEC) and reduction of quinones QA and QB under sunlight irradiation. | ||
Results and discussion
As a short summary of our previous report, nano-PDPB was obtained from the controlled polymerization of diphenylbutadiyne (DPB) in the confined space of soft templates by radiolysis as we described (Scheme 2).20 | ||
| Scheme 2 Soft template photopolymerization of diphenylbutadiyne. | ||
Transmission electron microscopy (TEM) images of nano-PDPB indicate nanofibers of diameter ∼20 nm and some tens of micrometers long (Fig. 1). The MALDI-TOF mass spectrometry technique indicates the presence of short oligomers with 5 to 8 repeating units of DPB (see ESI Fig. S2‡). While gel-permeation chromatography supports mainly a molar mass of nano-PDPB estimated around 1625 g mol−1 corresponding to the formulation of a degree of polymerisation of 8.20 The nano-PDPB in the solid state presents a broad absorption band covering most of the visible region. The Kubelka–Munk and Tauc plot analyses (see ESI Fig. S3‡ and 1, left) give a direct band-gap value of 2.37 eV. This value is in good agreement with the maximum of the photoemission band detected at 530 nm after laser flash excitation of an acetonitrile solution of nano-PDPB (see ESI Fig. S4‡) corresponding to a HOMO–LUMO gap of ca. 2.34 eV. From the cyclic voltammetry studies, the conduction band minimum and valence band maximum of the nano-PDPB were located within the potential window of −0.6 V and 1.4 V (vs. NHE), respectively.20 These band positions suggest that from a thermodynamic point of view nano-PDPB could act as a photocatalyst for the water splitting reaction (Fig. 1, right).
 | ||
| Fig. 1 (Left) Tauc plot; middle: TEM image of nano-PDPB; and (right) energy diagram representing the evaluated valence band (V.B.) and conduction band (C.B.) levels of the PDPB polymer with regard to the water splitting reactions. | ||
From a kinetic point of view, the transient absorption data indicate that the emissive singlet excited state is evolving to a long-lived charge separated state (see ESI Fig. S4‡). Irradiating a solution of PDPB in acetonitrile with visible light in the presence of a sacrificial electron donor such as triethanolamine (TEOA) and methyl viologen (MV2+) as an electron acceptor leads to the formation of a persistent blue solution, characteristic of the formation of the methyl viologen radical MV+˙ (Fig. 2a). Considering the potentials of TEOA (0.84–1.14 V/NHE)21 and MV2+ (−0.45 V/NHE)22 this result is consistent with the estimated band positions.
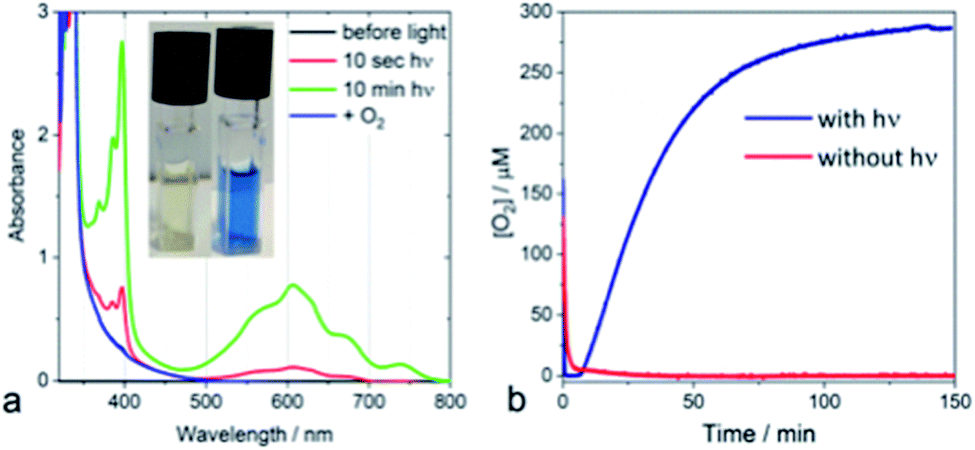 | ||
| Fig. 2 (a) Spectral changes induced in an Ar-purged solution of nano-PDPB, 100 μM MV2+, 10% TEOA in MeCN by illumination with blue light and after aeration (inset: formation of the blue MV+˙ radical). (b) Time courses of oxygen evolution from water measured by a Clark electrode. Blue curve: with nano-PDPB (synthesized in mesophases) and red curve: no irradiation. | ||
The photocatalytic activity of nano-PDPB was tested by suspending 2 mg of the orange solid heterogeneously in 2 mL of pure water (without the presence of any co-catalyst or sacrificial agents) in a water-cooled Clark electrode maintained at 20 °C and subject to visible light irradiation (λ ≥ 435 nm) under stirring (see ESI‡). The time course of the evolution of O2 is represented in Fig. 2b.
After a time lag of a couple of minutes, the evolution of O2 follows a sigmoid curve that levels off at a concentration of around 250 μM (saturated solution of O2 in water) after irradiation for roughly one hour. The evolved O2 was detected and quantified by GC-MS and the Clark electrode, respectively.23 No O2 production was observed in the control experiments without nano-PDPB (see ESI Fig. S5‡) or when the nano-PDPB suspension was left in the dark (Fig. 2b). Importantly, photocatalysis with H218O indicates the formation of 36O2 detected by GC-MS, providing clear proof that the O2 comes from the genuine oxidation of water (see ESI Fig. S6‡). We also found that the O2 evolution was a function of the intensity of the irradiation and the spectral window (see ESI Fig. S7‡). An important point to check before validating the intrinsic photocatalytic activity of an organic material as a photocatalyst is the insidious participation of traces of metal ions used during the synthetic process. We excluded the presence of metal ions upon examination using the EDX method (see ESI Fig. S1‡). To ascertain this result, inductively coupled plasma mass spectrometry (ICP-MS) was used to evaluate the presence of metal ions, more specifically of copper or palladium. These salts are commonly used in the coupling of terminal alkyne groups. No trace of copper was found within the limit of detection. In a control photocatalytic experiment, we deliberately added a copper(II) salt and we found a longer time delay and weaker photocatalytic activity for O2 release (see ESI Fig S8‡). This can be supported by a similar quenching mechanism as proposed for the g-C3N4 material.24 It has to be noted that no significant activity was obtained with bulk-PDPB (see ESI Fig. S9‡), indicating that the nano-structuration of the conjugated polymers is crucial for photocatalytic activity. A rough estimation of the internal quantum yield (photon-to-electron) in the wavelength interval 450–700 nm yielded a value of 12% (see ESI‡ for details). Surprisingly, no H2 gas could be detected under our experimental conditions (see ESI Fig. S10‡). This could appear intriguing as the band-gap structure diagram suggests that the reduction potential for the H+/H2 couple is indeed below the level of the conduction band of nano-PDPB. Such an observation can be assigned to kinetics issues for the formation of H2 at the surface of the nano-structured polymeric material. An archetypal example is bare TiO2, where H2 production under UV irradiation is very low and catalytic nanoparticles are needed for the formation of the H–H bond.25–27 As already pointed out in the literature, traces of palladium originating from the synthesis of the material are often involved as the catalytic species responsible for H2 production. In our case no H2 formation was observed and ICP-MS also rules out the presence of Pd (see ESI Table S1‡).
Inspired by the functions of PS II, we added a quinone derivative, dichlorobenzoquinone (DCBQ), to the mixture at different oxygen concentrations upon photocatalysis until the plateau for O2 evolution was reached. A rapid change in color of the solution from colorless to purple was observed with an intensity increasing with the concentration of O2 produced (Fig. 3a, ESI Fig. S11 and S18‡). This purple solution presents an absorption band with a maximum at 530 nm. This absorption feature is characteristic of the formation of a quinhydrone complex exhibiting a charge transfer band between reduced hydroquinone and quinone.28–32 The formation of such a redox pair provides unambiguous support for the charge accumulation on the nano-PDPB upon the water photo–oxidation reaction. The storage of electrons and protons on metal oxide semiconductors such as TiO2 for instance has recently opened new perspectives in solar energy conversion.33 This finding is of major interest, as nano-PDPB can be considered as a photocatalytic material where light-driven oxidation of water and dark phase chemical energy storage can be delayed in time.34–37 In a complementary test, we added silver nitrate to the nano-PDPB suspension in water when the plateau of O2 evolution was reached and the irradiation was ceased. Silver nanoparticles were found to decorate the nano-PDPB fibers, as confirmed by TEM images (Fig. 3b). This result further confirmed that the nano-PDPB is acting as the location for the charge accumulation upon photooxidation of water.
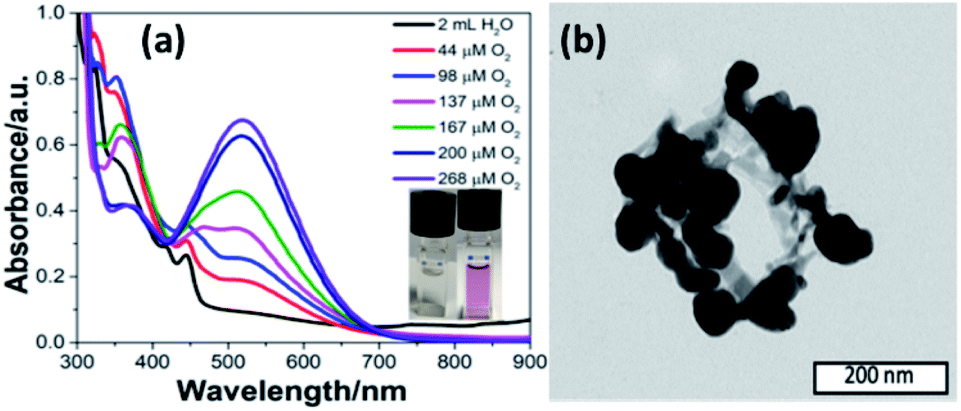 | ||
| Fig. 3 (a) UV-visible spectra of 1 mM DCBQ in 2 mL photo catalytic solution at different concentrations of evolved oxygen (inset: visible colour change upon formation of quinhydrone). (b) TEM image of Ag NPs decorating PDPB nanofibers after addition of AgNO3 ions in the dark after a photocatalytic run. | ||
With these findings, we reasoned that adding an exogenous electron acceptor may help to evacuate the built-up charges on the nano-PDPB and thereby would enhance the photocatalytic water oxidation reaction. In a first attempt, we added an excess of DCBQ at the beginning of the photocatalytic run. Accordingly, we determined a noticeable improvement in the photocatalytic O2 evolution (Fig. 4a). When an excess of sodium persulfate, an irreversible electron acceptor, was added followed by irradiation, we noticed a vertiginous increase in O2 production (Fig. 4a). With this very efficient electron acceptor the concentration of dissolved O2 increases to levels far above the 250 μM expected for an air-equilibrated solution within some minutes. This fast rate of production does not allow establishment of an equilibrium between dissolved O2 and the head space volume (see ESI Fig. S12‡). A control experiment with alternate periods of dark and illumination in the presence of S2O82− was carried out to rule out the decomposition of S2O82− with the release of O2 pathway (see ESI Fig. S13‡). The time-course of the O2 evolution curve clearly indicates that O2 is generated only in the light excitation phase while no O2 is released in the dark condition thus confirming that the liberated O2 comes from the light-driven oxidation of water. Control experiments with persulfate in the dark or irradiating the electron acceptor in the absence of nano-PDPB did not indicate the formation of any trace of O2 (see ESI Fig. S14‡). Noteworthily, in weakly buffered water we registered a noticeable drop in the pH of the solution from 7 to 2.5 upon the evolution of O2 in the presence of S2O82− ions corresponding to the occurrence of water oxidation with the release of protons. In contrast, no marked pH change was registered when quinone was present, indicating the consumption of the protons upon reduction of the quinone, as expected. Also in the absence of an exogenous electron acceptor, no marked variation of the pH was evident, thus providing more support to the fact that protons are being stored at the nano-PDPB. In an attempt to obtain more information about such a hypothetical storage of reducing equivalents on the PDPB, we measured changes in the FTIR spectra during illumination in the presence of H2O or D2O (see ESI Fig. S15‡). These data clearly point to noticeable changes in the spectral window of the C–C aromatic ring stretch region with bands appearing at 1350 and 1525 cm−1 corresponding to π-conjugated enyne and aromatic rings. The H/D isotopic effect can be tentatively attributed to the changes in the electron density together with the accumulations of protons/deuterons at the nano-PDPB. Interestingly, we also found that the presence of an electron acceptor from the start of the photocatalysis led to the disappearance of the lag period before the detection of O2. We attribute this observation to the more efficient accumulation of holes in the valence band upon rapid evacuation of the electrons from the conduction band resulting in reduced charge recombination.
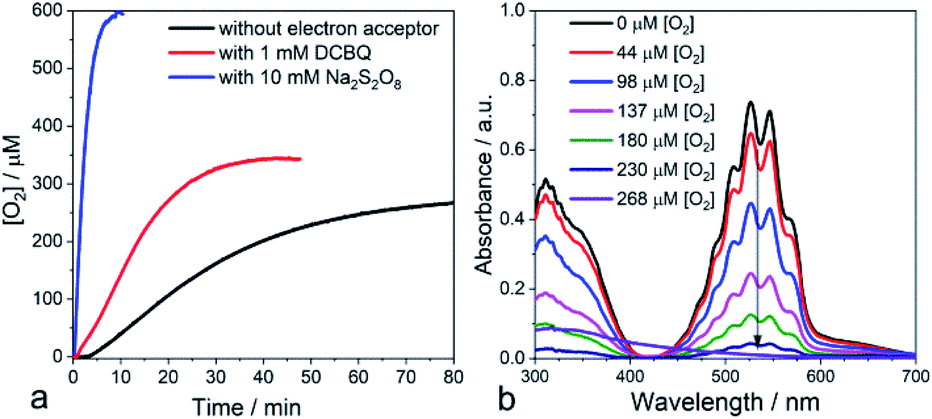 | ||
| Fig. 4 (a) Time courses of oxygen evolution from water measured by a platinum Clark electrode in the absence of an electron acceptor (black) and in the presence of DCBQ (red) and S2O82− as electron acceptors (blue). (b) Titration of H2O2 with KMnO4 at different concentrations of photoproduced O2 in the absence of an electron acceptor. | ||
Another important observation comes from the monitoring of the oxygen production when irradiation was ceased (in the absence of external electron acceptors). We consistently observed a small consumption of O2 (see ESI Fig. S16‡) while the temperature of the mixture was controlled. Actually, any drop in temperature upon stopping the irradiation would have led to a small increase in O2 in the aqueous phase. We assign this consumption to the partial reduction of dioxygen at the surface of the nano-PDPB by the accumulated photogenerated charges to form reduced oxygen species such as O2•−. It is important to notice that detection of sustained production of O2 in our system supports the fact that O2 release from oxidation of water largely outcompetes the partial O2 consumption due to its reduction to O2•−. Such reduced oxygen species are quite short-lived and are expected to undergo dismutation to form H2O2.38 We thus checked the presence of H2O2 by titration of the reaction solution with KMnO4.39 Interestingly, titration at the beginning of the photocatalytic experiment revealed only a negligible formation of H2O2. However, a titration realized during the time course of O2 evolution clearly attests to the presence of H2O2 by the fading of the prominent violet permanganate color, as shown in Fig. 4b (see also ESI Fig. S17 and S18‡). Further support for this attribution comes from the experiment with a sacrificial electron acceptor where no lag time for O2 production was found and no trace of H2O2 was detected after photocatalysis. These observations and the time course of O2 and H2O2 production (see ESI Fig. S18 and S13‡) suggest that the two-electron oxidation of H2O to H2O2, unfavorable also from an energetic point of view, can be disregarded. With a focus to access the recyclability of the nano-PDPB, we isolated the nano-PDPB after photocatalysis in the absence of an electron acceptor, air dried it and reused it in fresh N2 degassed water. As we can see (see ESI Fig. S19‡) for the same time lapses, the amount of O2 produced stays almost constant. These experiments suggest that nano-PDPB can be efficiently reused for repeated cycles without any appreciable loss of activity. A crucial concern for all materials that can perform the water oxidation reaction is their chemical alteration during the photocatalytic reaction. In this study, the mineralization of the nano-PDPB to CO or CO2 was excluded as none of these components was detected by gas chromatography (see ESI Fig S10‡). All gathered spectroscopic data, FTIR (see ESI Fig. S20‡), 1H and 13C NMR (see ESI Fig. S21 and S22‡) as well as MALDI/-TOF analysis (see ESI Fig. S2‡) and SEM images (see ESI Fig. S23‡) before and after completion of a photocatalytic cycle did not indicate any marked change, thereby attesting to the fact that nano-PDPB conserves the same chemical formulation and morphology.
Conclusions
In summary, we found that nano-PDPB, a nanostructured semiconducting conjugated polymer, can replicate the processes occurring in photosystem II, by oxidizing water to O2 under visible light irradiation and accumulating electrons on the nanostructured material recoverable later in time to reduce quinone.Photosystem II upon excitation by light splits water producing molecular dioxygen (2H2O → O2 + 4H+ + 4e−) on one side of the membrane and storing electrons and protons by the reduction of plastoquinone to plastoquinol (PQ + 2e− + 2H+ → PQH2) on the other. The plastoquinol then escapes the enzyme transporting electrons and protons to a chain of electron transfer proteins to be ultimately fixed on carbon dioxide (Scheme 1).40 Even after decades of effort, bioinspired molecular models of this enzyme have not yet replicated all these functions in one system. However, much has been accomplished with great success to model the separate parts of this multi-cofactor biological system.41,42
Of particular interest, nano-PDPB can realize these reactions without the need of a co-catalyst, and in the absence of sacrificial reagents.43 The formation of H2O2 can be assigned to the partial reduction of O2 acting as an escape channel of charge relief at the photocatalytic material.19 In all, these findings open new perspectives in the field of artificial photosynthesis to convert solar energy and store it in a chemical form without the need to separate the gaseous mixture produced by the water splitting reaction. Preliminary results also indicate that nano-PDPB can also oxidize natural water from a river source with the same efficiency (see ESI Fig. S24‡) and be used on a TiO2 photoanode (see ESI Fig. S25‡). These results will be further confirmed in upcoming studies. Many challenges are ahead of us to fully understand the fundamental charge transport dynamics to unravel the photophysical processes and mechanistic aspects of these carbon-based materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. P. is indebted to the IRS Université Paris-Saclay MOMENTOM program for a postdoctoral grant. Université Paris-Sud (ERM project) is acknowledged for financial support for the Cobalt-60 panoramic gamma source and LABEX CHARMMMAT and NANOSACLAY for technical support. This work was supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01. P. Mejanelle is acknowledged for the ICP-OES measurement.Notes and references
- K. Sanderson, Nature, 2008, 452, 400–402 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- M. Z. Rahman, K. Davey and C. B. Mullins, Adv. Sci., 2018, 5, 1800820 CrossRef PubMed.
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- Y. Xia, K. Xiao, B. Cheng, J. Yu, L. Jiang, M. Antonietti and S. Cao, ChemSusChem, 2020, 13, 1730–1734 CrossRef CAS.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef.
- L. Favereau, A. Makhal, Y. Pellegrin, E. Blart, J. Petersson, E. Goransson, L. Hammarstrom and F. Odobel, J. Am. Chem. Soc., 2016, 138, 3752–3760 CrossRef CAS PubMed.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed. Engl., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645 CrossRef CAS PubMed.
- Y. Wang, H. Li, J. Yao, X. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- C. Butchosa, P. Guiglion and M. A. Zwijnenburg, J. Phys. Chem. C, 2014, 118, 24833–24842 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- S. Matsuoka, H. Fujii, T. Yamada, C. Pac, A. Ishida, S. Takamuku, M. Kusaba, N. Nakashima and S. Yanagida, J. Phys. Chem. C, 1991, 95, 5802–5808 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed. Engl., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
- L. Wang, Y. Wan, Y. Ding, Y. Niu, Y. Xiong, X. Wu and H. Xu, Nanoscale, 2017, 9, 4090–4096 RSC.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- H. Sun and M. Z. Hoffman, J. Phys. Chem., 1994, 98, 11719–11726 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- L. C. Clark Jr, R. Wolf, D. Granger and Z. Taylor, J. Appl. Physiol., 1953, 6, 189–193 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- J. B. Joo, R. Dillon, I. Lee, Y. Yin, C. J. Bardeen and F. Zaera, Proc. Natl. Acad. Sci., 2014, 111, 7942 CrossRef CAS PubMed.
- A. L. Luna, E. Novoseltceva, E. Louarn, P. Beaunier, E. Kowalska, B. Ohtani, M. A. Valenzuela, H. Remita and C. Colbeau-Justin, Appl. Catal., B, 2016, 191, 18–28 CrossRef CAS.
- M. G. Méndez-Medrano, E. Kowalska, A. Lehoux, A. Herissan, B. Ohtani, D. Bahena, V. Briois, C. Colbeau-Justin, J. L. Rodríguez-López and H. Remita, J. Phys. Chem. C, 2016, 120, 5143–5154 CrossRef.
- T. Sakurai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 403–412 CrossRef CAS.
- J. E. Guillet, Pure Appl. Chem., 1991, 63, 917–924 CAS.
- M. Bouvet, B. Malezieux and P. Herson, Chem. Commun., 2006, 1751–1753, 10.1039/b600345a.
- J. Regeimbal, S. Gleiter, B. L. Trumpower, C.-A. Yu, M. Diwakar, D. P. Ballou and J. C. A. Bardwell, Proc. Natl. Acad. Sci., 2003, 100, 13779 CrossRef CAS PubMed.
- A. Pawlukojć, I. Natkaniec, G. Bator, E. Grech and L. Sobczyk, Spectrochim. Acta, Part A, 2004, 60, 2875–2882 CrossRef PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298 CrossRef CAS PubMed.
- T. Wilke, M. Schneider and K. Kleinermanns, Open J. Phys. Chem., 2013, 03(02), 6 Search PubMed.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403–409 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed.
- S. Er, C. Suh, M. P. Marshak and A. Aspuru-Guzik, Chem. Sci., 2015, 6, 885–893 RSC.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS.
- N. V. Klassen, D. Marchington and H. C. E. McGowan, Anal. Chem., 1994, 66, 2921–2925 CrossRef CAS.
- J. Messinger, W. Lubitz and J. R. Shen, Phys. Chem. Chem. Phys., 2014, 16, 11810–11811 RSC.
- L. Hammarström, Chem, 2016, 1, 515–518 Search PubMed.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879–17893 CrossRef.
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673–677 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Pr. Jean-Jacques Girerd for his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, additional reference experiments, spectroscopic and structural results. See DOI: 10.1039/d0sc02122a |
| This journal is © The Royal Society of Chemistry 2020 |