
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic details of the cobalt-mediated dehydrogenative dimerization of aminoquinoline-directed benzamides†
Li-Ping
Xu
 abc,
Elaine E. L.-N.
Liu
b,
John
Bacsa
b,
Cora E.
MacBeth
*b and
Djamaladdin G.
Musaev
*ab
abc,
Elaine E. L.-N.
Liu
b,
John
Bacsa
b,
Cora E.
MacBeth
*b and
Djamaladdin G.
Musaev
*ab
aCherry L. Emerson Center for Scientific Computation, Emory University, Atlanta, Georgia 30322, USA. E-mail: dmusaev@emory.edu
bDepartment of Chemistry, Emory University, Atlanta, Georgia 30322, USA. E-mail: cmacbet@emory.edu
cSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, 255000, China
First published on 19th May 2020
Abstract
Key mechanistic features of the cobalt-mediated and aminoquinoline-directed dehydrogenative aryl–aryl coupling were investigated computationally and experimentally. A series of CoII and CoIII complexes relevant to the proposed reaction cycle have been synthesized and characterized. Stoichiometric reactions and electrochemical studies were used to probe the role of different additives in the reaction pathway. Computationally, three different mechanisms, such as charge neutral, anionic, and dimetallic were explored. It is shown that the mono-metallic anionic and charge neutral mechanisms are the most favorable ones, among which the former mechanism is slightly more encouraging and proceeds via the: (a) concerted-metalation-deprotonation (CMD) of the first benzamide C–H bond, (b) PivOH-to-PivO− rearrangement, (c) CMD of the second benzamide C–H bond, (d) C–C coupling, (e) product formation facilitated by the amide nitrogen re-protonation, and (f) catalyst regeneration. The rate-determining step of this multi-step process is the C–C coupling step. The computational studies suggest that the electronics of both the aryl-benzamide and pyridine fragments of the aminoquinoline-benzamide ligand control the efficiency of the reaction.
Introduction
The prevalence of aryl–aryl and aryl-heteroaryl structural motifs in natural products, pharmaceutically active compounds, and functional materials has commanded the development of effective and inexpensive strategies for biaryl synthesis.1–3 Many widely used aryl–aryl coupling methodologies, however, require pre-activated substrates such as aryl halides and aryl organometallic reagents that generate undesirable byproducts.4 Therefore, the search for cost-effective and sustainable methods of biaryl synthesis remains an active field of chemical research. In this context, transition-metal-catalyzed dehydrogenative C–H/C–H coupling is a promising strategy.5–9 Many advances in this area of research have relied on the use of expensive second-row transition metal (e.g., palladium, rhodium, and ruthenium) complexes and thus limits their application in large-scale industrial processes.10–12 Therefore, catalysts that incorporate relatively inexpensive and earth-abundant first-row transition metals are attractive alternatives. Recent research by several groups has led to the discovery of multiple cobalt and copper promoted aryl–aryl and aryl-heteroaryl dimerization reactions.13–17Scheme 1a highlights a recent example of the Co-mediated homocoupling of aminoquinoline (AQ) benzamides.16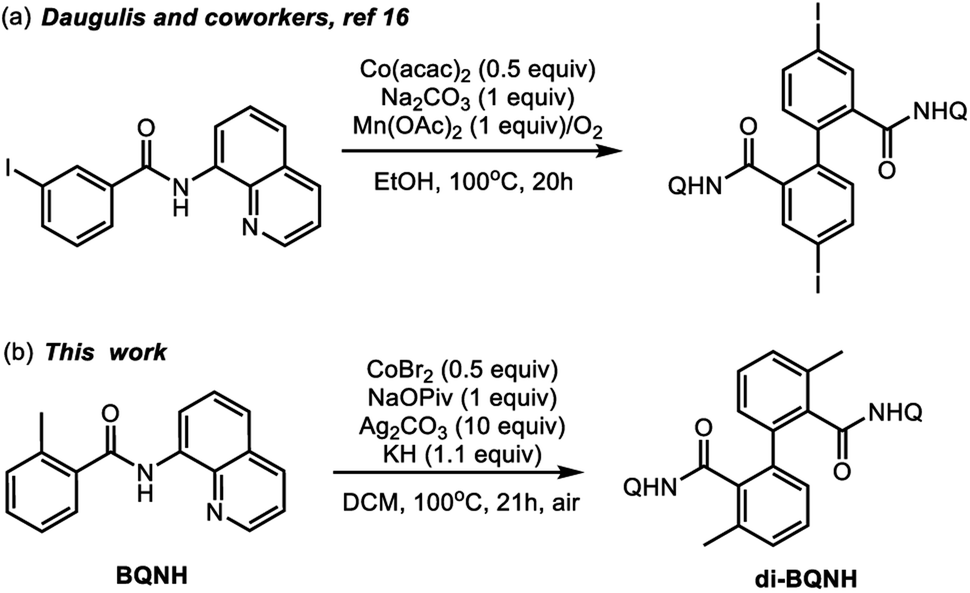 | ||
| Scheme 1 Representative of the cobalt-mediated dehydrogenative dimerization of aminoquinoline benzamide derivatives. | ||
In light of these synthetic advances, the key mechanistic features of the first-row transition metal mediated dehydrogenative coupling of AQ benzamides warrants comprehensive investigations. Advancing fundamental understanding of these transformations could aid in the design of more effective, selective, and cost-efficient catalysts for first-row transition metal mediated dehydrogenative biaryl synthesis. Here, intrigued by the initial reports by Daugulis and coworkers (Scheme 1a)16 on the cobalt-mediated homocoupling of AQ benzamides, we launched a joint experimental and computational study to elucidate the mechanism and controlling factors of this reaction.
This work adds to and compliments a growing number of mechanistic and theoretical studies exploring cobalt-mediated organic transformations that have highlighted multiple challenges involved in computational investigations of Co-complexes, including lower-lying multiple electronic and spin states and the important role of dispersive interactions.18–24
Results and discussion
Initial complex formation
Under the reported reaction conditions, the deprotonation of substrate (BQNH) and its bidentate coordination to a Co(II)-center of the pre-catalyst CoII(OPiv)2 is expected to be an initial step of the reaction. To validate this hypothesis, we calculated free energy of the reaction,| 2BQNH + CoII(OPiv)2 → CoII(BQN)2 + 2HOPiv |
which is found to be exergonic by 6.2 kcal mol−1. The calculations show that the neutral CoII(BQN)2 complex (1) has a distorted tetrahedral geometry with π–π stacking interactions between the aryl rings and quinoline moieties (Fig. 1). The calculated distances between the aryl rings and quinoline moieties are in range of 3.37–3.61 Å (see ESI†). The electronic ground state of this CoII(d7)-species is a high-spin quartet state with 2.92|e| α-spin on the Co-center. The doublet low-spin excited state of 1 has 0.99|e| α-spin on the Co-center and is 11.7 kcal mol−1 higher in free energy.
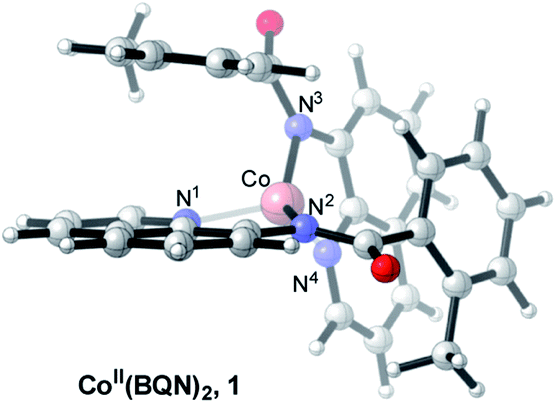 | ||
| Fig. 1 The calculated structure of initial intermediate CoII(BQN)2, 1. For its selected geometry parameters, see Fig. 2, as well as ESI.† | ||
To verify that postulated complex 1 is synthetically viable during the CoII mediated dehydrogenative aryl–aryl coupling reaction, we synthesized CoII(BQN) or Co(κ2-(2-methylbenzoyl)(quinolin-8-yl)amide)2 (1) as shown in Scheme 2.
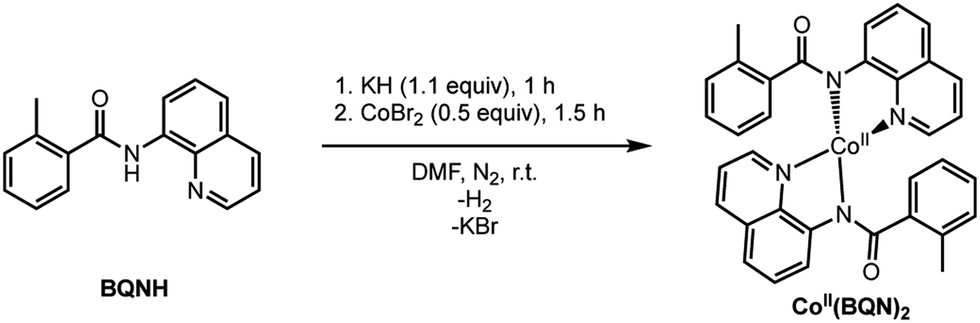 | ||
| Scheme 2 Synthetic protocol for synthesis of CoII(BQN)2, 1. | ||
This complex represents, to the best of our knowledge, the first reported four-coordinate CoII-aminoquinoline benzamide complex. The complex was characterized by 1H NMR, UV-visible spectroscopy, IR spectroscopy, magnetic moment, mass spectrometry, and X-ray crystallography (see ESI† for full Experimental details). Diffusion of petroleum ether into a concentrated dichloromethane solution of the complex yielded bright red crystals suitable for single-crystal X-ray diffraction (Fig. 2).
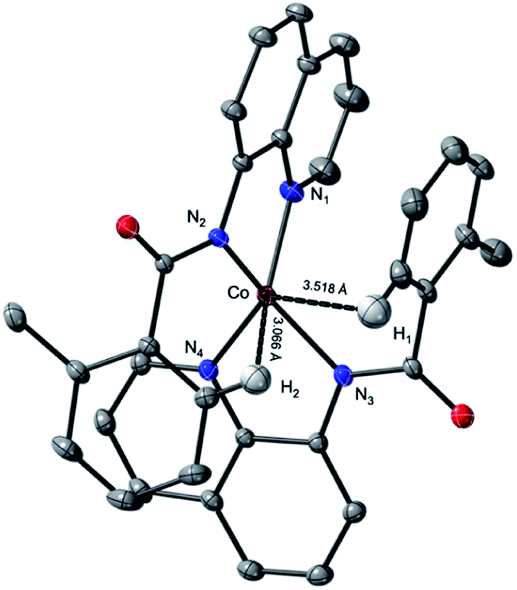 | ||
| Fig. 2 Thermal ellipsoid plot of CoII(BQN)2, (1). Ellipsoids drawn at 50% probability; H-atoms have been omitted for clarity with exception of H1 and H2 showing the intramolecular Co–H distances. Select distances (in brackets are given the calculated values): Co–N1: 2.024(1) [2.06] Å; Co–N2: 1.967(1) [2.00] Å; Co–N3: 1.967(1) [2.00] Å; Co–N4: 2.024(1) [2.06] Å. Select angles: N1–Co–N2: 83.1°; N1–Co–N4: 119.9°; N3–Co–N4: 82.9°; N2–Co–N4: 122.1°. | ||
The crystal structure reveals a bis-ligated Co(II) species with a distorted tetrahedral Co-center exhibiting a τ4 = 0.84 (where τ4 = 1 for a tetrahedral geometry, τ4 = 0 for square planar geometry).25 This geometry aligns the metal center with both of the aryl ortho C–H bonds. One of the hydrogen atoms is situated 3.066 Å from the cobalt and this close proximity poises the hydrogen for an agostic interaction with the metal center, which may help facilitate C–H bond activation.26,27 The X-ray structural data obtained for CoII(BQN)2 supports our computational findings that show the corresponding Co–H bond distance of 3.076 Å. The π–π stacking interactions between the aryl rings and quinoline moieties predicted in the computation are also present in the experimentally determined structure. There is evidence for π–π stacking from the way that the quinoline and aryl moieties are oriented in the complex. The two planes are approximately coplanar with a plane fold angle of 175 degrees and a twist angle of 177 degrees. The plane-to-plane-centroid distance is 3.429 Å. The calculated and measured C–N bond distances for 1 are also in good agreement. The paramagnetic susceptibility of CoII(BQN)2 was measured to be μeff = 4.41 μ using the Evans method.28 This experimental finding is consistent with the computed quartet (S = 3/2) ground electronic state of the complex.
UV-visible absorption spectroscopy of 1 in dichloromethane shows a metal-to-ligand charge transfer at 400 nm and three d–d transitions at 535, 590, and 617 nm (Fig. 3A). To probe redox profile of CoII(BQN)2 (1), we examined the complex using cyclic voltammetry. In acetonitrile (0.1 M TBAPF6), we observed a reversible, one-electron oxidation at E1/2 = −2.275 V (Fig. 3B) suggesting the complex should be readily oxidized. In DMF, however, complex 1 shows two quasi-reversible oxidation events at E1 = −2.20 V and E2 = −1.79 V (see Fig. 5A).
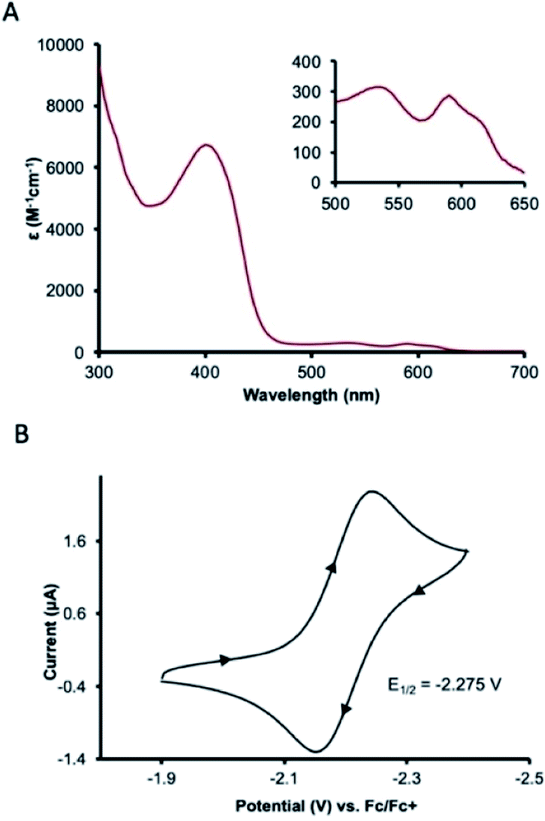 | ||
| Fig. 3 Characterization data for CoII(BQN)2 (1). (A) UV-visible absorption spectrum in CH2Cl2, inset: d–d transitions, and (B) cyclic voltammogram, scan rate 100 mV s−1 with 0.1 M TBAPF6 as the supporting electrolyte in CH3CN. Referenced vs. Fc/Fc+. | ||
To validate and provide an additional experimental support to this expectation, we synthesized and characterized AQ complexes of cobalt with both pivalate, PPh4[CoII(BQN)(PivO)2] (2), and acetate, PPh4[CoII(BQN)(OAc)2] (3) ligands (Scheme 3).
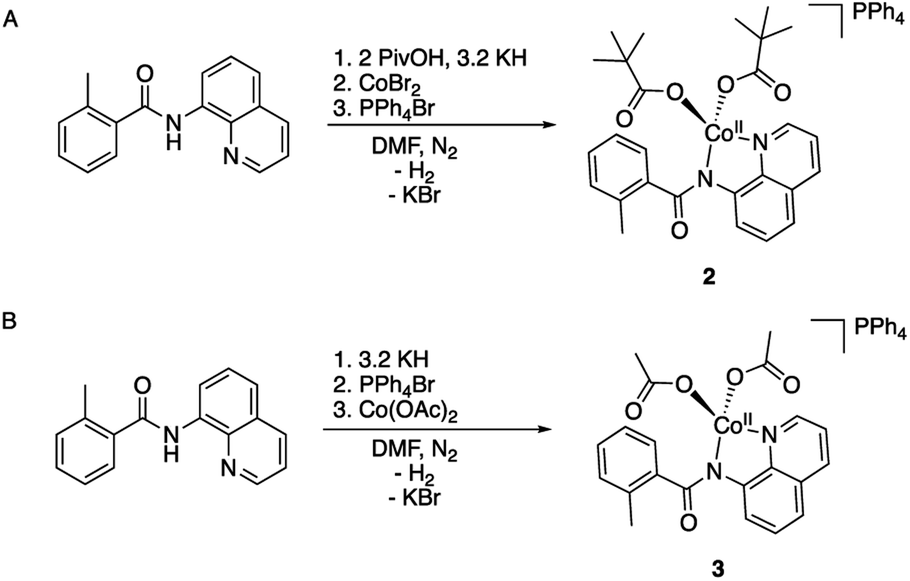 | ||
| Scheme 3 Synthesis of the carboxylate and aminoquinoline containing complexes (A) PPh4[CoII(BQN)(PivO)2] (2), and (B) PPh4[CoII(BQN)(OAc)2] (3). | ||
The crystal structures of complexes 2 and 3 (Fig. 4) have revealed that the carboxylate ligands coordinate to the cobalt center in a κ1-coordination mode. The Co(II) center in both 2 and 3 exhibits distorted tetrahedral geometries with τ4 values of 0.90 and 0.83, respectively.25 The paramagnetic susceptibility of these complexes was measured using the Evans' method and determined to be μeff = 4.40 μB and 4.20 μB, respectively consistent with S = 3/2 spin states. The absorbance spectra of 2 and 3 show a metal-to-ligand charge transfer at 378 nm and 381 nm, respectively. Complex 2 has a single d–d transition at 593 nm, while complex 3 has two d–d transitions at 498 nm and 591 nm. Attempts to isolate a five-coordinate cobalt complex with two AQ ligands and one carboxylate-type ligand, however, were unsuccessful. To further explore possible carboxylate binding, we carried out an electrochemical titration of complex 1 with exogenous acetate (Fig. 5A). This experiment shows a new irreversible redox event at E1/2 = −2.3 V, which is more negative than the initial oxidation event observed for complex 1 (E1/2 = −1.8 V), formation of a new cobalt species that is easier to oxidize. The electrochemical responses observed during the acetate titration of complex 1 are very similar to the cyclic voltammogram of isolated PPh4[CoII(BQN)(OAc)2] (3), Fig. 5B. Both species show events at roughly the same potential. This indicates that, with added acetate, complex 1 is experiencing an altered ligand environment with similar electrochemical properties to complex 3. Based on this data, we speculate that there is ligand exchange at the Co(II) center that allows a relatively labile conversion between complex 1 and 3. These results also suggest that carboxylate ligands play a key role in lowering the Co(II)/Co(III) potential.
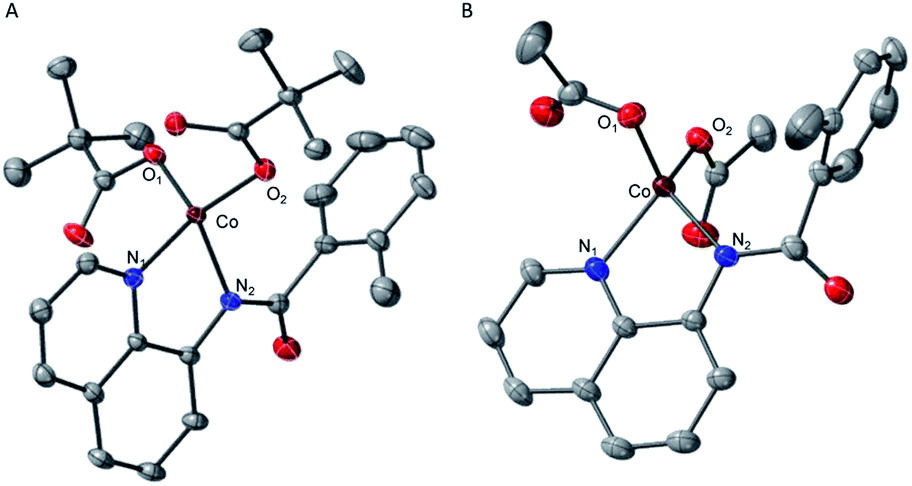 | ||
| Fig. 4 ORTEP diagrams of the single-crystal structures of (A) complex 2 and (B) complex 3. 50% ellipsoid probability; H atoms omitted for clarity. Select distances are: for complex 2, Co–N1, 2.035(1) Å; Co–N2, 2.007(1) Å; Co–O1, 1.954(1) Å; Co–O2, 1.950(1) Å, and for complex 3, Co–N1, 2.043(1) Å; Co–N2, 2.016(1) Å; Co–O1, 1.993(1) Å; Co–O2, 1.952(1) Å. | ||
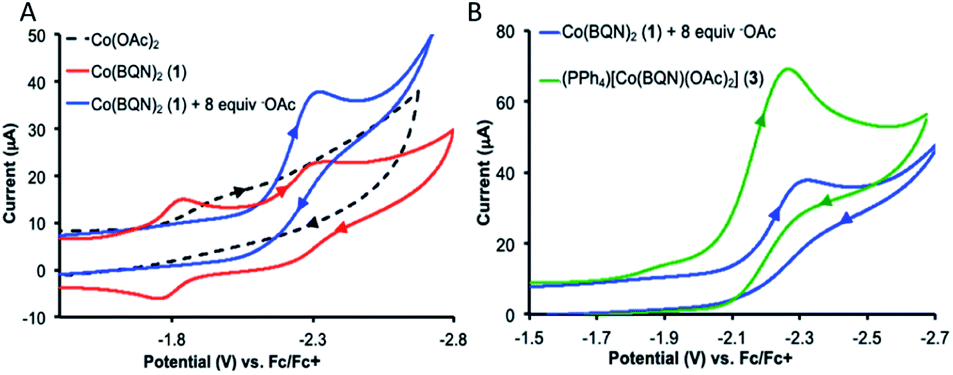 | ||
| Fig. 5 Cyclic voltammogram of (A) CoII(BQN)2 (1) titrated with additional acetate and (B) an overlay of the cyclic voltammogram of isolated PPh4[CoII(BQN)(OAc)2] (3) with the acetate titration of CoII(BQN)2 (1). Scan rate of 100 mV s−1, with 0.1 M TBAPF4 as the supporting electrolyte in dimethylformamide (DMF). Referenced vs. Fc/Fc+. | ||
To assess whether the complexes 2 (and 3) could facilitate the insertion of Co into the C–H bond to form an isolable complex with a Co–C bond, complex 2 was treated with ferrocenium tetrafluoroborate in the presence of an additional AQ ligand (Scheme 4). We choose to use an outer-sphere oxidant for this transformation to demonstrate that metal oxidants (e.g., Mn or Ag) are not required to facilitate the key C–H bond activation step. To our delight, this oxidation yielded complex 4 (Fig. 6) in good yield. Complex 4 is a six-coordinated Co(III) species with a Co–C bond (Fig. 6). Water occupies the sixth coordination site in this complex. As expected for six-coordinated Co(III) species, complex 4 is diamagnetic and can be readily characterized by proton NMR (see ESI† for full characterization details). Complex 4 adds to the small number of isolable Co(III)-aryl complexes prepared via direct C–H insertion.15,29,32–34
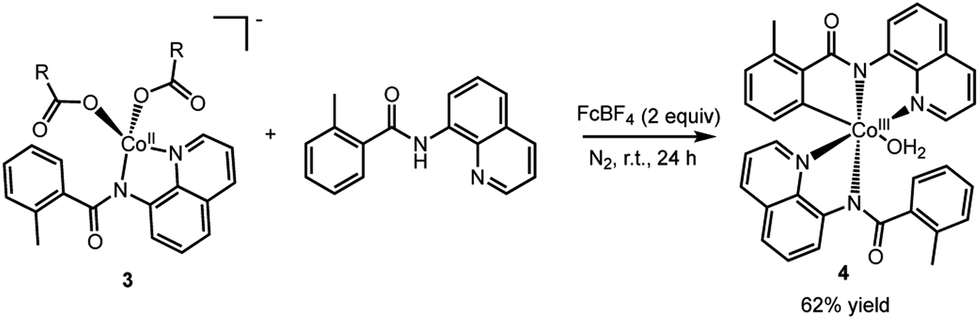 | ||
| Scheme 4 Oxidation of complex 3 to form the C–H bond activated Co(III) complex, 4, with a Co–C bond. | ||
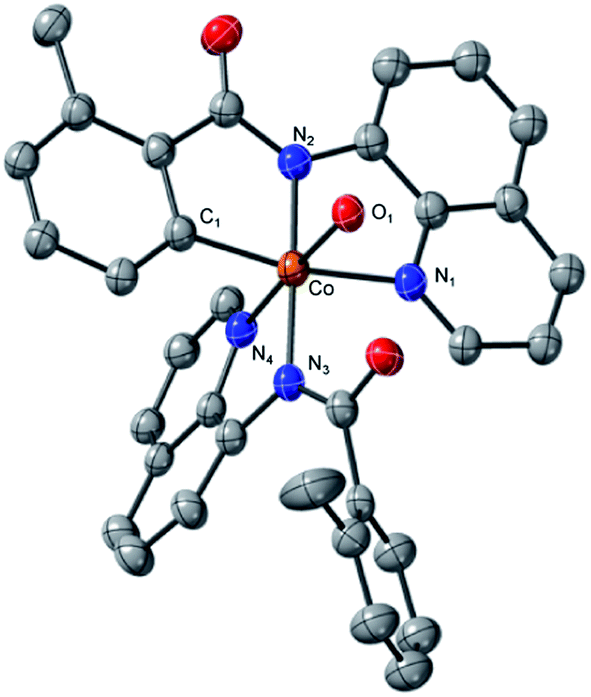 | ||
| Fig. 6 ORTEP diagram of complex 4. 50% ellipsoid probability; H atoms omitted for clarity. Select distances are: Co–C1. 1.928(7) Å; Co–N1, 2.029(6) Å; Co–N2, 1.893(6) Å; Co–N3, 1.981(6) Å; Co–N4, 1.903(6) Å; Co–O1, 1.959(5) Å. | ||
Inspired by these experimental findings, we extended our computational studies to explore the reactivity of [Co(BQN)2(PivO)], 1a. We found that this complex possesses a trigonal bipyramid geometry (see Fig. 7) at its lower-lying singlet, triplet, and quintet electronic states. As seen in Fig. 7, at the enthalpy level, singlet state of 1a (below, labeled as 1a-s) is lower by 1.4 and 2.0 kcal mol−1 than its triplet (1a-t) and quintet (1a-q) states, respectively. However, inclusion of entropy corrections makes the quintet state of 1a by 2.6 and 3.0 kcal mol−1 more favourable than its singlet and triplet states.35 In complex 1a-t the majority of two unpaired α-spins, ca. 1.48|e|, is located on the Co-center, while the remaining 0.52|e| spin is delocalized to the quinoline rings. In 1a-q, 2.67|e| spins are on the Co-center, and the remaining 1.03|e| spin is delocalized to the quinoline moieties. Taken together these spin and Mulliken charge analyses,36 we characterized complexes 1a-t and 1a-q as a (PivO)CoII(BQN)2ox (i.e. as a complex with CoII-center and BQN “radical-cation”) species.37
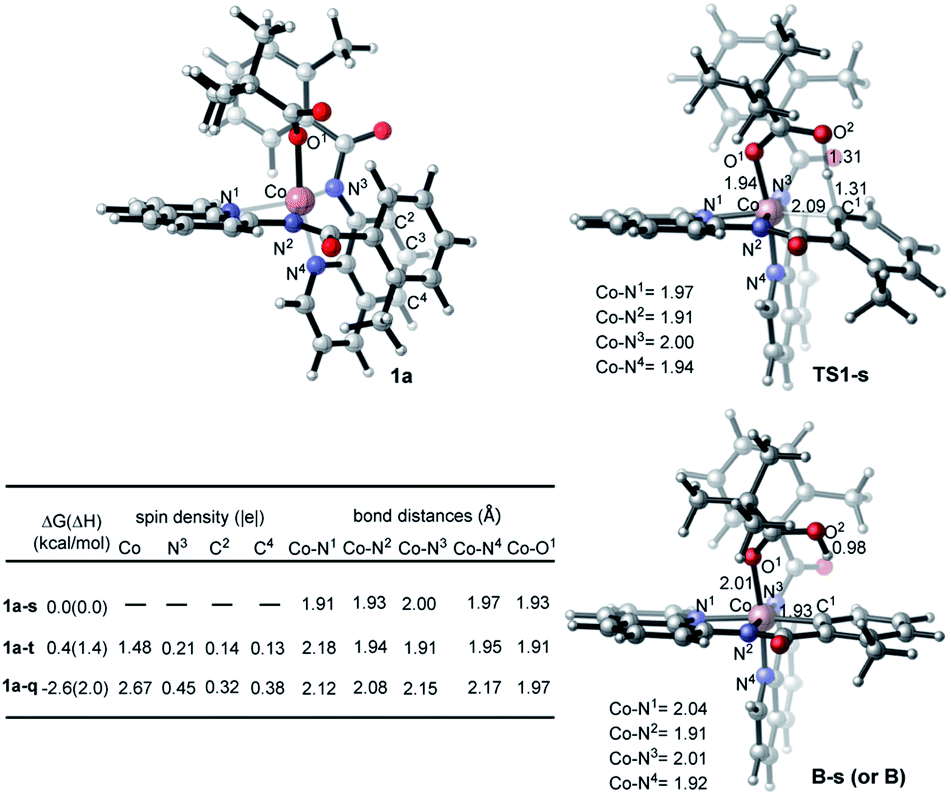 | ||
| Fig. 7 Relative energies, spin density distribution, and important geometry parameters of the singlet, triplet, and quintet states of the complex (PivO)Co(BQN)2, 1a, as well as geometry parameters of the first C–H deprotonation transition state TS1-s and product B-s. | ||
Proposed reaction cycle
Based on the above presented experiments showing feasibility of insertion of Co into the C–H bond in 2 (and 3) to form complex 4 with a Co–C bond, it is conceivable to expect that from the complex [(PivO)Co(BQN)2], 1a, the biaryl formation will proceed via the C–H bond deprotonation by PivO to yield intermediate B (see Scheme 5 and Fig. 8). Calculations show that this step of the reaction proceeds via the transition state TS1 (here, we also have studied the meta-CH activation, which is found to be less favorable and will not be discussed in details: see ESI† for more details). The calculated (the same electronic state-to-same electronic state) free energy barriers are 5.1, 21.6, and 23.6 kcal mol−1 for the singlet, triplet and quintet electronic states, respectively. The overall reaction, i.e., the transformation of 1a to B, is 6.6 kcal mol−1 exergonic, but by 1.7 and 23.1 kcal mol−1 endergonic for the singlet, and triplet and quintet state processes, respectively. Thus, the first C–H deprotonation proceeds through singlet transition state TS1-s with a relatively low energy barrier (5.1 kcal mol−1) and leads to the singlet state intermediate B-s. Therefore, below we discuss only the singlet state transition state, TS1-s, and product, B-s, while we include important structural and energetic data for the triplet and quintet states of TS1 and B in ESI (Fig. S1†).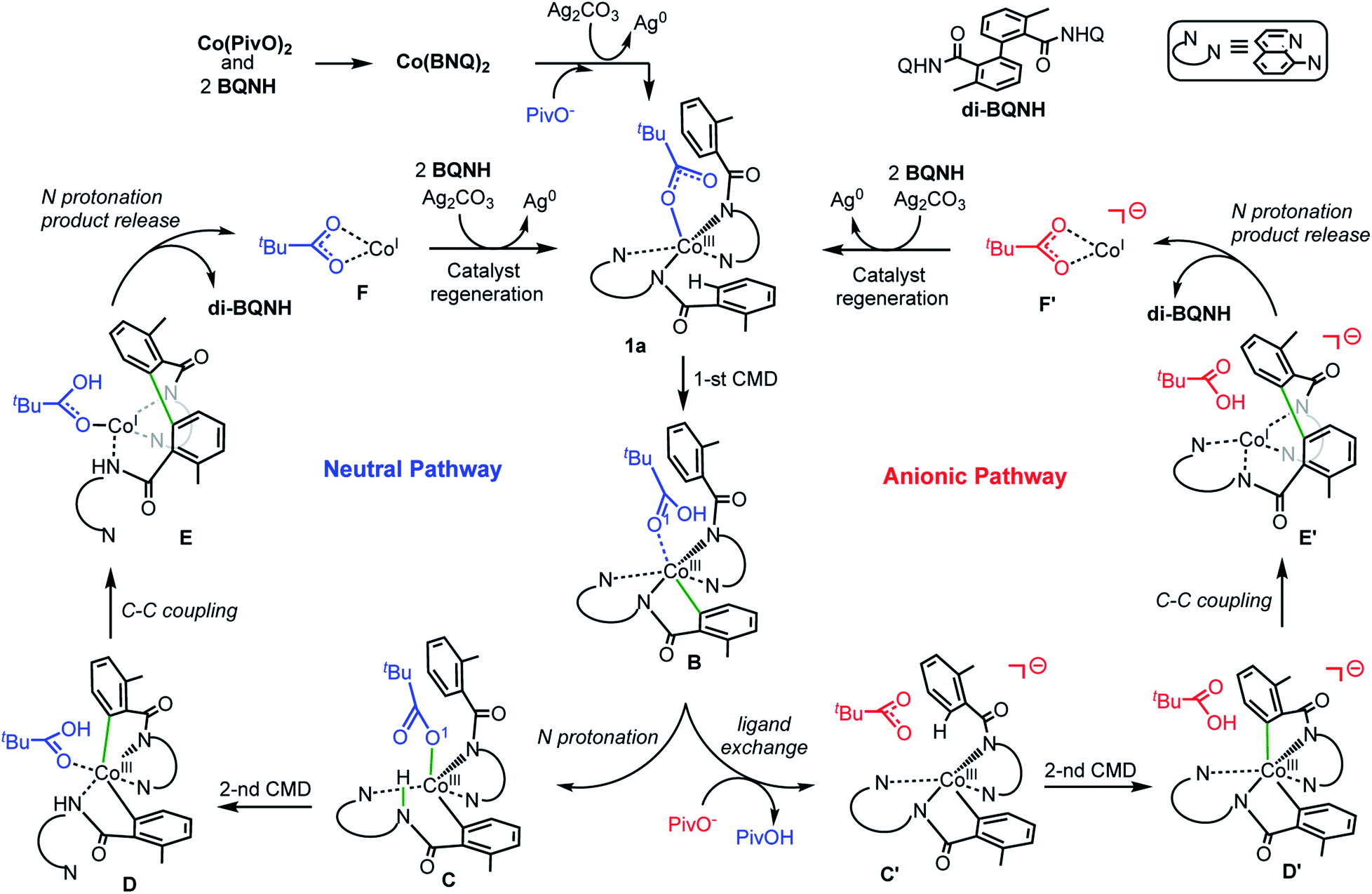 | ||
| Scheme 5 The proposed charge neutral and anionic catalytic cycles for the Co-mediated dehydrogenative dimerization of aminoquinoline directed benzamides. | ||
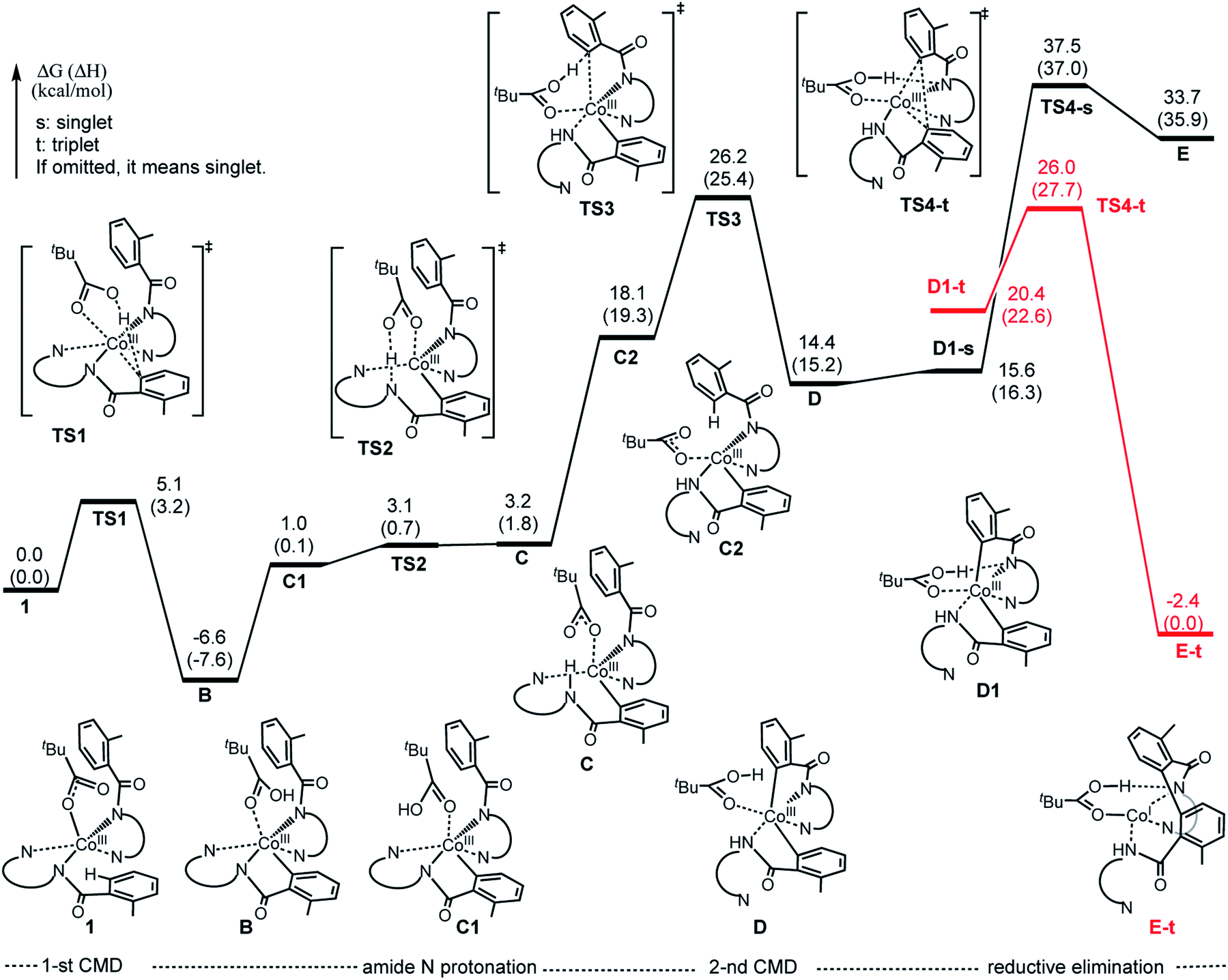 | ||
| Fig. 8 Potential energy surface for the charge neutral pathway of the Co-mediated dehydrogenative dimerization of aminoquinoline directed benzamides. | ||
As seen from Fig. 7, TS1-s is a concerted-metalation deprotonation transition state for the formation of Co–C1 (2.09 Å) and O2–H (1.31 Å), and cleavage of C1–H (1.31 Å) bonds. In the product complex B-s, the Co–C1 bond has already been formed with a 1.93 Å bond distance. Other geometric parameters around the Co center are changed only slightly (with some elongation of the Co–N1 and Co–O1 bonds) upon going from 1a-s to B-s.
From the complex B-s (for sake of simplicity, below we will call it B) the dimerization of AQ-directed benzamides may proceed via several pathways. Here, we investigated three of them, namely, the (a) charge neutral, (b) anionic, and (c) dimetallic pathways (Scheme 5). The charge neutral mechanism of this reaction involves: (a) the intramolecular amide N protonation by PivOH to yield intermediate C, (b) the second C–H bond activation by the coordinated PivO− to form D, (c) the C–C coupling in intermediate D to produce E, (d) the second amide N protonation to release the biaryl and (PivO)CoI complex (F), which in the next step undergoes oxidation and substrate binding to regenerate catalytic active species 1a-s. Alternatively, the anionic mechanism of the reported reaction includes the following steps: (a) the PivOH to PivO− transformation to generate anionic intermediate C′; (b) the second C–H bond deprotonation by the PivO− anion to form intermediate D′; (c) the C–C coupling to form intermediate E′; (d) the amide N-center protonation to release the aryl–aryl coupling product and complex (PivO)CoI (F′), which in the next step undergoes oxidation and substrate binding to regenerate catalytic active species 1a-s.
The dimetallic mechanism38–40 (not shown in Scheme 5) also starts from the intermediate B, but proceeds via the (a) BQN-to-PivO− ligand exchange (which, as shown above, is a facile process), (b) dimerization of the resulted (PivO)Co(BQN) to form the (BQN)Co(PivO)2Co(BQN) intermediate, and (c) C–C coupling to form biaryl product. Our extensive calculations show that this pathway of the studied reaction requires very high (∼47.1 kcal mol−1) energy barrier for the C–C coupling, and is unlikely to be a viable mechanism of the reported biaryl formation reaction. Therefore, here we will not discuss this mechanistic scenario, while we will include all calculated intermediates, transition states, and products into ESI (see Fig. S2 and S3†) along with a brief discussion.
Below, we discuss in details only the charge neutral and anionic mechanisms of the Co-promoted dehydrogenative dimerization of AQ-directed benzamides.
Charge neutral reaction pathway
The first step of the charge neutral mechanism of the studied biaryl formation reaction is the amide N-center protonation. This is a multistep process, and involves (a) rotation of PivOH around the Co–O1 bond, and (b) proton transfer from PivOH to the amide N-center leading to intermediate C. Overall, the B → C transformation is endergonic and requires 9.8 kcal mol−1 free energy (see Fig. 8, as well as ESI†).In structure C, in order to deprotonate the ortho C–H bond of the second benzamide, base PivO− should rotate closer to the “going to be deprotonated” C–H bond. This C → C2 isomerization is endergonic by 14.9 kcal mol−1 because of: (a) disruption one of the Co–N (pyridine) bonds to accommodate the Co–O bond, and (b) cleavage of the (O⋯H–N) hydrogen bond in C2 (see Fig. 8). The second C–H bond deprotonation in C2 proceeds through singlet state transition state TS3 (see Fig. 9) with energy barrier of 8.1 kcal mol−1 and is exergonic by 3.7 kcal mol−1 for formation of singlet state intermediate D. Thus, overall reaction B → TS3 → D requires of 32.8 kcal mol−1 energy barrier and is endergonic by 21.0 kcal mol−1, calculated relative to the most stable intermediate B (see Fig. 8). One should note that the C–H bond deprotonation of the second benzamide may also proceed via the “outer-sphere” deprotonation mechanism, i.e. by the weakly coordinated (through the O⋯H–C hydrogen bond) PivO− anion.
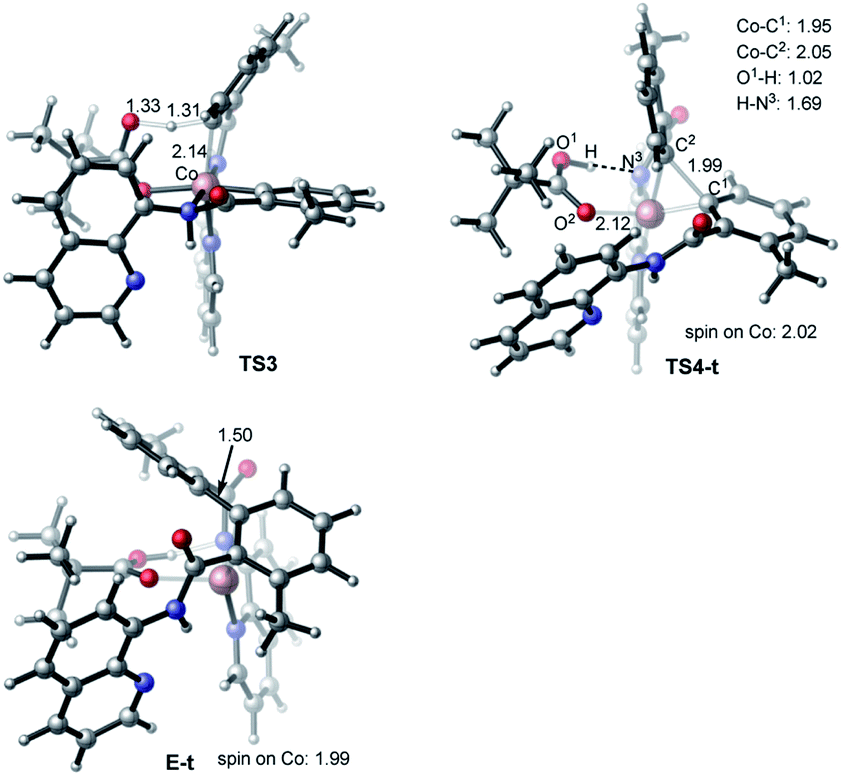 | ||
| Fig. 9 Geometries and spin densities of key transition states and product (bond distances are given in Å) of charge neutral pathway of the Co-mediated dehydrogenative dimerization of aminoquinoline directed benzamides. | ||
We located two “outer-sphere” C–H bond deprotonation transition states, but both of them are higher in free energy than transition state TS3 that associates with the “inner-sphere” C–H bond deprotonation. Therefore, here we will not discuss those “outer-sphere” transition states, as well as the “outer-sphere” deprotonation mechanism in more details (we included those structures and related discussion into the ESI†). As our analyses show, the overall 1a → B → C → D transformation is redox neutral. Furthermore, all reported intermediates and transition states involved in these transformations have a singlet ground electronic states, except pre-reaction complex 1a which has a nearly-degenerated singlet, triplet and quintet states. From the intermediate D the C–C reductive elimination occurs. In course of this process the oxidation state of the Co-center changes from formal 3+ to formal 1+. Since CoI(d8) species tend to having triplet ground states, it is critical to investigate both singlet and triplet electronic states of the C–C coupling transition state TS4.
As seen in Fig. 8, the reaction D (singlet) → TS4-s requires a 23.1 kcal mol−1 energy barrier. Furthermore, the performed IRC calculations (from the singlet transition state TS4-s, see ESI†) shows that before undergoing the C–C coupling, intermediate D rearranges to isomer D1, where the PivO–H bond is rotated toward the Co-bound amide nitrogen instead of the aryl group. This D (singlet) → D1 (singlet) isomerization is only a 1.2 kcal mol−1 endergonic process (Fig. 8).
Gratifyingly, the triplet state of the C–C coupling transition state, i.e.TS4-t (see Fig. 8 and 9), lies by 11.5 kcal mol−1 lower than singlet transition state TS4-s. In other words, the C–C coupling from the singlet state intermediate D will occur at the triplet transition state TS4-t. Thus, the C–C coupling from intermediate D requires a singlet–triplet seam of crossing before the energetically most favorable triplet C–C coupling transition state TS4-t: at the singlet–triplet crossing point of the potential energy surface system switches from its singlet electronic state to the triplet electronic state via the spin-flip phenomena. The performed IRC calculations at TS4-t connects it with the C–C coupling product, the triplet state complex E-t (see Fig. 8 and 9), where the C(sp2)–C(sp2) bond is already formed with a 1.50 Å bond distance. Analyses show that in complex E-t almost all of the two unpaired α-spins (namely 1.99|e| spins) are located on the Co-center: thus, complex E-t is the high-spin Co(I)-complex. Its singlet excited state lies 36.1 kcal mol−1 higher in energy and, therefore, will not be discussed in details. To summarize, the C–C reductive elimination in the singlet state intermediate D, i.e.D (singlet) → E-t transformation, proceeds via 11.6 kcal mol−1 free energy barrier and is exergonic by 16.8 kcal mol−1.41 However, as seen in Fig. 8, overall C–C coupling requires a 32.6 kcal mol−1 free energy barrier and is 4.2 kcal mol−1 endergonic, relative to the most stable intermediate B.
In complex E-t, the protonation of the nitrogen of the second amide by PivOH facilitates release of the C–C coupling product and formation of the (PivO)CoI species, which later undergoes oxidation by the silver salts, coordinates substrates and regenerates the catalytic active species 1a. Because the later steps of the reaction are facile events, here we did not examine them in detail.
In summary, as seen in Fig. 8, the two most energy demanding steps of the Co-mediated dehydrogenative dimerization of AQ-directed benzamides, i.e., reaction 1a → E-t, proceeding via the charge neutral pathway, are the C–H bond deprotonation of the second benzamide (at the singlet transition state TS3) and the C–C reductive elimination (at the triplet transition state TS4-t), which require 32.8 and 32.6 kcal mol−1 free energy barriers (calculated relative to the most stable intermediate B), respectively. The overall reaction B → E-t, is endergonic by 4.2 kcal mol−1.
Anionic reaction pathway
Alternatively, the anionic mechanism of the Co-promoted dehydrogenative dimerization of AQ-directed benzamides is initiated from intermediate B and starts with the PivOH to PivO- transformation. In general, this transformation may occur via multiple mechanisms, including (but not limited to) PivOH-to-PivO− ligand exchange and proton shuttling between solvent and coordinated PivOH ligand. Here we only briefly studied the PivOH-to-PivO− ligand exchange, which may proceed via either a dissociative-associative or concerted substitution pathways.The dissociative-associative pathway starts by dissociation of PivOH with a 7.6 kcal mol−1 free energy, and follows by coordination of anion PivO− to form anionic Co-species C′. This process is exergonic by 10.6 kcal mol−1 (relative to the pre-reaction complex B, Fig. 10). Here, we assumed that PivO− is freely available from the reaction mixture. As seen in Fig. 11, in complex C′, the Co-center adopts a well-defined octahedral geometry, with three nitrogen atoms (N1, N2, and N3) and one phenyl carbon (C1) in the plane, and one pivalate oxygen atom (O1) and one quinoline nitrogen atom (N4) in apical positions (Fig. 11). The relatively small, 7.6 kcal mol−1, free energy required for this process can be considered as an upper-limit of the energy required for the PivOH-to-PivO− ligand exchange. Thus, this step of the reported reaction is a facile process, therefore, here we did not discuss it in more details. It should be noted that recently Gong, Wu, and Zhang have reported an anionic chiral CoIII complex that is critical in an asymmetric C(sp3)–H functionalization.42 This complex resembles the above presented intermediate C′.
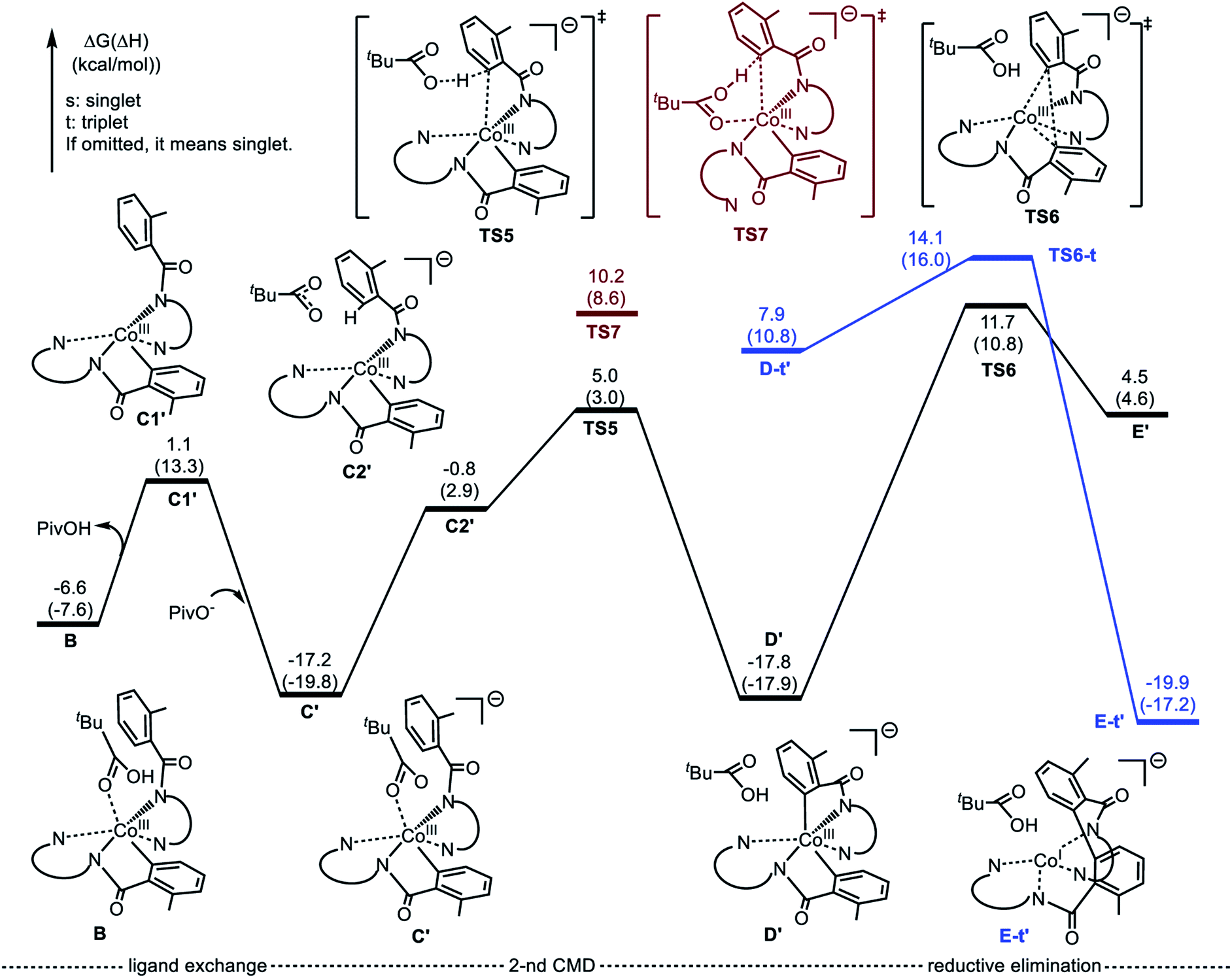 | ||
| Fig. 10 Potential energy surface for the anionic mechanism of the Co-mediated dehydrogenative aminoquinoline directed benzamide dimerization. | ||
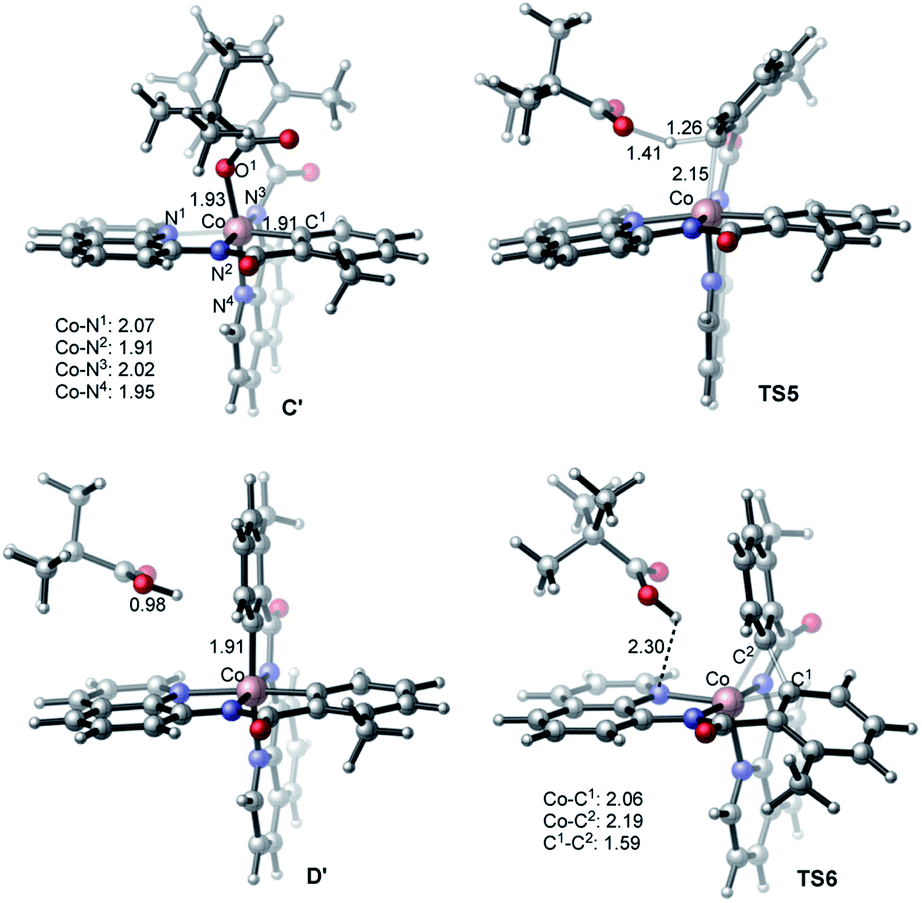 | ||
| Fig. 11 Geometries and spin densities of key intermediates and transition states (bond distances are given in Å) of anionic pathway of the studied Co-mediated dehydrogenative aminoquinoline directed benzamide dimerization. | ||
The elementary reaction initiated from complex C′ is the C–H bond activation of the second benzamide, which may proceed via either “inner-sphere” (at the transition state TS7) or “outer-sphere” (at the transition state TS5) pathways (see Fig. 11). Calculations show that the “inner-sphere” transition state TS7 is 5.2 kcal mol−1 unfavourable compared to the “outer-sphere” transition state TS5 (see Fig. 10). Therefore, we concluded that the C–H bond activation of the second benzamide in C′ will proceed via the “outer-sphere” pathway with an activation barrier of 22.2 kcal mol−1, relative to the energetically most stable intermediate C′. The performed IRC calculations connect TS5 with the product complex D′, which lies only 0.6 kcal mol−1 lower than intermediate C′. As seen from Fig. 10, the overall process of 1a → C′ → TS5 → D′ is a redox neutral (i.e. cobalt stays at its formal 3+ oxidation state), and exergonic by 17.8 kcal mol−1. Noteworthy, the calculations of the lower-lying singlet, triplet and quintet electronic states of all identified transition states and intermediates of this reaction show that all of them have a singlet ground state(see ESI† for details).
As we may expect, the next step of the reaction, which is the C–C reductive elimination, is a two-electron CoIII → CoI reduction process. Calculations show that this C–C coupling occurs at the singlet state transition state TS6 (see Fig. 10 and 11) with 29.5 kcal mol−1 free energy barrier, calculated relative to the pre-reaction complex D′. Since the product of this step is the Co(I)-complex E′ with a triplet ground electronic state, again, we expect the singlet-to-triplet seam of crossing, but this time, after (or vicinity of) the singlet state transition state TS6. As seen in Fig. 10, C–C reductive elimination is only 2.1 kcal mol−1 exergonic, calculated relative to the singlet D′ complex. Entire anionic catalytic cycle, namely, 1a → C′ → TS5 → D′ → E′, is exergonic by 19.9 kcal mol−1 and occurs with rate-limiting 29.5 kcal mol−1 free energy barrier for the C–C coupling. Same as in the complex E-t, discussed above, the complex E-t′ undergoes a multiple kinetically and thermodynamically facile transformations (here we did not examine them in detail) and regenerates the catalytic active species 1a.
Thus, at the B3LYP-D3BJ/BS1 level of theory (see “Computational details” section at the end of this paper) the calculated rate-determining steps of the charge neutral (cn) mechanism are the second C–H bond activation (with a ΔC–H(cn) = 32.8 kcal mol−1) and C–C coupling (ΔC–C(cn) = 32.6 kcal mol−1) steps. For the anionic (a) mechanism it is the C–C coupling (ΔC–H(a) = 29.5 kcal mol−1). In other words, at the B3LYP-D3BJ/BS1 level of theory, the anionic mechanism of the Co-mediated dimerization of AQ-directed benzamides requires around 3.3 kcal mol−1 smaller energy than the charge neutral pathway. To validate impact of the used density functional to the calculated rate-limiting steps of the charge neutral and anionic mechanisms of the Co-mediated dehydrogenative dimerization of AQ-directed benzamides we recalculated geometries and energies of the C–H activation and C–C coupling transition states and associated pre-reaction complexes at the M06/BS1 level of theory (see ESI† for details). We found that at the M06/BS1 level rate-determining step of the charge neutral pathway is the second C–H bond activation with a 31.3 kcal mol−1 activation barrier, since a barrier required for its C–C coupling step reduced to 24.1 kcal mol−1 [from the 32.6 kcal mol−1 calculated at the B3LYP-D3BJ/BS1 level of theory]. In the meantime, the rate-limiting C–C coupling free energy barrier for the anionic pathway is reduced to 24.4 kcal mol−1. Thus, even at the M06/BS1 level the anionic mechanism of the Co-mediated dimerization of AQ-directed benzamides is more favourable (now, by 6.9 kcal mol−1). We also have validated the impact of the used basis sets (i.e. BS1) to the calculated critical energy barriers. For this reason, we performed single-point energy calculations (at the previously optimized geometries, at the B3LYP-D3BJ/BS1 and M06/BS1 levels of theory, respectively) of the C–H activation and C–C coupling transition states and associated pre-reaction complexes of the charge neutral and anionic pathways of the of the Co-mediated dehydrogenative dimerization of AQ-directed benzamides at the B3LYP-D3BJ/BS2 and M06/BS2 levels, where basis set BS2 = [6-311++G(d,p)] (for all atoms except Co) + SDD (and associated ECP for Co) and is larger than BS1 (see ESI† for details). We found that at the M06/BS2//M06/BS1 level, the rate-determining C–H bond activation step of the charge neutral pathway requires a 31.8 kcal mol−1 free energy barrier. However, the free energy barrier required for the rate-limiting C–C coupling for the anionic pathway increased by 2.1 kcal, i.e. to 26.5 kcal mol−1.
These findings empowered us to conclude that regardless of the used density functional (i.e. either B3LYP-D3BJ or M06) and basis sets (i.e. either BS1 or BS2), the studied Co-mediated dehydrogenative dimerization of the AQ-directed benzamides will proceed via the kinetically and thermodynamically favourable anionic mechanism (see Scheme 5).
Since the rate-limiting step of the favourable anionic mechanism of the studied biaryl formation reaction is the C–C coupling in the CoIII complex D′ [(PivOH)Co(BQN)2]−, it is conceivable to expect that the electronics of the aryl groups of benzamides as well as the directing AQ ligands will have significant impact to the calculated reductive elimination barriers, and, consequently, to the reaction outcome. This expectation is consistent with the experimental findings by Daugulis and coworkers.16,17 In order to test this hypothesis we calculated the C–C coupling barriers for series of AQ-benzamides with different para-substituted aryls, as well as with different para-substituted pyridines of the AQ ligand.
As seen in Fig. 12a, in general, the calculated C–C coupling barriers correlate reasonable well (with a linear correlation coefficient R2 = 0.86) with the electronic nature (Hammett σp value)43,44 of the substituent X (where for X = NH2, OH, Me, H, Cl and Br): AQ-benzamides with the electron donating substituents X at the para position of aryl groups (i.e. with electron-rich aryls) have lower activation barriers.
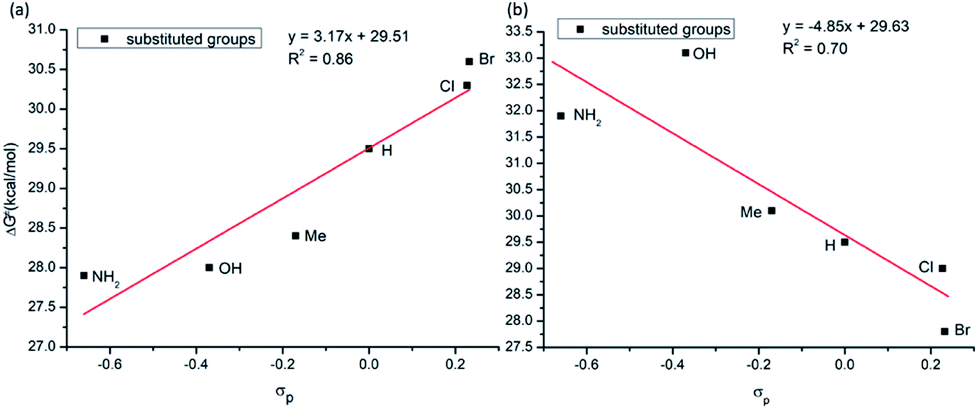 | ||
| Fig. 12 Linear correlation between activation barrier and Hammett parameter (σp) of the used substituents (a) para-substituted aryl group; (b) para-substituted pyridine of the AQ ligand. | ||
Change in electronics of the AQ ligands is expected to impact the C–C coupling barriers via the different mechanism. Indeed, AQs ligands coordinate to Co-center and provide much needed stability to the Co-(bis)aryl framework. It is expected that electron-rich AQs (i.e. with the electron-donating (EDG) substituents) would stabilize the Co–AQ bonding: as a result, will retard the C–C coupling. On the contrary, electron-poor AQs (i.e., with the electron-withdrawing (EWG) substituent) will decrease the Co–AQ bonding and may promote the C–C coupling. We have validated this hypothesis by introducing multiple substitution on the para-position pyridine ring of the coordinated AQ ligand (Fig. 12b) and calculated the C–C coupling barriers. In general, as it was expected, the activation barriers are higher for AQs with electron-donating substituents, but are lower for AQs with electron-withdrawing substituent (see Fig. 12b). Based on these extensive analyses, we have found that AQ-benzamide with NH2 substituent on the aryl ring and Br-substituent on the pyridine ring of AQ moiety may have a better reactivity (i.e. lowest activation barrier, 25.6 kcal mol−1).
Formation of the CoIII(PivO)(BQN–BQN) and CoII(BQN–BQN) complexes
Above, we have demonstrated, both computationally and experimentally, that deprotonation of substrate BQNH by the Co-coordinated pivalates of CoII(PivO)2 is a facile process under the utilized reaction conditions. Therefore, the presence of intermediate species such as Co(BQN)2, 1, [Co(PivO)(BQN)2], 1a, and PPh4[CoII(PivO)2(BQN)] (2) are expected to be prevailing Co-complexes at the earlier stages of the reaction, i.e., when the concentration of the substrate BQNH is still high. However, upon increase of concentration of biaryl product (i.e. di-BQNH) and reduce of concentration of substrate BQNH the former could act as substrate and that will lead to formation of the Co-complexes with the deprotonated di-BQNH molecules, i.e. with di-BQN. This hypothesis gets an additional support from the computation of energy of the reaction:| di-BQNH + CoII(OPiv)2 → CoII(di-BQN) + 2HOPiv |
showing that it is only 0.7 kcal mol−1 endergonic. Furthermore, previously, the Daugulis lab45 had reported AQ-directed biaryl homocoupling products in relatively high yields.
To validate this hypothesis, we experimentally endeavored to determine visibility of formation of CoII–(di-BQN) species during the reaction of Co-mediated dehydrogenative aryl–aryl coupling in the AQ-directed benzamides.33,45,46 To that end, we isolated and characterized complexes 5, 6, and 7 (Fig. 13, for details of experimental procedures and discussion of these species see ESI†). We also wish to mention previous example of this type of complex reported by Xie et al. in 2016.15 In this report Xie et al. were able to isolate a structure with the biaryl coupling and acetylacetanoate in the 5th and 6th coordination spheres.
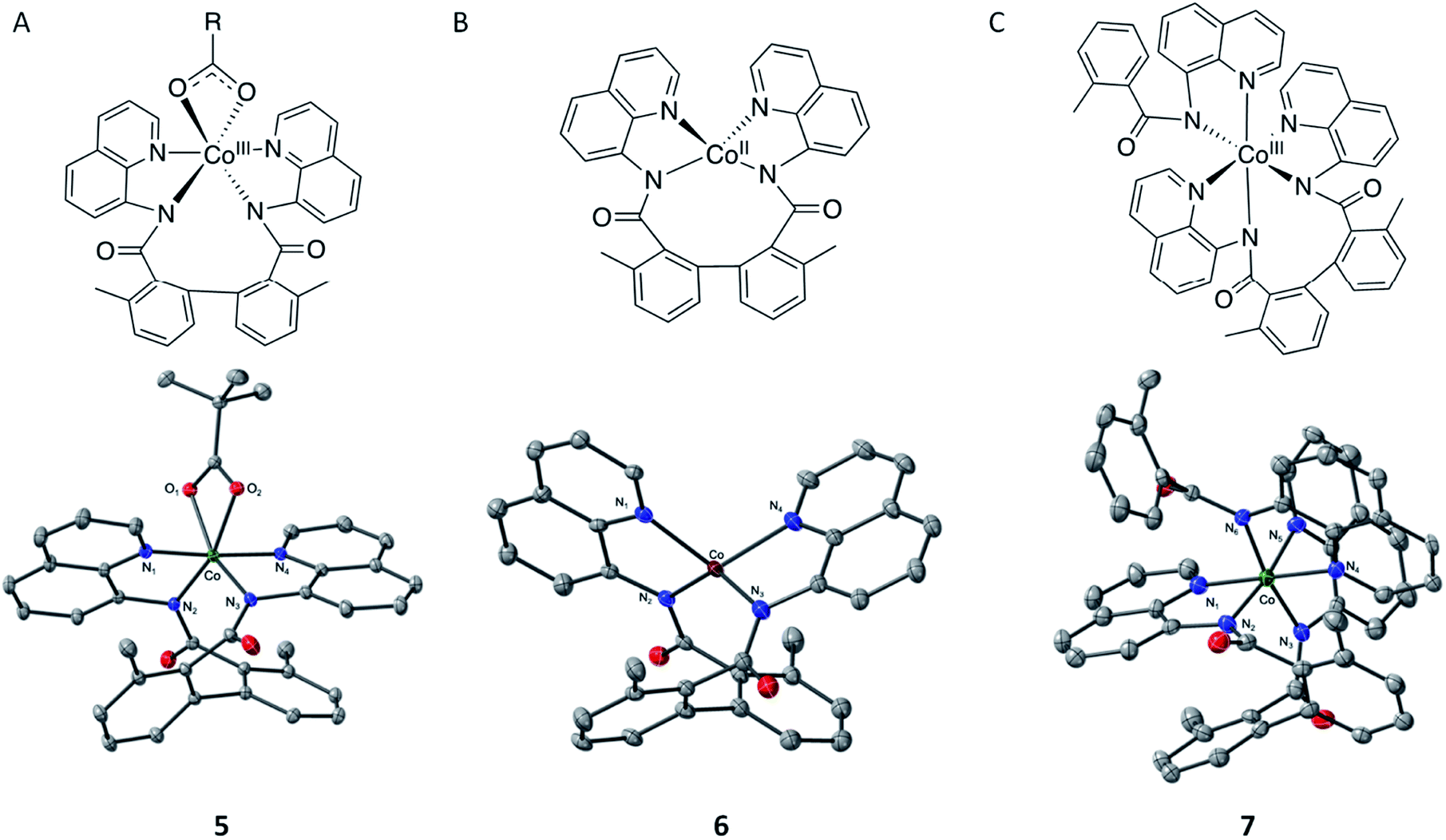 | ||
| Fig. 13 2D and thermal ellipsoid diagrams of the single-crystal structures of complexes (PivO)CoIII(BQN–BQN), (5), CoII(BQN–BQN), (6), and (BQN)CoIII(BQN–BQN), (7). 50% ellipsoid probability; H-atoms omitted for clarity. Complex 5 select distances: Co–N1, 1.913(1) Å; Co–N2, 1.940(1) Å; Co–N3, 1.918(1) Å; Co–N4, 1.917(1) Å, Co–O1, 1.977(1) Å; Co–O2, 1.969(1) Å. Complex 6 select distances: Co–N1, 2.028(5) Å; Co–N2, 1.971(5) Å; Co–N3, 1.973(5) Å; Co–N4, 2.026(4) Å. τ4 = 0.726. Complex 7 select distances: Co–N1, 1.920(5) Å; Co–N2, 1.998(5) Å; Co–N3, 2.004(6) Å, Co–N4, 1.937(6) Å, Co–N5 1.993(5) Å, Co–N6 1.951(3) Å. | ||
Conclusions
In summary, we report joint experimental and theoretical studies on key mechanistic features of the Co-mediated and AQ-directed dehydrogenative aryl–aryl coupling of benzamides, and synthesized and characterized several key intermediates of the predicted reaction mechanism. We find that:(a) The initial step of this reaction is a deprotonation of AQ of the substrate BQNH by the CoII-coordinated carboxylates and formation of the bis-ligated CoII(BQN)2, 1, species. Complex 1 was isolated and fully characterized by multiple experimental techniques.
(b) The ligand environment of the CoII-center in 1 is mobile and could undergo a facile BQN-to-PivO ligand exchange, even coordinate an additional carboxylate ligand. Here, we predicted the (PivO)Co(BQN)2, 1a, to be an active species in the reaction. This species was characterized as a high-spin (PivO)CoII(BQN)2ox (i.e., with CoII-center and BQN “radical-cation” ligands) species, at its quintet and triplet states.
(c) Among the three possible mechanisms of the studied reaction, including the dimetallic, charge neutral and anionic mechanisms, the anionic one is shown to be most favorable: it involves (see Scheme 6): (a) the concerted-metalation-deprotonation (CMD) of the first benzamide C–H bond, (b) the PivOH-to-PivO− rearrangement, (c) the CMD of the second benzamide C–H bond, (d) C–C coupling, (e) product formation (facilitated by the amide nitrogen protonation), and (f) catalyst regeneration via another equivalent of substrate and silver salts (or other oxidants).
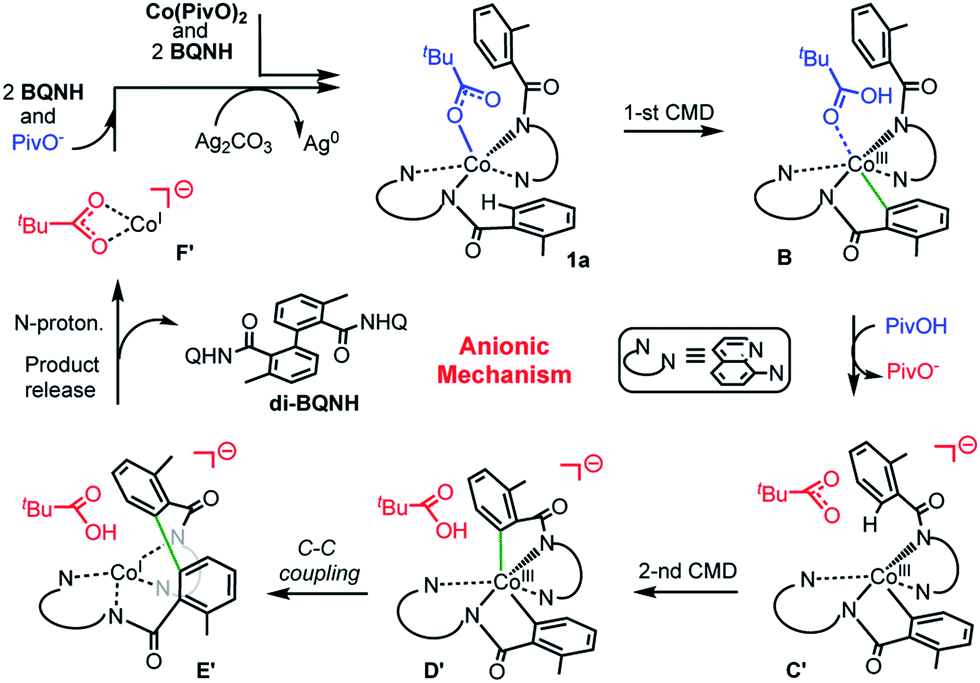 | ||
| Scheme 6 Schematic presentation of intermediates and transition states of the most favorable anionic mechanism of the Co-mediated dehydrogenative aminoquinoline directed benzamide dimerization. | ||
(d) The rate-determining step of the overall reaction is the C–C coupling that requires 29.5 kcal mol−1 free energy barrier. It is predicted that the electron-rich aryl of benzamide, as well as the electron-poor pyridine of AQ of the aminoquinoline-benzamide ligand will reduce the rate-limiting C–C coupling barrier and facilitate the Co-mediated and AQ-directed dehydrogenative aryl–aryl coupling of benzamides.
Experimental and computational procedures
Computational details
Geometry optimizations and frequency calculations of all reported structures were performed with the Gaussian 09 suite of programs47 at the B3LYP-D3BJ/BS1 level of theory with the corresponding Hay-Wadt effective core potential for Co,48 and Grimme's empirical dispersion-correction (D3) with Becke-Johnson (BJ) damping for B3LYP.49 Here, BS1 is a basis set including Lanl2dz for Co and 6-31G(d,p) for all other atoms. Frequency analysis was used to characterize each minimum with zero imaginary frequency and each transition state (TS) structure with only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed for all TSs to ensure their true nature. Bulk solvent effects were incorporated for all calculations (including geometry optimizations) using the self-consistent reaction field polarizable continuum model (IEF-PCM).50,51 As a solvent we chose dichloromethane. The reported thermodynamic data were computed at the 298.15 K temperature and 1 atm pressure. Various spin states were considered for all key species, while below, the most stable ones are discussed. 3D geometries were drawn by the CYLview software.52 Unless otherwise stated, the energies given as ΔG(ΔH) are in kcal mol−1. In the presented calculations, we used aminoquinoline (AQ) benzamide (BQNH, Scheme 1b) as a substrate and Co(OPiv)2 as the metal precursor.General experimental procedures
All manipulations were carried out using standard Schlenk techniques or conducted in an MBraun Labmaster 130 drybox under a nitrogen atmosphere. All reagents used were purchased from commercial vendors and used as received unless otherwise noted. 1H NMR spectra of cobalt complexes were recorded on an INova 400 MHz spectrometer at ambient temperature. 1H NMR spectrum of (2-methylbenzoyl)(quinolin-8-yl)amide and all 13C NMR were recorded on an INova 600 MHz spectrometer at ambient temperature. 1H chemical shifts were referenced to the residual solvent peak (CDCl3: δH = 7.26 ppm, CD2Cl2: δH = 5.32 ppm). UV-visible absorption spectra were recorded on a Cary50 spectrophotometer using 1.0 cm quartz cuvettes. Solution-state magnetic moments were measured using Evans' method.28,53 X-ray diffraction studies were carried out in the X-ray Crystallography Laboratory at Emory University on a Rigaku XtaLAB Synergy diffractometer. Cyclic voltammetry experiments were carried out using a CH Instruments (Austin, TX) Model 660C potentiostat. All experiments were conducted in CH2Cl2, MeCN, or DMF with 0.10 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. Electrochemical experiments were conducted in a three-component cell consisting of a Pt-wire auxiliary electrode, a non-aqueous reference electrode (Ag/AgCl), and a glassy-carbon working electrode. The used synthetic procedures of the prepared molecules were presented in ESI† (in details) and are briefly stated in the presented reaction schemes. We also included to ESI† details of the general procedure for the reaction condition screen for biaryl homocoupling of 1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982). D.G.M. gratefully acknowledges the NSF MRI-R2 grant (CHE-0958205) and the use of the resources of the Cherry Emerson Center for Scientific Computation at Emory University. L. P. Xu acknowledges the National Science Foundation of China (NSFC 21702126) and China Scholarship Council for support.Notes and references
- G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed.
- M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC.
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed.
- K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS PubMed.
- D. G. Pintori and M. F. Greaney, Org. Lett., 2011, 13, 5713 CrossRef CAS PubMed.
- S. Oi, Y. Ogino, S. Fukita and Y. Inoue, Org. Lett., 2002, 4, 1783 CrossRef CAS PubMed.
- X. Chen, G. Dobereiner, X.-S. Hao, R. Giri, N. Maugel and J.-Q. Yu, Tetrahedron, 2009, 65, 3085 CrossRef CAS.
- G. Tan, S. He, X. Huang, X. Liao, Y. Cheng and J. You, Angew. Chem., Int. Ed., 2016, 55, 10414 CrossRef CAS PubMed.
- Y. Xie, D. Xu, W.-W. Sun, S.-J. Zhang, X.-P. Dong, B. Liu, Y. Zhou and B. Wu, Asian J. Org. Chem., 2016, 5, 961 CrossRef CAS.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2015, 17, 1204 CrossRef CAS PubMed.
- T. T. Nguyen, L. Grigorjeva and O. Daugulis, ACS Catal., 2016, 6, 551 CrossRef CAS PubMed.
- C. Hou, J. Jiang, S. Zhang, G. Wang, Z. Zhang, Z. Ke and C. Zhao, ACS Catal., 2014, 4, 2990 CrossRef CAS.
- X.-K. Guo, L.-B. Zhang, D. Wei and J.-L. Niu, Chem. Sci., 2015, 6, 7059 RSC.
- Q. Jiang and T. R. Cundari, Comput. Theor. Chem., 2017, 1105, 97 CrossRef CAS.
- H. Li, J. V. Obligacion, P. J. Chirik and M. B. Hall, ACS Catal., 2018, 8, 10606 CrossRef CAS PubMed.
- S.-B. Wu, T. Zhang, L. W. Chung and Y.-D. Wu, Org. Lett., 2019, 21, 360 CrossRef CAS PubMed.
- K. Yamazaki, Y. Kommagalla, Y. Ano and N. Chatani, Org. Chem. Front., 2019, 6, 537 RSC.
- T. H. Meyer, J. C. A. Oliveira, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202002258.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 9, 955 RSC.
- K. Kanamori, W. E. Broderick, R. F. Jordan, R. D. Willett and J. I. Legg, J. Am. Chem. Soc., 1986, 108, 7122 CrossRef CAS.
- W. E. Broderick, K. Kanamori, R. D. Willett and J. I. Legg, Inorg. Chem., 1991, 30, 3875 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC.
- L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed.
- J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990 CrossRef CAS PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS.
- O. Planas, C. J. Whiteoak, V. Martin-Diaconescu, I. Gamba, J. M. Luis, T. Parella, A. Company and X. Ribas, J. Am. Chem. Soc., 2016, 138, 14388 CrossRef CAS PubMed.
- N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361 CrossRef CAS PubMed.
- S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493 CrossRef CAS.
- It should be mentioned that all attempts trying to locate open-shell singlet species failed and they all converged to close-shell singlet ones.
- Mulliken charge analysis show that the electron density of the quinolone moiety has been changed from −0.7|e| in free quinolone anion species to 0.18|e| in 1a-q, which clearly indicates an electron transfers from quinolone to the cobalt atom.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705 RSC.
- T. Inatomi, Y. Koga and K. Matsubara, Molecules, 2018, 23, 140 CrossRef PubMed.
- D. G. A. Verhoeven, H. A. Negenman, A. F. Orsino, M. Lutz and M.-E. Moret, Inorg. Chem., 2018, 57, 10846 CrossRef CAS PubMed.
- We also investigated several other possible C–C coupling pathways, such as (1) PivOH removal then C–C coupling, and (2) the second amide nitrogen protonation prior to the C–C coupling. Since all these alternative pathways require much more energy barriers, we included them into the ESI.†.
- H.-J. Jiang, X.-M. Zhong, J. Yu, Y. Zhang, X. Zhang, Y.-D. Wu and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 1803 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Kommagalla, Y. Aihara and N. Chatani, Chem. Commun., 2016, 52, 10129 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- C. Y. Legault, CYLview, Université de Sherbrooke, https://www.cylview.org/, 2009 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and the detailed crystallographic information for complexes 1–7. CCDC (1) 1848074, (2) 1862102, (3) 1862295, (4) 1988219, (5) 1848433, (6) 1850726, (7) 1862101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02066d |
| This journal is © The Royal Society of Chemistry 2020 |