
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxalamides by acceptorless dehydrogenative coupling of ethylene glycol and amines and the reverse hydrogenation catalyzed by ruthenium†
You-Quan
Zou‡§
 a,
Quan-Quan
Zhou‡
a,
Yael
Diskin-Posner
b,
Yehoshoa
Ben-David
a and
David
Milstein
*a
a,
Quan-Quan
Zhou‡
a,
Yael
Diskin-Posner
b,
Yehoshoa
Ben-David
a and
David
Milstein
*a
aDepartment of Organic Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: david.milstein@weizmann.ac.il
bChemical Research Support, Weizmann Institute of Science, Rehovot 76100, Israel
First published on 22nd June 2020
Abstract
A sustainable, new synthesis of oxalamides, by acceptorless dehydrogenative coupling of ethylene glycol with amines, generating H2, homogeneously catalyzed by a ruthenium pincer complex, is presented. The reverse hydrogenation reaction is also accomplished using the same catalyst. A plausible reaction mechanism is proposed based on stoichiometric reactions, NMR studies, X-ray crystallography as well as observation of plausible intermediates.
Introduction
Oxalamide skeletons are prevalent in many biologically active molecules and pharmaceuticals, such as Lixiana (1, Fig. 1), widely used as an anticoagulant.1 Oxalamide 2 shows promising antiviral activity of entry inhibitors targeting the CD4-binding site of HIV-1.2 Additionally, oxalamides are employed as robust ligands in copper catalyzed cross coupling reactions (e.g., oxalamides 3–5).3 Besides these, oxalamides are also popular in food processing as flavoring agents (oxalamide 6)4 and in drug delivery as a synthon of organosilica nanoparticles (oxalamide 7).5 Given the importance of oxalamides, there is considerable interest in the development of novel, efficient protocols for their preparation.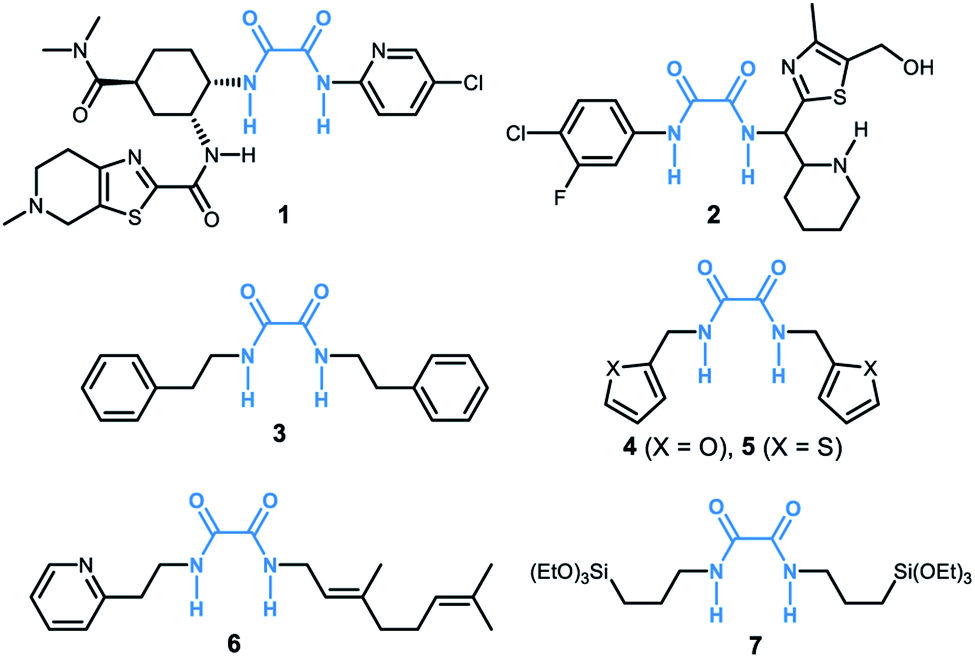 | ||
| Fig. 1 Representative examples of functional molecules containing an oxalamide moiety. | ||
Conventional methods for the synthesis of oxalamides are largely based on the reaction of oxalic acid with thionyl chloride to form oxalyl chloride followed by treatment with amines (Fig. 2a).3,6 Other methods such as oxidative carbonylation of amines using carbon monoxide7a,b and aminolysis of oxalates7c,d also lead to the formation of oxalamides. However, these methods are either not atom economic, generate stoichiometric amounts of waste, or involve acrid or toxic agents. Therefore, the development of atom-economic green and sustainable methods for the efficient construction of oxalamides is highly desirable and remains an important goal both in chemical and pharmaceutical industries.
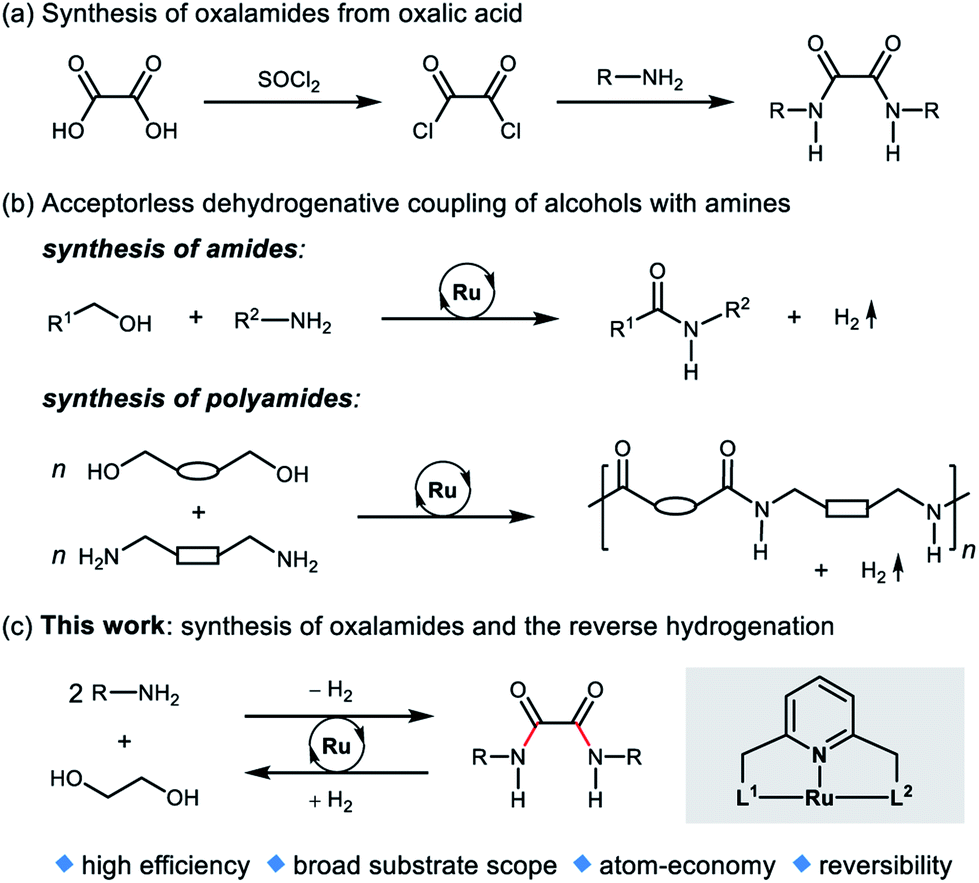 | ||
| Fig. 2 (a) Conventional synthesis of oxalamides, (b) dehydrogenative amide bond formation, and (c) this work. | ||
In 2007, we reported the ruthenium catalyzed dehydrogenative coupling of alcohols with amines leading to the environmentally benign synthesis of amides with H2 liberation as the sole byproduct (Fig. 2b).8 Guan's group9a and our group9b also demonstrated the ruthenium pincer complex catalyzed dehydrogenative coupling of diols and diamines, to form polyamides. Other Ru-catalyzed systems for the synthesis of amides from amines and alcohols were subsequently reported.10 Recently, we,11a–d Prakash11e and Liu groups11f have developed several liquid organic hydrogen carrier systems based on amide bond formation and the reverse hydrogenation reactions.
Ethylene glycol (EG) is an inexpensive and convenient feedstock in industry,12 which can be accessed from renewable biomass-derived hydrocarbons.13 Very recently, we disclosed a reversible liquid organic hydrogen carrier system based on EG, capable of chemically loading and unloading hydrogen.14 In this system, EG undergoes acceptorless dehydrogenative esterification to oligoesters, and then the oligoesters are hydrogenated back to EG by using a ruthenium pincer catalyst. As part of our ongoing research program on green and sustainable homogeneous catalysis, we herein report an atom-economic and environmentally benign strategy for the direct synthesis of oxalamides via acceptorless dehydrogenative coupling of EG with amines (Fig. 2c). A potential challenge is the coordination ability of oxalamides as chelating ligands,3 which could possibly result in product inhibition.
We envisioned that dehydrogenative coupling of EG and the amine will first form an α-hydroxy amide. Subsequently, the resulting α-hydroxy amide would react with another molecule of amine to form the desired oxalamide via a similar catalytic cycle. In addition, the reverse hydrogenation of oxalamides back to EG and amines was also investigated using the same catalyst. To the best of our knowledge, there has hitherto been no report on acceptorless dehydrogenative synthesis of oxalamides from EG and amines, the only byproduct being hydrogen gas, valuable by itself.
Results and discussion
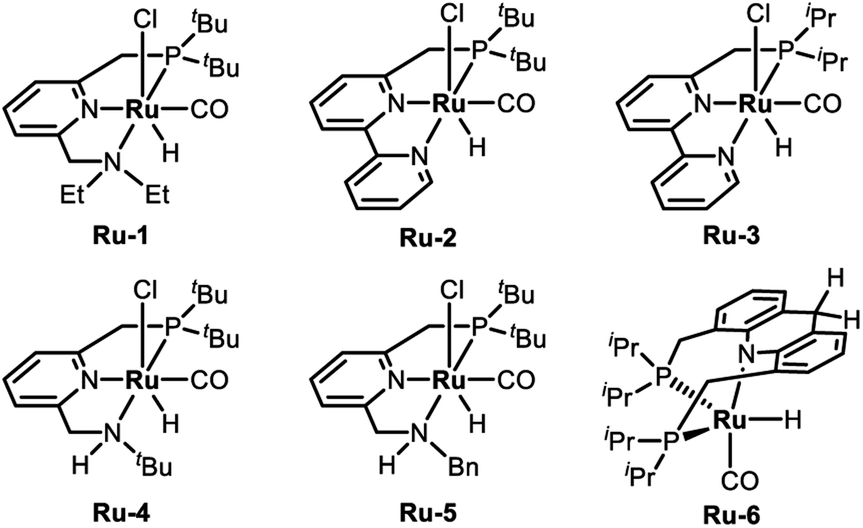
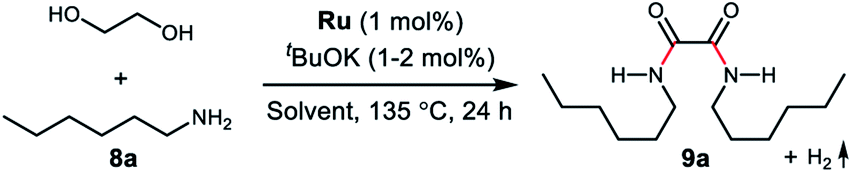
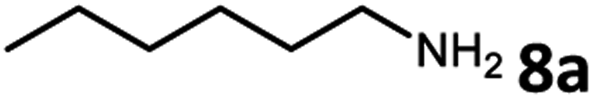
To examine the feasibility of the acceptorless dehydrogenative coupling of ethylene glycol and amines, our initial studies were focused on the reaction of EG and hexan-1-amine (8a). At the beginning, we tested the catalytic efficiency of ruthenium pincer complex Ru-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 which upon deprotonation forms the dearomatized complex and catalyzes the synthesis of amides and polyamides from amines and alcohols.8 The reaction did indeed take place using Ru-1 (1 mol%) and tBuOK (1 mol%) at 135 °C in a mixture of toluene and dimethoxyethane (v/v = 1/1) in a closed system, affording the desired oxalamide 9a in 26% isolated yield after 24 hours (Table 1, entry 1). Using the bipyridine-derived catalysts Ru-216 and Ru-3,16 the isolated yield of 9a was improved to 50% (entries 2 and 3). Employing the PNNH-based complexes Ru-417 and Ru-517 resulted in the case of Ru-5 a considerable yield of 9a to 77% using 2 mol% of tBuOK (entry 5). Encouraged by this promising result, we next evaluated the solvent effect (entries 6–9). Using toluene as a sole solvent with Ru-5 as the catalyst 9a was isolated in 86% yield (entry 6). The dearomatized acridine-based complex Ru-6, previously used in the esterification of EG,14 only afforded product 9a in 27% yield (entry 10). Accordingly, the optimal reaction conditions for the acceptorless dehydrogenative coupling are as depicted in entry 6: 1 mol% of Ru-5 as catalyst, 2 mol% of tBuOK as base, 135 °C in 2.0 mL of toluene.
15 which upon deprotonation forms the dearomatized complex and catalyzes the synthesis of amides and polyamides from amines and alcohols.8 The reaction did indeed take place using Ru-1 (1 mol%) and tBuOK (1 mol%) at 135 °C in a mixture of toluene and dimethoxyethane (v/v = 1/1) in a closed system, affording the desired oxalamide 9a in 26% isolated yield after 24 hours (Table 1, entry 1). Using the bipyridine-derived catalysts Ru-216 and Ru-3,16 the isolated yield of 9a was improved to 50% (entries 2 and 3). Employing the PNNH-based complexes Ru-417 and Ru-517 resulted in the case of Ru-5 a considerable yield of 9a to 77% using 2 mol% of tBuOK (entry 5). Encouraged by this promising result, we next evaluated the solvent effect (entries 6–9). Using toluene as a sole solvent with Ru-5 as the catalyst 9a was isolated in 86% yield (entry 6). The dearomatized acridine-based complex Ru-6, previously used in the esterification of EG,14 only afforded product 9a in 27% yield (entry 10). Accordingly, the optimal reaction conditions for the acceptorless dehydrogenative coupling are as depicted in entry 6: 1 mol% of Ru-5 as catalyst, 2 mol% of tBuOK as base, 135 °C in 2.0 mL of toluene.
|
|
||||
|---|---|---|---|---|
| Entrya | Ru | t BuOK | Solvent | Yieldb (%) |
| a Reaction conditions: ethylene glycol (1.0 mmol), 8a (3.0 mmol), Ru catalyst (1 mol%), tBuOK (1–2 mol%), and solvent (2.0 mL) at 135 °C (bath temperature) for 24 hours in a closed system. b Isolated yield. DME: dimethoxyethane; THF: tetrahydrofuran; Bn: benzyl. Bold row is indicative of optimal conditions. | ||||
| 1 | Ru-1 | 1 mol% | Toluene/DME | 26 |
| 2 | Ru-2 | 1 mol% | Toluene/DME | 50 |
| 3 | Ru-3 | 1 mol% | Toluene/DME | 40 |
| 4 | Ru-4 | 2 mol% | Toluene/DME | 37 |
| 5 | Ru-5 | 2 mol% | Toluene/DME | 77 |
| 6 | Ru-5 | 2 mol% | Toluene | 86 |
| 7 | Ru-5 | 2 mol% | DME | 71 |
| 8 | Ru-5 | 2 mol% | THF | 84 |
| 9 | Ru-5 | 2 mol% | 1,4-Dioxane | 81 |
| 10 | Ru-6 | — | Toluene | 27 |
|
||||
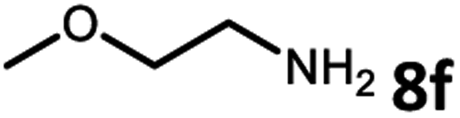
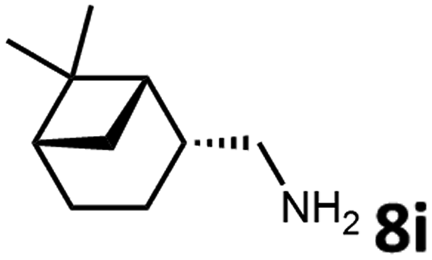
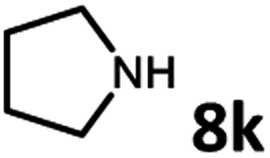
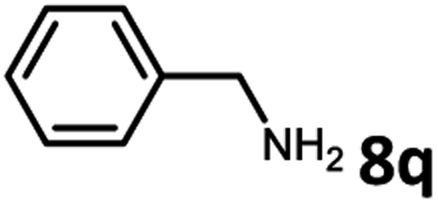
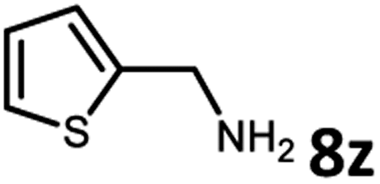
With the optimized reaction conditions in hand, we next examined the generality of this catalytic dehydrogenative coupling system using various amines. As shown in Table 2, various aliphatic amines such as hexan-1-amine (8a), butan-1-amine (8b), pentan-1-amine (8c), heptan-1-amine (8d), dodecan-1-amine (8e) as well as 2-methoxyethan-1-amine (8f) smoothly dehydrogenatively coupled with ethylene glycol to give the corresponding oxalamides 9a–9f in good to excellent isolated yields (66–96%). tert-Butyl(2-aminoethyl)carbamate (8g) and N1,N1-dimethylpropane-1,3-diamine (8h) reacted with ethylene glycol forming the desired products in 53% (9g) and 73% (9h) yields. It is worth noting that (−)-cis-myrtanylamine (8i) and (+)-dehydroabietylamine (8j) could also be applied in this reaction generating oxalamides 9i and 9j in 84% and 70% yields, respectively. The oxalamidation procedure was compatible with secondary amines as well, as shown in the reactions of pyrrolidine and morpholine to form oxalamides 9k (89% yield) and 9l (83% yield). As a limitation of the system, α-branched primary amines such as cyclohexanamine (8m), cycloheptanamine (8n) and (S)-1-phenylethan-1-amine (8o) produced the desired oxalamide products with lower yields (14–32%). The less nucleophilic aniline exhibited low reactivity and only delivered N1,N2-diphenyloxalamide (9p) in 12% yield using 50 mol% of base. Benzylamines (8q–8v) reacted well, and various electron-withdrawing (8r–8t) and electron-donating groups (8u–8v) on the phenyl group did not interfere with the reaction efficiency, furnishing the desired products 9q–9v in good yields (73–97% yields). It is worth mentioning that pyridine-containing amine 8w also worked quite well and the desired product 9w was isolated in 67% yield. Moreover, oxalamides 3–5 which were reported to act as ligands,3 were obtained in 76–89% yields. Compared to previously reported methods, this protocol is more atom-economic, sustainable and efficient.
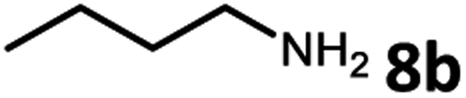
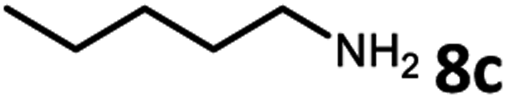
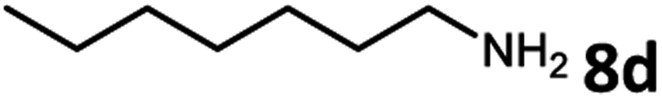
Amines are an important class of compounds widely used in agrochemicals, pharmaceuticals and organic synthesis.18 Hydrogenation of amide bonds represents a green and straightforward method to access amines. Although efficient hydrogenation of amides to amines and alcohols catalyzed by pincer complexes was reported by several groups,19 there is only one example of catalytic hydrogenation of oxalamides (60 bar hydrogen gas, 160 °C).7b Interestingly, the oxalamides synthesized by us via acceptorless dehydrogenative coupling of EG and amines could be fully hydrogenated back to the amines and ethylene glycol using the same catalyst Ru-5. As shown in Table 3, in the presence of 1 mol% of Ru-5, 4 mol% of tBuOK and 40 bar of hydrogen gas at 135 °C in 2.0 mL of toluene, oxalamides 9a–9d (entries 1–4), 9f, 9i, 9k (entries 5–7), 9q–9v (entries 8–13) and 3–5 (entries 14–16) were efficiently hydrogenated to form the corresponding amines and ethylene glycol in excellent yields within 24 hours (85–99% yields).
|
|
|||
|---|---|---|---|
| Entrya | 9, 3–5 | 8 | Yieldb (%) |
| a Reaction conditions: oxalamide (0.25 mmol), Ru-5 (1 mol%), tBuOK (4 mol%), H2 (40 bar) and toluene (2.0 mL) at 135 °C (bath temperature) for 24 hours. b Yields were determined by 1H NMR of the crude reaction mixture using mesitylene as an internal standard. | |||
| 1 | 9a |
|
95 |
| 2 | 9b |
|
95 |
| 3 | 9c |
|
97 |
| 4 | 9d |
|
98 |
| 5 | 9f |
|
86 |
| 6 | 9i |
|
92 |
| 7 | 9k |
|
85 |
| 8 | 9q |
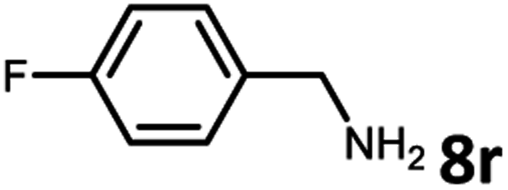
|
97 |
| 9 | 9r |
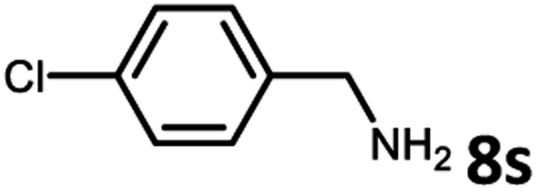
|
99 |
| 10 | 9s |
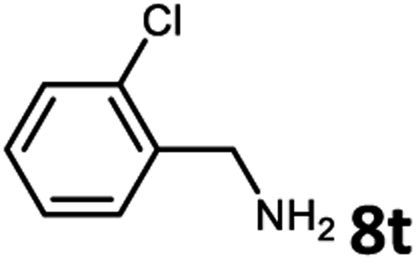
|
96 |
| 11 | 9t |
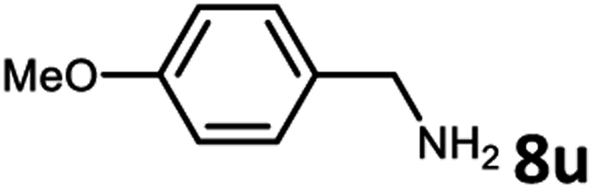
|
99 |
| 12 | 9u |
|
99 |
| 13 | 9v |
|
99 |
| 14 | 3 |
|
95 |
| 15 | 4 |
|
98 |
| 16 | 5 |
|
95 |
To get some insight into the reaction mechanism, complex Ru-5 was treated with 1.1 equivalents of tBuOK in 0.5 mL of THF at room temperature (Scheme 1a), resulting in immediate a color change of the transparent yellow solution to a homogeneous red brown solution, which exhibited a doublet at δ = 110.73 ppm (2JP–H = 15 Hz) in the 31P{1H} NMR spectrum in THF (Scheme 2b). Performing the reaction in d8-THF showed that the N–H proton disappeared and the two CH2 groups of the P-arm and N-arm were still present, clearly indicating that the deprotonation took place at the N–H bond and complex 10 was formed (see ESI and Fig. S6–S9† for details). The hydride resonance of 10 appeared at δ = −18.13 ppm (doublet, 2JP–H = 40.0 Hz) in the 1H NMR (see ESI, Fig. S6† for details). Using 2.2 equivalents of tBuOK also produced complex 10 together with the formation of a new species at δ = 124.0 ppm (broad singlet) in the 31P{1H} NMR, probably attributable to the doubly-deprotonated complex, as we observed before with the PNNH complex Ru-4.17
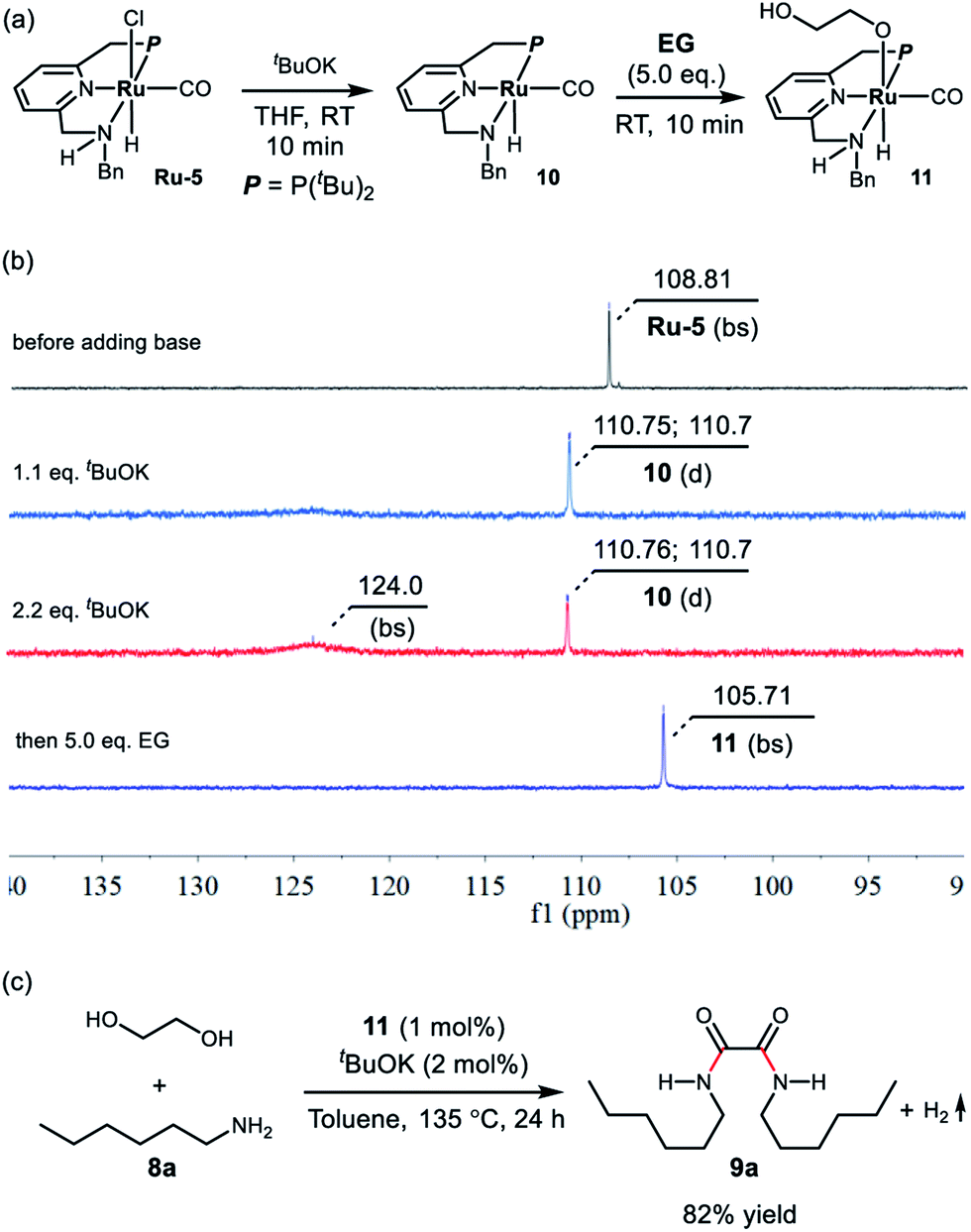 | ||
| Scheme 1 (a) Deprotonation of Ru-5 and activation of ethylene glycol by complex 10, (b) the corresponding 31P{1H} NMR spectra, and (c) the catalytic performance of complex 11. | ||
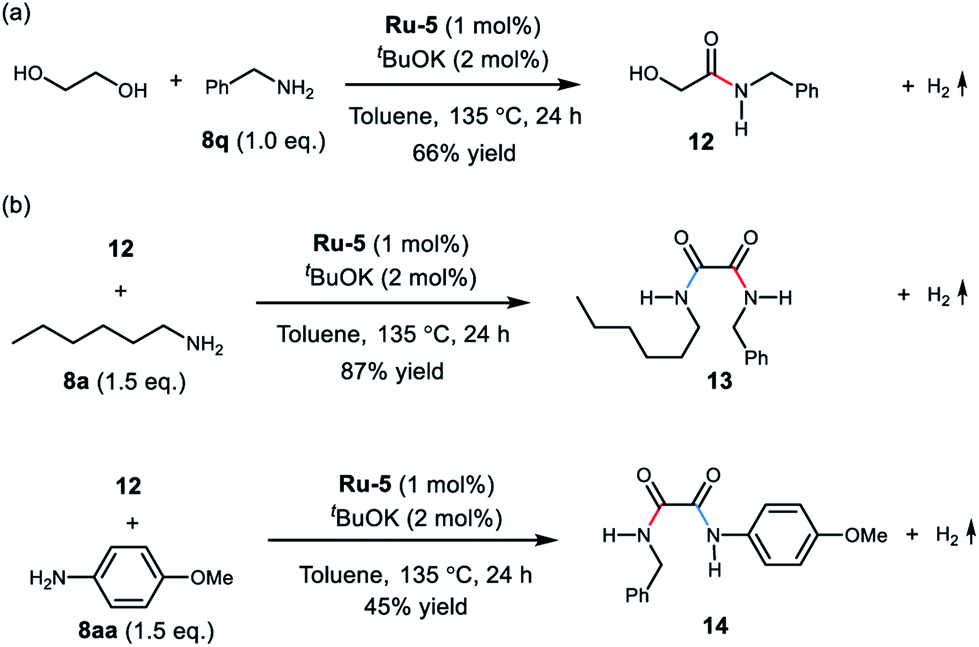 | ||
| Scheme 2 (a) Synthesis of the α-hydroxy amide 12, and (b) the mixed oxalamides 13 and 14. | ||
Upon treatment with 5.0 equivalents of ethylene glycol the above reaction mixture (either from 1.1 equivalents or 2.2 equivalents of base), a reddish brown solution was formed, generating complex 11, resulting from ethylene glycol addition to complex 10. Complex 11 exhibited a broad singlet at δ = 105.71 ppm in the 31P{1H} NMR in THF (Scheme 1b) and in the 1H NMR spectrum the hydride shifted downfield to −16.03 ppm (doublet, 2JH–P = 24.0 Hz), consistent with the alkoxide group of EG located trans to the hydride of complex 11 (see Fig. S10† for details). The IR spectrum of 11 showed a strong carbonyl absorption band at 1899 cm−1 (Fig. S14†). Upon slow evaporation of a solution of 11 in a mixture of THF and pentane, crystals suitable for X-ray diffraction were formed. As shown in Fig. 3, a neutral distorted octahedral complex was generated with the alkoxide group of EG coordinated to the ruthenium center. Upon heating complex 11 at 110 °C in d8-toluene for 25 minutes, a new species which gave rise to signal at δ = 9.59 ppm in the crude 1H NMR, which might be attributed to the formation of glycolaldehyde (see ESI, Fig. S15† for details). Interestingly, using complex 11 as a catalyst, the desired product 9a was isolated in 82% yield, indicating that 11 is a possible catalytic intermediate (Scheme 1c).
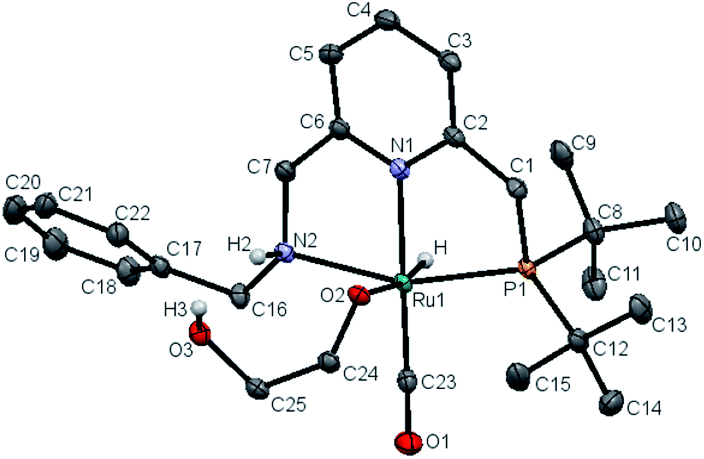 | ||
| Fig. 3 X-ray crystal structure of complex 11. Atoms are presented as thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity except the hydride, H2 and H3. Selected bond lengths (Å) and angles (deg.): Ru(1)–C(23), 1.829(19); Ru(1)–N(1), 2.106(15); Ru(1)–N(2), 2.195(15); Ru(1)–O(2), 2.190(13); Ru(1)–P(1), 2.280(4); Ru(1)–H, 1.58(3); C(23)–Ru(1)–N(1), 178.10(7); N(2)–Ru(1)–P(1), 158.90(4); N(1)–Ru(1)–O(2), 80.98(5). | ||
Interestingly, upon treatment EG with 1.0 equivalent of benzylamine under the optimal conditions, the mono-amidation product 12 was isolated in 66% yield (Scheme 2a). The α-hydroxy- amide 12 was also observed in the reaction of EG with two equivalents of benzylamine (see ESI, Fig. S16–S19† for details). These results support formation of the oxalamide product via an α-hydroxyamide intermediate. Moreover, reaction of amide 12 with hexan-1-amine (8a) afforded the mixed oxalamide 13 in 87% yield (Scheme 2b). The mixed aryl/alkyl oxalamide 14 was also successfully synthesised using p-anisidine as the arylamine coupling partner (45% yield). These results highlight the scope of this method for the synthesis of mixed oxalamides.
Based on the experimental results and previous work,8,11 we propose a possible reaction mechanism for the ruthenium homogeneously catalyzed acceptorless dehydrogenative coupling of ethylene glycol and amines to form oxalamides. As outlined in Scheme 3, deprotonation of Ru-5 by tBuOK leads to complex 10, which adds ethylene glycol to generate the alkoxide species 11via metal ligand cooperation.20 Although there is no vacant coordination site cis to the alkoxide ligand in complex 11, hydride elimination can take place forming intermediate 15via two alternative pathways. One involves the full dissociation of the alkoxide from 11, followed by hydride abstraction from it by the ruthenium center;21 the other possibility is that proton and hydride directly transfer from ethylene glycol to deprotonated complex 10 without the formation of alkoxide 11.22 The hydride elimination might also occur through the N-side arm dissociation. Subsequently, hydrogen evolution from complex 15 takes place, followed by coordination of the generated glycolaldehyde,23 affording the saturated amido intermediate 16. Reaction of 16 with the amine forms the hemiacetal intermediate 17 followed by release of α-hydroxy amide to regenerate the trans-dihydride complex 15. The formed α-hydroxy amide reacts with another molecule of amine to form the corresponding oxalamide 9via a similar pathway. Finally, complex 15 releases a second molecule of hydrogen, regenerating complex 10 which then re-enters the catalytic cycle.
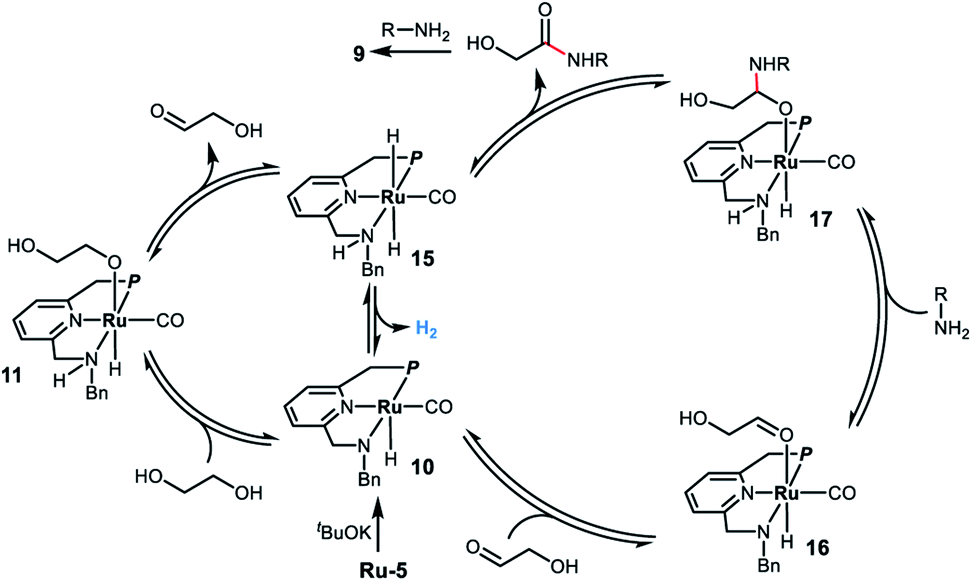 | ||
| Scheme 3 Proposed reaction mechanism for the ruthenium-catalyzed acceptorless dehydrogenative coupling of EG and amines to form oxalamides. | ||
Conclusions
In conclusion, we have developed the acceptorless dehydrogenative coupling of ethylene glycol and amines with H2 evolution, catalyzed by a ruthenium pincer complex, enabling the synthesis of oxalamides in an unprecedented highly atom-economic and sustainable manner. The reverse hydrogenation reaction was also accomplished, resulting in full hydrogenation of the generated oxalamides back to the corresponding amines and ethylene glycol, using the same catalyst. A plausible catalytic mechanism is proposed, supported by stoichiometric reactions, NMR studies, X-ray crystallography as well as the observation of some plausible intermediates. Further detailed mechanistic investigations and applications of this methodology are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (ERC AdG 692775). D. M. holds the Israel Matz Professorial Chair of Organic Chemistry. Y.-Q. Z. acknowledges the Sustainability and Energy Research Initiative (SAERI) of Weizmann Institute of Science for a research fellowship.Notes and references
- J. T. Njardarson, Poster: Top 200 Pharmaceutical Products by Retail Sales in 2018 Search PubMed.
- (a) F. Curreli, S. Choudhury, I. Pyatkin, V. P. Zagorodnikov, A. K. Bulay, A. Altieri, Y. D. Kwon, P. D. Kwong and A. K. Debnath, J. Med. Chem., 2012, 55, 4764–4775 CrossRef CAS PubMed; (b) F. Curreli, Y. D. Kwon, H. Zhang, Y. Yang, D. Scacalossi, P. D. Kwong and A. K. Debnath, Antimicrob. Agents Chemother., 2014, 58, 5478–5491 CrossRef PubMed.
- (a) Z. Chen, Y. Jiang, L. Zhang, Y. Guo and D. Ma, J. Am. Chem. Soc., 2019, 141, 3541–3549 CrossRef CAS PubMed; (b) W. Zhou, M. Fan, J. Yin, Y. Jiang and D. Ma, J. Am. Chem. Soc., 2015, 137, 11942–11945 CrossRef CAS PubMed; (c) S. De, J. Yin and D. Ma, Org. Lett., 2017, 19, 4864–4867 CrossRef CAS PubMed; (d) M. Fan, W. Zhou, Y. Jiang and D. Ma, Org. Lett., 2015, 17, 5934–5937 CrossRef CAS PubMed; (e) G. G. Pawar, H. Wu, S. De and D. Ma, Adv. Synth. Catal., 2017, 359, 1631–1636 CrossRef CAS; (f) V. S. Chan, S. W. Krabbe, C. Li, L. Sun, Y. Liu and A. J. Nett, ChemCatChem, 2019, 11, 5748–5753 CrossRef CAS; (g) D. V. Morarji and K. K. Gurjar, Organometallics, 2019, 38, 2502–2511 CrossRef CAS.
- K. Abdelmajid and W. Cornelis, WO 2011095533 A1, 2011.
- (a) J. G. Croissant, Y. Fatieiev, K. Julfakyan, J. Lu, A. Emwas, D. H. Anjum, H. Omar, F. Tamanoi, J. I. Zink and N. M. Khashab, Chem.–Eur. J., 2016, 22, 14806–14811 CrossRef CAS PubMed; (b) Y. Fatieiev, J. G. Croissant, K. Julfakyan, L. Deng, D. H. Anjum, A. Gurinov and N. M. Khashab, Nanoscale, 2015, 7, 15046–15050 RSC.
- D. Ma, W. Zhou, M. Fan, H. Wu, J. Yin and S. Xia, US Pat. 20180207628 A1, 2018.
- (a) R. Mancuso, D. S. Raut, N. D. Ca', F. Fini, C. Cargagna and B. Gabriele, ChemSusChem, 2015, 8, 2204–2211 CrossRef CAS PubMed; (b) K. Dong, S. Elangovan, R. Sang, A. Spannenberg, R. Jackstell, K. Junge, Y. Li and M. Beller, Nat. Commun., 2016, 7, 12075 CrossRef PubMed; (c) K. Şeker, D. Bariş, N. Arslan, Y. Turgut, N. Pirinççioğlu and M. Toğrul, Tetrahedron: Asymmetry, 2014, 25, 411–417 CrossRef; (d) B. P. Woods, M. Orlandi, C.-Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688–5691 CrossRef CAS PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- (a) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159–1161 CrossRef CAS PubMed; (b) B. Gnanaprakasam, E. Balaraman, C. Gunanathan and D. Milstein, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1755–1765 CrossRef CAS.
- (a) L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef PubMed; (b) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667–2670 CrossRef CAS PubMed; (c) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162–1167 CrossRef CAS; (d) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (e) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS; (f) D. Srimani, E. Balaraman, P. Hu, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2013, 355, 2525–2530 CrossRef CAS.
- (a) P. Hu, E. Fogler, Y. Diskin-Posner, M. A. Iron and D. Milstein, Nat. Commun., 2015, 6, 6859 CrossRef CAS PubMed; (b) P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 1061–1064 CrossRef CAS PubMed; (c) A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, J. Am. Chem. Soc., 2018, 140, 7453–7457 CrossRef CAS PubMed; (d) Y. Xie, P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2019, 58, 5105–5109 CrossRef CAS PubMed; (e) J. Kothandaraman, S. Kar, R. Sen, A. Goeppert, G. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 2549–2552 CrossRef CAS PubMed; (f) Z. Shao, Y. Li, C. Liu, W. Ai, S.-P. Luo and Q. Liu, Nat. Commun., 2020, 11, 591 CrossRef CAS PubMed.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- D. Milstein, E. Balaraman, C. Gunanathan, B. Gnanaprakasam and J. Zhang, US Pat. 20130281664 A1, 2013.
- E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem.–Eur. J., 2014, 20, 15727–15731 CrossRef CAS PubMed.
- (a) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (b) P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Amines, Aliphatic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed; (b) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240–4242 CrossRef CAS PubMed; (c) J. M. John and S. H. Bergens, Angew. Chem., Int. Ed., 2011, 50, 10377–10380 CrossRef CAS PubMed; (d) J. R. Cabrero-Antonino, E. Alberico, H. J. Drexler, W. Baumann, K. Junge, H. Junge and M. Beller, ACS Catal., 2016, 6, 47–54 CrossRef CAS; (e) N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS; (f) V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spanneberg, K. Junge, H. Junge and M. Beller, Chem. Sci., 2017, 8, 3576–3585 RSC; (g) N. Gorgas and K. Kirchner, Acc. Chem. Res., 2018, 51, 1558–1569 CrossRef CAS PubMed; (h) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- (a) H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed; (b) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed; (d) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (e) K. Sordakis, C. Tang, L. Vogt, H. Junge, P. J. Dyson and M. Beller, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- O. Blum and D. Milstein, J. Organomet. Chem., 2000, 593–594, 479–484 CrossRef CAS.
- H. Li, X. Wang, F. Huang, G. Lu, J. Jiang and Z.-X. Wang, Organometallics, 2011, 30, 5233–5247 CrossRef CAS.
- The reaction of glycolaldehyde dimer 1,4-dioxane-2,5-diol with 8a also produced product 9a in 62% isolated yield under optimal conditions, which further supports glycolaldehyde as a likely intermediate in this transformation.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1957818. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02065f |
| ‡ You-Quan Zou and Quan-Quan Zhou contributed equally. |
| § Present address: Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. |
| This journal is © The Royal Society of Chemistry 2020 |