
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of an asparaginyl endopeptidase for chemo-enzymatic peptide and protein labeling†
T. M. Simon
Tang
 ,
Davide
Cardella
,
Alexander J.
Lander
,
Xuefei
Li
,
Jorge S.
Escudero
,
Yu-Hsuan
Tsai
and
Louis Y. P.
Luk
*
,
Davide
Cardella
,
Alexander J.
Lander
,
Xuefei
Li
,
Jorge S.
Escudero
,
Yu-Hsuan
Tsai
and
Louis Y. P.
Luk
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: lukly@cardiff.ac.uk
First published on 12th May 2020
Abstract
Asparaginyl endopeptidases (AEPs) are ideal for peptide and protein labeling. However, because of the reaction reversibility, a large excess of labels or backbone modified substrates are needed. In turn, simple and cheap reagents can be used to label N-terminal cysteine, but its availability inherently limits the potential applications. Aiming to address these issues, we have created a chemo-enzymatic labeling system that exploits the substrate promiscuity of AEP with the facile chemical reaction between N-terminal cysteine and 2-formyl phenylboronic acid (FPBA). In this approach, AEP is used to ligate polypeptides with a Asn–Cys–Leu recognition sequence with counterparts possessing an N-terminal Gly–Leu. Instead of being a labeling reagent, the commercially available FPBA serves as a scavenger converting the byproduct Cys–Leu into an inert thiazolidine derivative. This consequently drives the AEP labeling reaction forward to product formation with a lower ratio of label to protein substrate. By carefully screening the reaction conditions for optimal compatibility and minimal hydrolysis, conversion to the ligated product in the model reaction resulted in excellent yields. The versatility of this AEP-ligation/FPBA-coupling system was further demonstrated by site-specifically labeling the N- or C-termini of various proteins.
Introduction
There has been a vast expansion in the toolkit of protein bioconjugation,1–3 drawing on expertise from both biological4–6 and organic7–12 chemistry. Protein-based approaches offer the ability to function efficiently under mild reaction conditions.5,6 Transferase,13–15 oxidoreductase,16–18 ligase,19,20 transpeptidase2,4,21–27 and (split-)intein28–30 have been applied for protein bioconjugation. Nevertheless, adapting these enzymes or proteins for synthetic applications, which deviate from their natural function, often results in technical issues. Whereas stability and solubility of intein-fused constructs are extremely case-dependent,28–30 reversible enzymatic reactions need to be suppressed by a large excess of label24,31 or unstable substrate that has inherent limitation to where bioconjugation takes place.32–34 In contrast, chemical approaches with commercially available reagents are simple to perform and have become standard practice.4,8–10 However, efficiency and selectivity of these reactions relies on the availability of specific residues, which varies greatly among proteins.2,35 These descriptions apply to the bioconjugation reaction mediated by the enzyme asparaginyl endopeptidase (AEP) and the chemical labeling of N-terminal cysteine by 2-formyl phenylboronic acid (FPBA). Here, we combine their strengths together, such that site-specific protein labeling can be achieved at the terminus of choice (N or C).Many asparaginyl endopeptidases (AEPs) from plants possess transpeptidase function and present as ideal biocatalysts for protein labeling (i.e., intermolecular ligation).24,26,27,36–38 AEP hydrolyzes the C-terminal amide bond of an internal asparagine or aspartate residue (P1) and subsequently mediate ligation to the N-terminus of an incoming nucleophile peptide (Fig. 1).26,36,39–41 Some important examples of AEP include butelase 1 and OaAEP1, and their excellent kinetic properties and relatively short recognition sequence (3 amino acids, P1–P1′–P2′) are the key advantages. Protein substrates including GFP, ubiquitin, ompA, DARPin, maltose binding proteins and nanobodies have previously been modified by AEPs, whereas unnatural amino acids, click handles, modified residues, polyethylene glycol, fluorophores, biotin and drug molecules have been used as labels.24,26,31,32,42–45 However, similar to other transpeptidases, bioconjugation by AEP requires a relatively large excess of labeling agents, with respect to the protein.31 This becomes a major hurdle for expensive or non-commercially available labels (e.g., isotopic, radioactive and fluorescent labels).46–49
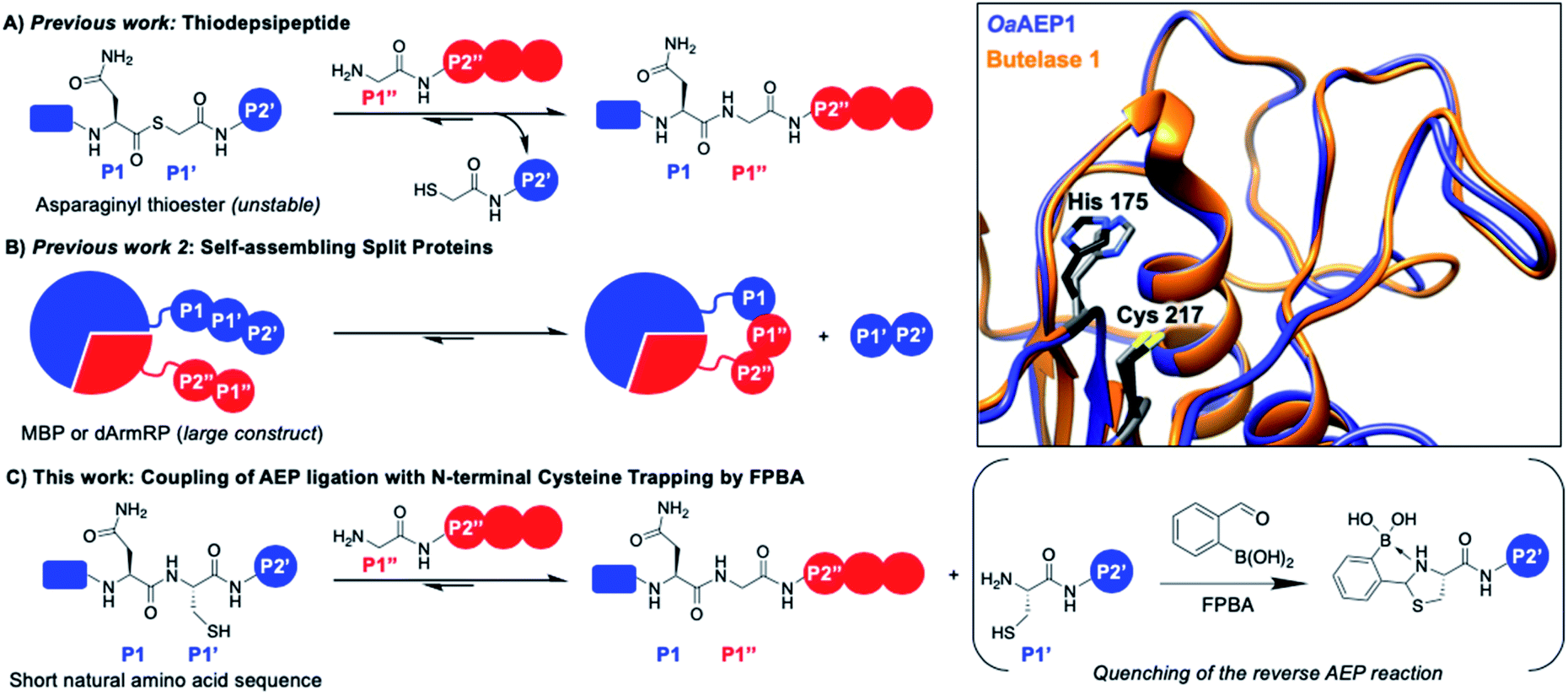 | ||
| Fig. 1 AEP catalyzes the hydrolysis of the amide bond between amino acids P1 (Asx) and P1′ followed by the ligation of the α-amine of P1′′. Various approaches have been reported to drive the reaction equilibrium towards product formation, including the use of (A) thiodepsipeptides32 and (B) self-assembling protein domains.48,49 (C) Here, a chemo-enzymatic approach that incorporates N-terminal cysteine trapping by formylphenyl boronic acid (FPBA) was reported. (Inset) Crystal structure overlay of the active sites of OaAEP1 (orange, 5H0I) and butelase 1 (blue, 6DHI), with the catalytic diad of OaAEP1 and butelase 1 highlighted in black and grey, respectively. Residues are numbered according to OaAEP1. | ||
One valuable feature of AEPs is their relatively relaxed substrate specificity, which has been used to improve the enzyme-catalyzed bioconjugation reaction.26,36,38,39,41,48,49 Asparaginyl thiodepsipeptides have been used to develop irreversible butelase 1-mediated labeling (Fig. 1A). However, these alternative substrates have extremely short half-lives (k1/2 ≈ 45–75 min at pH 6.5) and effective labeling requires four to five equivalent excess of label, added in small portions.32,50 Moreover, labeling with the use thiodepsipeptide is limited to the N-terminus.32 Other solutions include the use of split proteins which have inherent affinity towards each other (Fig. 1B),48,49 but it requires the insertion of a large split-protein domain. Rehm et al. has illustrated that the reactivity difference between two recognition sequences at P2' (Asn–Gly–Leu versus Asn–Gly–Val) can be exploited to avoid hydrolysis catalyzed by OaAEP1, a homologue that carries significant sequence identity to butelase 1 (66%) (Fig. 1 inset).24 However, a large excess of labeling agents (20–120 equivalents, relative to the protein substrate) for intermolecular ligation was used. It has also been demonstrated Asn–Cys–Leu (P1–P1′–P2′) can be recognized for ligation by OaAEP1;48,49 such a reaction will generate byproduct that carries a N-terminal cysteine (Cys–Leu) whose reactivity can be exploited to develop an irreversible AEP-reaction and has yet to be explored.
The 1,2-aminothiol functionality of N-terminal cysteine contains two nucleophilic centers, and thus it can react with aldehydes to form thiazolidinones.51–54 2-Formyl phenylboronic acid (FPBA) is one such electrophile that reacts with N-terminal cysteine with exquisite selectivity and efficiency.53,54 With biomolecular rate constant measured up to 105 M−1 s−1, FPBA and its derivatives have been used for polypeptide labeling.53,54 Nevertheless, preparing proteins with a N-terminal cysteine is case-dependent and can be a challenging task.30,55–59 While successful examples have been reported, the N-terminal cysteine can readily react during gene expression (e.g. pyruvate) limiting the potential FPBA labeling reaction.30,55–59 Instead, this cheap and commercially available reagent can be used as a scavenger for AEP catalysis. We propose that the non-natural secondary amine motif formed between FPBA and N-terminal cysteine is unlikely to be a reactive substrate for AEP catalysis, and thus the byproduct of the enzymatic reaction Cys–Leu can be potentially trapped by the addition of FPBA. To this end, an irreversible AEP-labeling system can be achieved, such that bioconjugation can take place at the terminus of choice (N or C) with minimal modification using a lower ratio of label to protein (Fig. 1C).
Here, we develop a chemo-enzymatic protein labeling strategy, whereby the intermolecular ligation by AEP is coupled to a FPBA reaction that quenches the ligation byproduct. The P1–P1′–P2′ recognition sequence used for AEP labeling is Asn–Cys–Leu, and FPBA is added as a scavenger that reacts with the 1,2-aminothiol motif of Cys–Leu (Fig. 1C).53,54 Consequently, the AEP-mediated peptide ligation can be driven forward, whilst lowering the amount of labeling reagents used. By carefully tuning the amount of FPBA, pH, temperature and reaction time, the newly developed labeling system has minimal hydrolysis for the label, protein substrate and product. In our model peptide ligation reactions, this chemo-enzymatic approach proceeds in excellent yields (up to 95%). We have also proved our concept by labeling the N- or C-terminus of proteins of different sequences.
Results and discussion
Preparation of OaAEP1–C247A
The AEP variant derived from Oldenlandia affinis OaAEP1–C247A was chosen due to the availability of its recombinant procedure, and this variant was also reported to have activity superior to that of the wild type and comparable to that of butelase 1 (under their respective optimized reaction conditions) (Fig. 1 inset).24,39 However, in the literature, a prodomain was included during gene expression, and thus enzyme activation under acidic condition was required.24,26,37 Although, in agreement with the literature,26,37 the activity of the activated enzyme can be mildly improved (∼15%, ESI Fig. S1†) after subjecting the activated OaAEP1–C247A to cation exchange chromatography, a lengthy preparation protocol is not desirable. With a view on practicality, we have simplified the enzyme preparation protocol by expressing the gene of the active enzyme fused with a ubiquitin and a hexahistidine tag. The enzyme yield is comparable to existing approaches (∼2 mg vs. 1.8 mg, see ESI† for more information),26 but the steps involved are reduced. Furthermore, this simplified enzyme construct showed comparable kinetic behaviors to the acid activated OaAEP1–C247A (for comparisons between the kcat and KM values, see Table S1†)24,39 and was shown to mediate protein bioconjugation via the chemo-enzymatic approach described below.Substrate specificity of OaAEP1–C247A
The hydrolysis and nucleophile peptide profiles for peptide cyclization by OaAEP1 has been previously reported.24,26,37,39 To complement these reports, here we present the hydrolysis profile for intramolecular OaAEP1–C247A (Fig. 2). The model ligation reaction between CFRAN![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) L (where X at P1′ position is any of the 20 amino acids, 50 μM) and GLGGIR (250 μM, 5 equivalents) was performed at pH 5.0 with 0.1 μM of enzyme (1
L (where X at P1′ position is any of the 20 amino acids, 50 μM) and GLGGIR (250 μM, 5 equivalents) was performed at pH 5.0 with 0.1 μM of enzyme (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 enzyme to substrate ratio, Fig. 2A). Specificity at the P1′ position is rather relaxed; OaAEP1–C247A was able to hydrolyze the peptide bond between asparagine and all 20 amino acids except proline (Fig. 2A). Similar to the previous work that characterized the wild-type OaAEP1,48,49 Asn–Cys–Leu can also be recognized by the C247A variant, thus allowing us to develop the proposed coupling between AEP catalysis and cysteine/FPBA reaction (Fig. 1C). In contrast, specificity at the P2′ position is more restricted. When CFRANG was used, the enzyme prefers large hydrophobic residues such as Phe, Ile, Leu, Met and Trp (Fig. 2B). Furthermore, echoing the report by Rehm et al.,24 our hydrolysis profile also illustrates that a Val residue at the P2′ position results in poor hydrolytic activity. The P2′ preference of OaAEP1–C247A also resembles the P2′′ acceptor profile of butelase 1, an AEP with significant sequence identity (66%) (Fig. 1; inset).26,36 Based on previous studies of OaAEP1,24,26,37,39 G and L at P1′′ and P2′′ represent one of the most effective combination for the nucleophile peptide. Nevertheless, other combinations of residues can also be used, as long as they do not interfere with the reactivity of FPBA (e.g., avoid N-terminal cysteine) and are suitable for the specific AEP variant used.39
:500 enzyme to substrate ratio, Fig. 2A). Specificity at the P1′ position is rather relaxed; OaAEP1–C247A was able to hydrolyze the peptide bond between asparagine and all 20 amino acids except proline (Fig. 2A). Similar to the previous work that characterized the wild-type OaAEP1,48,49 Asn–Cys–Leu can also be recognized by the C247A variant, thus allowing us to develop the proposed coupling between AEP catalysis and cysteine/FPBA reaction (Fig. 1C). In contrast, specificity at the P2′ position is more restricted. When CFRANG was used, the enzyme prefers large hydrophobic residues such as Phe, Ile, Leu, Met and Trp (Fig. 2B). Furthermore, echoing the report by Rehm et al.,24 our hydrolysis profile also illustrates that a Val residue at the P2′ position results in poor hydrolytic activity. The P2′ preference of OaAEP1–C247A also resembles the P2′′ acceptor profile of butelase 1, an AEP with significant sequence identity (66%) (Fig. 1; inset).26,36 Based on previous studies of OaAEP1,24,26,37,39 G and L at P1′′ and P2′′ represent one of the most effective combination for the nucleophile peptide. Nevertheless, other combinations of residues can also be used, as long as they do not interfere with the reactivity of FPBA (e.g., avoid N-terminal cysteine) and are suitable for the specific AEP variant used.39
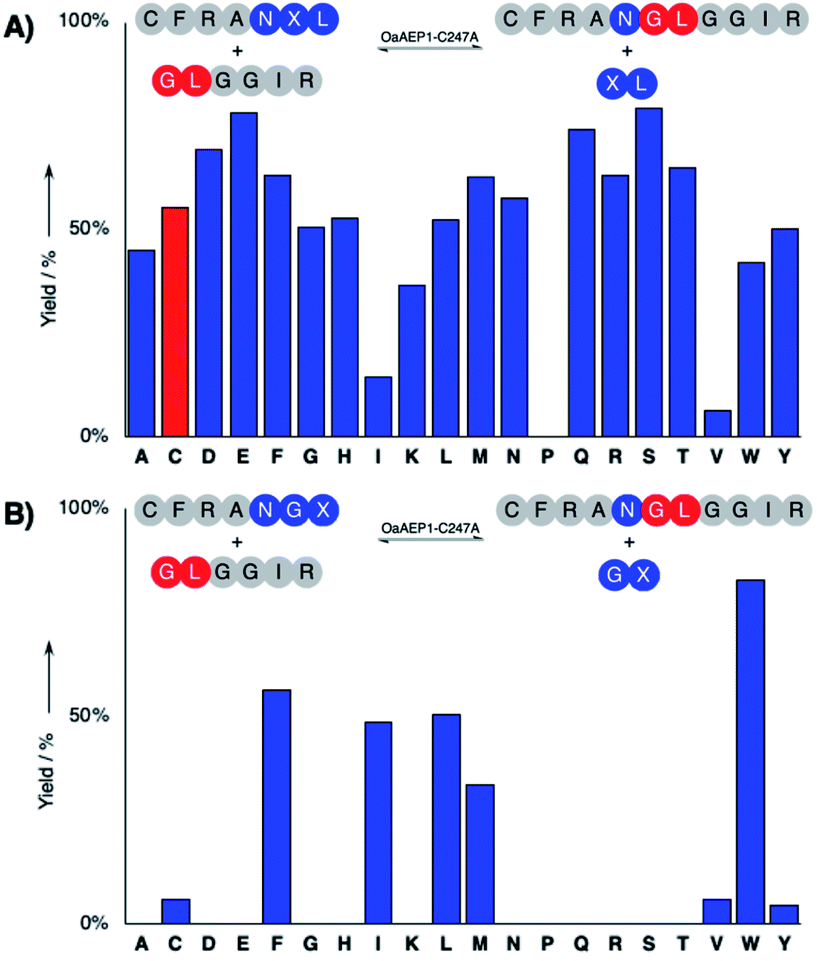 | ||
| Fig. 2 Hydrolysis profile for peptide ligation catalyzed by OaAEP1–C247A. (A) The ligation of CFRANXL to GLGGIR, with the investigated alternative recognition sequence (Asn–Cys–Leu) colored in red, and (B) the profile using CFRANGX, where X is any of the 20 amino acids. | ||
Kinetic characterizations of OaAEP1–C247A and FPBA conjugation
The aim of this work is to quench the reactivity of the P1′–P2′ byproduct (Cys–Leu) generated from the AEP labeling reaction by including the electrophile FPBA (Fig. 1C). Consequently, the compatibility between AEP catalysis and cysteine/FPBA reaction was investigated. A model thiazolidine formation reaction between FPBA and a N-terminal cysteine peptide CFRANGL was monitored by a reported UV spectroscopic assay.53,54 Gratifyingly, the bioconjugation reaction took place at all examined pH's with rate constants ranging from 0.7 × 103 M−1 s−1 at pH 5.0 to 6.0 × 103 M−1 s−1 at pH 7.0 (Fig. S2†). For OaAEP1–C247A catalysis, the pH rate profile indicates that the catalytic turnover constant (kcat) can be as high as 7.5 s−1 at pH 5.0 and decreases to ∼1 s−1 at pH 7.5 at 20 °C. Since the Michaelis constant (KM) remains largely unchanged, the catalytic efficiency (kcat/KM) was found to be within a three-fold difference from pH 5.0 to 7.5 (2.8 to 0.9 × 104 M−1 s−1) (Fig. S3†). In the other words, the two reactions are kinetically compatible at the examined pH range (5.0–7.0). Even at a low concentration (e.g., μM to mM range), the rate of thiazolidine formation is compatible to the catalytic turnover for OaAEP1–C247A.Intermolecular peptide ligation mediated by OaAEP1–C247A
One consideration is that AEPs are able to catalyze hydrolysis of internal asparagine (and aspartate) residues using water as the nucleophile.26,37,38,43 Consequently, the label, substrate or product could be hydrolyzed to yield unwanted side-products. Using the intermolecular ligation reaction between the peptides LFRANCLK and GLGGIR (Table 1; eqn (1)) as a model, we have isolated several key variables that influence the extent of hydrolysis by OaAEP1–C247A.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | pH | X | Z | GZGGIRb (equiv.) | Time (h) | Conversion (%) | |
| (−) FPBA | (+) FPBA | ||||||
| a All reactions were carried out in triplicate with OaAEP1–C247A (0.3 μM), LFRANXLK (300 μM) and GZGGIR (300–6000 μM) at 20 °C. OaAEP1–C247A prepared from acid activation of the zymogen and from the simplified construct were used. b Equivalents of labeling peptide used relative to the peptide substrate, LFRANXLK. c 50 mM NaOAc buffer with 50 mM NaCl, 1 mM EDTA, 0.5 mM TCEP. d 50 mM MES buffer with 50 mM NaCl, 1 mM EDTA, 0.5 mM TCEP. e 10–15%. f 5–10%. g <5% of the undesired hydrolysis product observed. | |||||||
| 1 | 5.0c | Cys | Leu | 1.2 | 3 | 53e | 89e |
| 2 | 5.2c | 3 | 54f | 95f | |||
| 3 | 5.5d | 3 | 49f | 85f | |||
| 4 | 4 | 48f | 90f | ||||
| 5 | 5.7d | 3 | 51g | 78g | |||
| 6 | 4 | 50g | 94g | ||||
| 7 | 5.7d | Ala | 3 | 67g | 61g | ||
| 8 | 4 | 70g | 67g | ||||
| 9 | 5.7d | Cys | Val | 1.0 | 4 | 24f | 45f |
| 10 | 1.5 | 33f | 55f | ||||
| 11 | 2.0 | 32f | 66f | ||||
| 12 | 5.0 | 65g | >95g | ||||
| 13 | 10.0 | >95g | >95g | ||||
| 14 | 20.0 | >95g | >95g | ||||
The pH of the reaction was kept at ≥5.0 to minimize the undesired hydrolytic reaction (Table 1; entry 1–6. Table S2;† entry 42–43, 56–57, 62–63, 72–73 for reactions tested at pH 4.5–5.7); similar observations were made in the studies of AEP from other plant species.36,40 Hydrolysis was found to be further minimized when the temperature was kept at ≤20 °C (Table S2;† entry 46–53 and 92–96 for reactions tested at 20 and 37 °C). Also, reaction time needed to be carefully screened in order to strike a balance between hydrolysis and product formation (Table S2;† entry 58–59). While AEP can hydrolyze peptide upon prolonged incubation,36,40 FPBA was also found to slow the process of peptide ligation. This is likely because of reversible interactions with either the enzyme or the nucleophilic peptide (e.g., forming iminium ion; Table 1; entry 3–6 and entry 80–85 in Table S2†). Based on the +100 polypeptide reactions tested here, the reaction time was kept in between 2 and 18 h, with a mode average of 4 h.24 Lastly, a relatively small excess of the nucleophilic peptide (1.2–2 equivalents) was used to further diminish hydrolysis (Tables 1 and 2 and entry 30–32 in Table S1†).
Under the optimized conditions, 94% of the ligated product LFRANGLGGIR was obtained in 4 h at pH 5.7 using 1.2 equivalent of the nucleophilic peptide, when FPBA was included in the system (Table 1; entry 6). In contrast, when FPBA was excluded, product conversion was stalled at ∼50% (Table 1; entry 1–6). For the cysteine-free peptide LFRANALK, no significant difference in ligation yield was observed by adding FPBA (Table 1; entry 7–8). Also, the enzymatic activity difference between the P1–P1′–P2′ recognition sequences Asn–Cys–Leu and Asn–Gly–Leu was only about 10% (Fig. 2A). Together, these observations indicate that the increase of reaction yield (>40%, Table 1) is caused by the coupling between the FPBA reaction and AEP ligation.
Recently, it has been demonstrated that hydrolysis can be avoided by using polypeptides that carry Gly–Val as the P1′′–P2′′ nucleophile.24 To examine if this concept can be applied to the chemo-enzymatic strategy described here, an alternative peptide GVGGIR was tested in the model ligation reaction (Table 1; entries 9–14).24 Independent of FPBA addition, excellent conversion (>95%) was observed using 10–20 equivalents of peptides. However, when only 1–5 equivalents of GVGGIR were used, conversion was found to be ∼1.5 to 2-fold higher in the reactions that included FPBA. Nevertheless, product conversion was notably lower when compared to those where GLGGIR were used, thus suggesting that Gly–Val nucleophile is less suitable for the chemo-enzymatic approach. Consequently, nucleophilic peptides with Gly–Leu at the N-terminus were used for protein bioconjugation.
OaAEP1–C247A mediated protein bioconjugation
The newly developed AEP-ligation/FPBA-coupling approach was employed for site-specific C-terminal labeling. A linker containing Asn–Cys–Leu was added to the C-terminus of enhanced green fluorescent protein (eGFP), and ligation reactions with the biotinylated peptide GLGGZ (where Z is biotinylated lysine) were performed. Estimated by LC-MS analysis,10,24,60 the yield of the C-terminally modified protein increased up to 1.5-fold when FPBA is included (Table S3†), and up to 92% of the C-terminally modified protein could be achieved using two equivalents of the biotin label (Table 2 and Fig. 3). Similar findings were observed in the labeling of other monomeric and multimeric proteins. 85% of β-lactamase was modified at the C-terminus under the same reaction condition (Fig. 3). As demonstrated here and in previous works,27,37 AEP functions from pH ∼5.0–7.5, but some proteins may become unstable even under mildly acidic environments. Hence, the presented labeling system was also tested with the engineered lumazine synthase AaLS-13, a macromolecular complex composed of 360 protein subunits which is prone to precipitation at pH below 7.0.63–65 Up to 75% of the AaLS-13 subunits were found to be labeled under neutral pH condition (Fig. 3). While it is of little doubt that full conversion can be achieved by using a slightly higher equivalent of labeling peptide (Table 2), reactions performed here were capped at two equivalents to demonstrate the effectiveness of this approach at a relatively low label-to-protein ratio. In a similar fashion, ubiquitin bearing a C-terminal Asn–Cys–Leu sequence could be labeled with the biotinylated peptide GLGGZ (Fig. 3). These findings illustrate that the chemo-enzymatic approach is suitable for a range of proteins with different sequence and biophysical properties, whilst lowering the ratio of label to protein substrate needed, and is complementary to the existing site-specific C-terminal modification technologies.12,24,32,42–44,60,66,67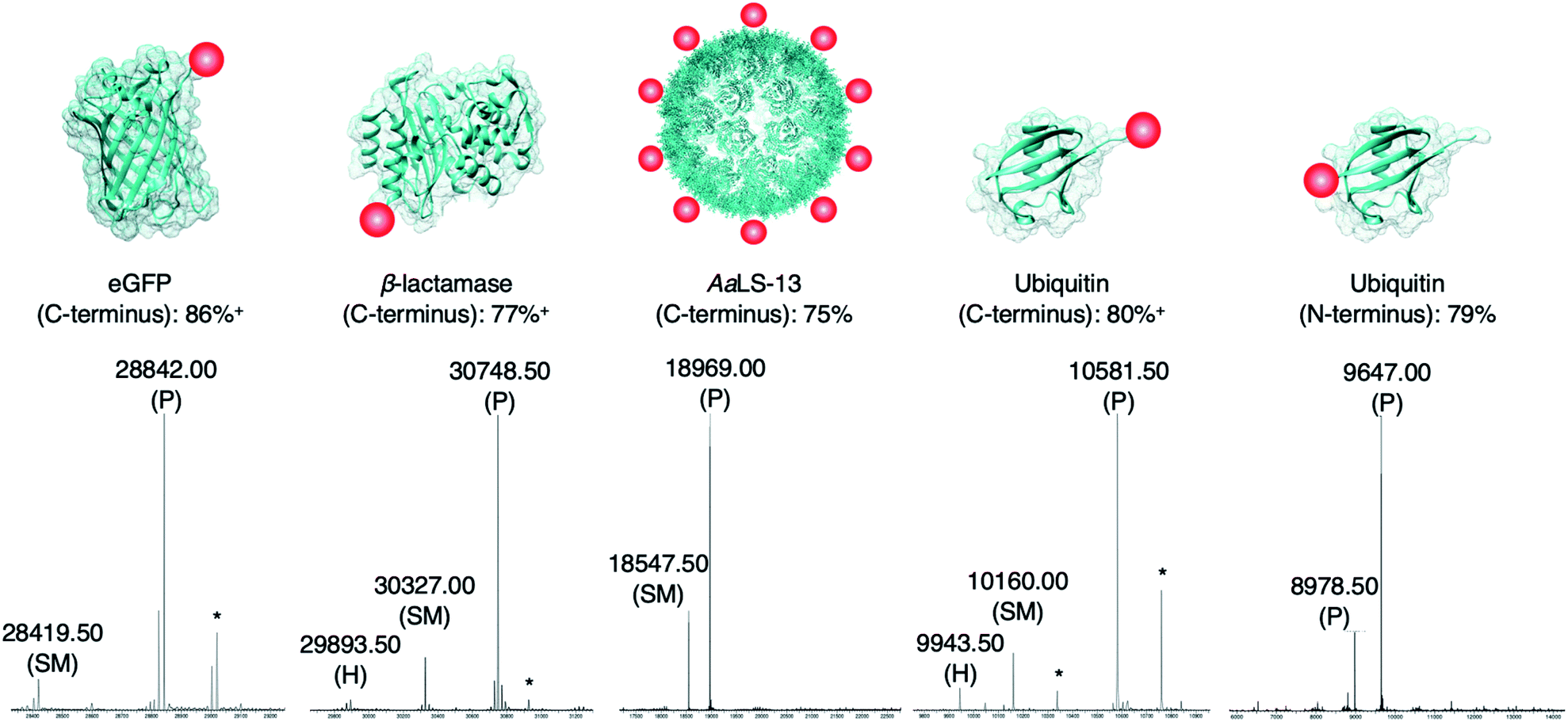 | ||
| Fig. 3 Protein modification. Proteins (100 μM) modified with a short linker and the recognition sequence (Asn–Cys–Leu) for OaAEP1 were incubated with biotin labeled peptide (200 μM), OaAEP1-C247A (0.25 μM) and FPBA (200 μM). The reactions were quenched with 1 M HCl then analyzed by UPLC-MS. SM refers to starting material and P to product. (*) Denotes peaks correlating to α-N-gluconoylation of recombinant protein starting material and products (+178 Da).61,62 (+) Denotes conversion averaged from repeated experiments, associated errors are reported in Fig. S5, S6 and S8.† Asp-to-Ala mutation was introduced to eGFP in order to avoid undesired hydrolytic reaction (Table S3†). The corresponding chromatograms and full mass spectra of the UPLC-MS analysis are reported in Fig. S5–S9.† Other than the species reported above, there is no evidence of hydrolyzed peptides or formation of any other byproducts. Quenching by the addition of 1 M HCl was used solely for the purpose of obtaining a precise reaction time during optimization. | ||
Since only natural amino acids are needed for recognition, the chemo-enzymatic method was also applied for site-specific N-terminus labeling. Ubiquitin containing an extra Gly–Leu sequence at the N-terminus was recombinantly prepared as the substrate, and biotin-labeled peptide bearing the recognition sequence Asn–Cys–Leu at the C-terminus (i.e., biotin-ATRNCL) was synthesized for labeling (see ESI†). 79% of ubiquitin was labeled at the N-terminus using two equivalents of the biotinylated label when FPBA was included (Fig. 3). This finding is complementary to the previous work in which thiodepsipeptide was used,32 but issue surrounding the stability of label was not observed here. It should be noted that all the proteins used here contained other internal asparagine and aspartate residues. However, they are neither hydrolyzed nor modified with only one exception. An Asp235Ala mutation at the solvent-exposed internal site of eGFP (11 residues from the C-terminus) was needed (Fig. S5 and Table S4†).68 In the other words, accessibility plays a critical role in AEP-based modification, dictating both the reaction yield and side reactions.
Conclusions
The presented work combines the advantages of chemical and enzymatic labeling, creating a bioconjugation system with features that could not be achieved by either method alone. Transpeptidases are appealing tools for bioconjugation,5,6,13,14,16,21,26,39,42 but their reactions are reversible, and thus a large ratio of labeling agents to protein substrate is needed to achieve high conversion.21,23,32,53,54,60,69 While the use of backbone-modified labels such as (thio)depsipeptides has improved yields of reactions catalyzed by sortase,33,34 subtiligase,70,71 trypsiligase22,25 and AEPs,32 stability of these alternative labels varies significantly and can be difficult to prepare.50 Furthermore, the backbone-modified approach is largely limited to N-terminal labeling.32,33,70 On the other hand, FPBA is a commercially available reagent that offers a fast, selective and simple method for modification,4,53,54 but preparation of proteins with a free N-terminal cysteine is not universally trivial, as it is prone to side reaction (oxidation and thiazolidinone formation).30,55–59 However, when the AEP catalysis and FPBA bioconjugation are combined together, a system that enables both N- and C-terminal ligation with the use of stable labeling agents is developed.32Provided that the AEP-ligation/FPBA-coupling method lowers the ratio of label to protein substrate, it is particularly applicable when expensive or non-commercially available labels are used.46–49 While some of the proteins labeled here, including eGFP, beta-lactamase and AaLS13, contain cysteine residues (in oxidized or reduced form), it should be noted that extension of this approach might require preliminary investigations, as addition of cysteine residue could potentially destabilize some proteins,72,73 particularly those that contains significant number of disulfide bonds (e.g., anti- and nano-bodies).72,73 Since the activity of OaAEP1 is limited to neutral or acidic conditions,37 studies towards the use of FPBA to enhance other enzymatic labeling strategies may be worthwhile. The development in this technology complements the use of existing transpeptidases such as sortase, as the differences in substrate specificity may be fully exploited in combination to develop orthogonal ligation strategies.23,24,44 In summary, the pairing of enzymatic transpeptidation with well-established chemical reactions offers a versatile and efficient approach to the preparation of tailored protein constructs.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to thank Professor Donald Hilvert at ETH, Zurich, for the AaLS-13 plasmid. We would also like to thank the analytical service at the Cardiff University School of Chemistry for their support and EPSRC (EP/L027240/1) for the HR-LCMS resources. We would like to thank for the financial support provided by BBSRC (BB/T015799/1), Leverhulme Trust (RPG-2017-195), Royal Society (RG170187) and Wellcome Trust (202056/Z/16/Z) for Louis Y. P. Luk as well as the UKRI PhD studentship for T. M. Simon Tang (1928909). We dedicate this article to Professor Stephen BH Kent.Notes and references
- S. B. Gunnoo and A. Madder, Bioconjugation – using selective chemistry to enhance the properties of proteins and peptides as therapeutics and carriers, Org. Biomol. Chem., 2016, 14, 8002–8013 RSC
.
- N. Stephanopoulos and M. B. Francis, Choosing an effective protein bioconjugation strategy, Nat. Chem. Biol., 2011, 7, 876 CrossRef CAS PubMed
- J. N. deGruyter, L. R. Malins and P. S. Baran, Residue-Specific Peptide Modification: A Chemist's Guide, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed
- C. B. Rosen and M. B. Francis, Targeting the N terminus for site-selective protein modification, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS PubMed
- M. Schmidt, A. Toplak, P. J. L. M. Quaedflieg and T. Nuijens, Enzyme-mediated ligation technologies for peptides and proteins, Curr. Opin. Chem. Biol., 2017, 38, 1–7 CrossRef CAS PubMed
- Y. Zhang, K.-Y. Park, K. F. Suazo and M. D. Distefano, Recent progress in enzymatic protein labelling techniques and their applications, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC
- K. Lang and J. W. Chin, Bioorthogonal Reactions for Labeling Proteins, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed
- L. B. Knudsen, P. F. Nielsen, P. O. Huusfeldt, N. L. Johansen, K. Madsen, F. Z. Pedersen, H. Thøgersen, M. Wilken and H. Agersø, Potent Derivatives of Glucagon-like Peptide-1 with Pharmacokinetic Properties Suitable for Once Daily Administration, J. Med. Chem., 2000, 43, 1664–1669 CrossRef CAS PubMed
- A. Orlova, A. Jonsson, D. Rosik, H. Lundqvist, M. Lindborg, L. Abrahmsen, C. Ekblad, F. Y. Frejd and V. Tolmachev, Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting Affibody molecule–albumin-binding domain fusion protein, J. Nucl. Med., 2013, 54, 961–968 CrossRef CAS PubMed
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed
- R. J. Spears, R. L. Brabham, D. Budhadev, T. Keenan, S. McKenna, J. Walton, J. A. Brannigan, A. M. Brzozowski, A. J. Wilkinson, M. Plevin and M. A. Fascione, Site-selective C–C modification of proteins at neutral pH using organocatalyst-mediated cross aldol ligations, Chem. Sci., 2018, 9, 5585–5593 RSC
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials, Nat. Chem., 2017, 10, 205–211 CrossRef PubMed
- C. Kulkarni, M. Lo, J. G. Fraseur, D. A. Tirrell and T. L. Kinzer-Ursem, Bioorthogonal Chemoenzymatic Functionalization of Calmodulin for Bioconjugation Applications, Bioconjugate Chem., 2015, 26, 2153–2160 CrossRef CAS PubMed
- J. Grünewald, H. E. Klock, S. E. Cellitti, B. Bursulaya, D. McMullan, D. H. Jones, H.-P. Chiu, X. Wang, P. Patterson, H. Zhou, J. Vance, E. Nigoghossian, H. Tong, D. Daniel, W. Mallet, W. Ou, T. Uno, A. Brock, S. A. Lesley and B. H. Geierstanger, Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Phosphopantetheinyl Transferases, Bioconjugate Chem., 2015, 26, 2554–2562 CrossRef PubMed
- R. Viswanathan, G. R. Labadie and C. D. Poulter, Regioselective covalent immobilization of catalytically active glutathione S-transferase on glass slides, Bioconjugate Chem., 2013, 24, 571–577 CrossRef CAS PubMed
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Introducing genetically encoded aldehydes into proteins, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, A Pictet-Spengler ligation for protein chemical modification, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed
- R. Kudirka, R. M. Barfield, J. McFarland, A. E. Albers, G. W. de Hart, P. M. Drake, P. G. Holder, S. Banas, L. C. Jones, A. W. Garofalo and D. Rabuka, Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation, Chem. Biol., 2015, 22, 293–298 CrossRef CAS PubMed
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed
- S. A. Slavoff, I. Chen, Y.-A. Choi and A. Y. Ting, Expanding the Substrate Tolerance of Biotin Ligase through Exploration of Enzymes from Diverse Species, J. Am. Chem. Soc., 2008, 130, 1160–1162 CrossRef CAS PubMed
- L. Chen, J. Cohen, X. Song, A. Zhao, Z. Ye, C. J. Feulner, P. Doonan, W. Somers, L. Lin and P. R. Chen, Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency, Sci. Rep., 2016, 6, 31899 CrossRef CAS PubMed
- S. Liebscher, M. Schöpfel, T. Aumüller, A. Sharkhuukhen, A. Pech, E. Höss, C. Parthier, G. Jahreis, M. T. Stubbs and F. Bordusa, N-Terminal Protein Modification by Substrate-Activated Reverse Proteolysis, Angew. Chem., Int. Ed., 2014, 53, 3024–3028 CrossRef CAS PubMed
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1, Bioconjugate Chem., 2018, 29, 3245–3249 CrossRef CAS PubMed
- F. B. H. Rehm, T. J. Harmand, K. Yap, T. Durek, D. J. Craik and H. L. Ploegh, Site-Specific Sequential Protein Labeling Catalyzed by a Single Recombinant Ligase, J. Am. Chem. Soc., 2019, 141, 17388–17393 CrossRef CAS PubMed
- C. Meyer, S. Liebscher and F. Bordusa, Selective Coupling of Click Anchors to Proteins via Trypsiligase, Bioconjugate Chem., 2016, 27, 47–53 CrossRef CAS PubMed
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed
- J. Du, K. Yap, L. Y. Chan, F. B. H. Rehm, F. Y. Looi, A. G. Poth, E. K. Gilding, Q. Kaas, T. Durek and D. J. Craik, A bifunctional asparaginyl endopeptidase efficiently catalyzes both cleavage and cyclization of cyclic trypsin inhibitors, Nat. Commun., 2020, 11, 1575 CrossRef CAS PubMed
- N. H. Shah and T. W. Muir, Inteins: nature's gift to protein chemists, Chem. Sci., 2014, 5, 446–461 RSC
- F. Pinto, E. L. Thornton and B. Wang, An expanded library of orthogonal split inteins enables modular multi-peptide assemblies, Nat. Commun., 2020, 11, 1529 CrossRef CAS PubMed
- L. Y. P. Luk, J. J. Ruiz-Pernía, A. S. Adesina, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, Chemical
Ligation and Isotope Labeling to Locate Dynamic Effects during Catalysis by Dihydrofolate Reductase, Angew. Chem., Int. Ed., 2015, 54, 9016–9020 CrossRef CAS PubMed
- G. K. T. Nguyen, Y. Qiu, Y. Cao, X. Hemu, C.-F. Liu and J. P. Tam, Butelase-mediated cyclization and ligation of peptides and proteins, Nat. Protoc., 2016, 11, 1977–1988 CrossRef CAS PubMed
- G. K. T. Nguyen, Y. Cao, W. Wang, C. F. Liu and J. P. Tam, Site-Specific N-Terminal Labeling of Peptides and Proteins using Butelase 1 and Thiodepsipeptide, Angew. Chem., Int. Ed., 2015, 54, 15694–15698 CrossRef CAS PubMed
- D. J. Williamson, M. E. Webb and W. B. Turnbull, Depsipeptide substrates for sortase-mediated N-terminal protein ligation, Nat. Protoc., 2014, 9, 253–262 CrossRef CAS PubMed
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Efficient N-Terminal Labeling of Proteins by Use of Sortase, Angew. Chem., Int. Ed., 2012, 51, 9377–9380 CrossRef CAS PubMed
- C. D. Spicer and B. G. Davis, Selective chemical protein modification, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed
- J. Haywood, J. W. Schmidberger, A. M. James, S. G. Nonis, K. V. Sukhoverkov, M. Elias, C. S. Bond and J. S. Mylne, Structural basis of ribosomal peptide macrocyclization in plants, Elife, 2018, 7, e32955 CrossRef PubMed
- K. S. Harris, R. F. Guarino, R. S. Dissanayake, P. Quimbar, O. C. McCorkelle, S. Poon, Q. Kaas, T. Durek, E. K. Gilding, M. A. Jackson, D. J. Craik, N. L. van der Weerden, R. F. Anders and M. A. Anderson, A suite of kinetically superior AEP ligases can cyclise an intrinsically disordered protein, Sci. Rep., 2019, 9, 10820 CrossRef PubMed
- M. A. Jackson, E. K. Gilding, T. Shafee, K. S. Harris, Q. Kaas, S. Poon, K. Yap, H. Jia, R. Guarino, L. Y. Chan, T. Durek, M. A. Anderson and D. J. Craik, Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases, Nat. Commun., 2018, 9, 2411 CrossRef CAS PubMed
- G. K. T. Nguyen, S. J. Wang, Y. B. Qiu, X. Hemu, Y. L. Lian and J. P. Tam, Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed
- F. B. Zauner, E. Dall, C. Regl, L. Grassi, C. G. Huber, C. Cabrele and H. Brandstetter, Crystal Structure of Plant Legumain Reveals a Unique Two-Chain State with pH-Dependent Activity Regulation, Plant Cell, 2018, 30, 686–699 CrossRef CAS PubMed
- A. M. James, J. Haywood, J. Leroux, K. Ignasiak, A. G. Elliott, J. W. Schmidberger, M. F. Fisher, S. G. Nonis, R. Fenske, C. S. Bond and J. S. Mylne, The macrocyclizing protease butelase 1 remains autocatalytic and reveals the structural basis for ligase activity, Plant J., 2019, 98, 988–999 CAS
- J. M. Antos, J. Ingram, T. Fang, N. Pishesha, M. C. Truttmann and H. L. Ploegh, Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation, Curr. Protoc. Protein Sci., 2017, 89, 15.3.1–15.3.19 Search PubMed
- R. L. Yang, Y. H. Wong, G. K. T. Nguyen, J. P. Tam, J. Lescar and B. Wu, Engineering a Catalytically Efficient Recombinant Protein Ligase, J. Am. Chem. Soc., 2017, 139, 5351–5358 CrossRef CAS PubMed
- Y. Cao, G. K. T. Nguyen, J. P. Tam and C.-F. Liu, Butelase-mediated synthesis of protein thioesters and its application for tandem chemoenzymatic ligation, Chem. Commun., 2015, 51, 17289–17292 RSC
- X. Bi, J. Yin, G. K. T. Nguyen, C. Rao, N. B. A. Halim, X. Hemu, J. P. Tam and C.-F. Liu, Enzymatic Engineering of Live Bacterial Cell Surfaces Using Butelase 1, Angew. Chem., Int. Ed., 2017, 56, 7822–7825 CrossRef CAS PubMed
- L. Karmani, P. Levêque, C. Bouzin, A. Bol, M. Dieu, S. Walrand, T. Vander Borght, O. Feron, V. Grégoire, D. Bonifazi, C. Michiels, S. Lucas and B. Gallez, Biodistribution of 125I-labeled anti-endoglin antibody using SPECT/CT imaging: Impact of in vivo deiodination on tumor accumulation in mice, Nucl. Med. Biol., 2016, 43, 415–423 CrossRef CAS PubMed
- R. Marega, E. A. Prasetyanto, C. Michiels, L. De Cola and D. Bonifazi, Fast Targeting and Cancer Cell Uptake of Luminescent Antibody-Nanozeolite Bioconjugates, Small, 2016, 12, 5431–5441 CrossRef CAS PubMed
- K. M. Mikula, L. Krumwiede, A. Plückthun and H. Iwaï, Segmental isotopic labeling by asparaginyl endopeptidase-mediated protein ligation, J. Biomol. NMR, 2018, 71, 225–235 CrossRef CAS PubMed
- K. M. Mikula, I. Tascón, J. J. Tommila and H. Iwaï, Segmental isotopic labeling of a single-domain globular protein without any refolding step by an asparaginyl endopeptidase, FEBS Lett., 2017, 591, 1285–1294 CrossRef CAS PubMed
- B. Dang, T. Kubota, K. Mandal, F. Bezanilla and S. B. H. Kent, Native Chemical Ligation at Asx–Cys, Glx–Cys: Chemical Synthesis and High-Resolution X-ray Structure of ShK Toxin by Racemic Protein Crystallography, J. Am. Chem. Soc., 2013, 135, 11911–11919 CrossRef CAS PubMed
- H. Bagum, B. R. Shire, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Bicyclic Lactams Derived from Serine or Cysteine and 2-Methylpropanal, Synlett, 2020, 31, 378–382 CrossRef CAS
- R. M. Jagtap, S. R. Shaikh, R. G. Gonnade, S. Raheem, M. A. Rizvi and S. K. Pardeshi, Cyanuric-Chloride-Mediated Synthesis of 2-Aryl-3-tert-butoxycarbonyl-thiazolidine-4-carboxylic Acid Anilides: Mechanistic, X-Ray Crystal Structures and Cytotoxicity Studies, ChemistrySelect, 2019, 4, 12534–12546 CrossRef CAS
- A. Bandyopadhyay, S. Cambray and J. Gao, Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation, Chem. Sci., 2016, 7, 4589–4593 RSC
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalisation, Chem. Sci., 2016, 7, 5052–5058 RSC
- I. E. Gentle, D. P. De Souza and M. Baca, Direct Production of Proteins with N-Terminal Cysteine for Site-Specific Conjugation, Bioconjugate Chem., 2004, 15, 658–663 CrossRef CAS PubMed
- K. Pane, M. Verrillo, A. Avitabile, E. Pizzo, M. Varcamonti, A. Zanfardino, A. Di Maro, C. Rega, A. Amoresano, V. Izzo, A. Di Donato, V. Cafaro and E. Notomista, Chemical Cleavage of an Asp–Cys Sequence Allows Efficient Production of Recombinant Peptides with an N-Terminal Cysteine Residue, Bioconjugate Chem., 2018, 29, 1373–1383 CrossRef CAS PubMed
- J. R. Frost, N. T. Jacob, L. J. Papa, A. E. Owens and R. Fasan, Ribosomal Synthesis of Macrocyclic Peptides in Vitro and in Vivo Mediated by Genetically Encoded Aminothiol Unnatural Amino Acids, ACS Chem. Biol., 2015, 10, 1805–1816 CrossRef CAS PubMed
- J. Lee, A. J. Boersma, M. A. Boudreau, S. Cheley, O. Daltrop, J. Li, H. Tamagaki and H. Bayley, Semisynthetic Nanoreactor for Reversible Single-Molecule Covalent Chemistry, ACS Nano, 2016, 10, 8843–8850 CrossRef CAS PubMed
- T. W. Muir, Semisynthesis of proteins by expressed protein ligation, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed
- J. M. Antos, G.-L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W.-L. Popp and H. L. Ploegh, Site-Specific N- and C-Terminal Labeling of a Single Polypeptide Using Sortases of Different Specificity, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed
- M. C. Martos-Maldonado, C. T. Hjuler, K. K. Sørensen, M. B. Thygesen, J. E. Rasmussen, K. Villadsen, S. R. Midtgaard, S. Kol, S. Schoffelen and K. J. Jensen, Selective N-terminal acylation of peptides and proteins with a Gly–His tag sequence, Nat. Commun., 2018, 9, 3307 CrossRef PubMed
- K. F. Geoghegan, H. B. F. Dixon, P. J. Rosner, L. R. Hoth, A. J. Lanzetti, K. A. Borzilleri, E. S. Marr, L. H. Pezzullo, L. B. Martin, P. K. LeMotte, A. S. McColl, A. V. Kamath and J. G. Stroh, Spontaneous α-N-6-Phosphogluconoylation of a “His Tag” in Escherichia coli: The Cause of Extra Mass of 258 or 178 Da in Fusion Proteins, Anal. Biochem., 1999, 267, 169–184 CrossRef CAS PubMed
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Directed Evolution of a Protein Container, Science, 2011, 331, 589–592 CrossRef PubMed
- R. Zschoche and D. Hilvert, Diffusion-Limited Cargo Loading of an Engineered Protein Container, J. Am. Chem. Soc., 2015, 137, 16121–16132 CrossRef CAS PubMed
- T. Beck, S. Tetter, M. Künzle and D. Hilvert, Construction of Matryoshka-Type Structures from Supercharged Protein Nanocages, Angew. Chem., Int. Ed., 2015, 54, 937–940 CrossRef CAS PubMed
- B. Wu, H. J. Wijma, L. Song, H. J. Rozeboom, C. Poloni, Y. Tian, M. I. Arif, T. Nuijens, P. J. L. M. Quaedflieg, W. Szymanski, B. L. Feringa and D. B. Janssen, Versatile Peptide C-Terminal Functionalization via a Computationally Engineered Peptide Amidase, ACS Catal., 2016, 6, 5405–5414 CrossRef CAS
- S. Baer, J. Nigro, M. P. Madej, R. M. Nisbet, R. Suryadinata, G. Coia, L. P. T. Hong, T. E. Adams, C. C. Williams and S. D. Nuttall, Comparison of alternative nucleophiles for Sortase A-mediated bioconjugation and application in neuronal cell labelling, Org. Biomol. Chem., 2014, 12, 2675–2685 RSC
- A. Royant and M. Noirclerc-Savoye, Stabilizing role of glutamic acid 222 in the structure of Enhanced Green Fluorescent Protein, J. Struct. Biol., 2011, 174, 385–390 CrossRef CAS PubMed
- E. Jacob and R. Unger, A tale of two tails: why are terminal residues of proteins exposed?, Bioinformatics, 2007, 23, e225–e230 CrossRef CAS PubMed
- A. M. Weeks and J. A. Wells, Engineering peptide ligase specificity by proteomic identification of ligation sites, Nat. Chem. Biol., 2017, 14, 50–57 CrossRef PubMed
- H. A. I. Yoshihara, S. Mahrus and J. A. Wells, Tags for labeling protein N-termini with subtiligase for proteomics, Bioorg. Med. Chem. Lett., 2008, 18, 6000–6003 CrossRef CAS PubMed
- R. C. Cumming, N. L. Andon, P. A. Haynes, M. Park, W. H. Fischer and D. Schubert, Protein Disulfide Bond Formation in the Cytoplasm during Oxidative Stress, J. Biol. Chem., 2004, 279, 21749–21758 CrossRef CAS PubMed
- J. Futami, A. Miyamoto, A. Hagimoto, S. Suzuki, M. Futami and H. Tada, Evaluation of irreversible protein thermal inactivation caused by breakage of disulphide bonds using methanethiosulfonate, Sci. Rep., 2017, 7, 12471 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02023k |
| This journal is © The Royal Society of Chemistry 2020 |