
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic nanoscale metal–organic framework harnesses hypoxia for effective cancer radiotherapy and immunotherapy†
Kaiyuan
Ni‡
a,
Guangxu
Lan‡
a,
Yang
Song
a,
Ziyang
Hao
a and
Wenbin
Lin
 *ab
*ab
aDepartment of Chemistry, The University of Chicago, Chicago, IL 60637, USA. E-mail: wenbinlin@uchicago.edu
bDepartment of Radiation and Cellular Oncology, Ludwig Center for Metastasis Research, The University of Chicago, Chicago, IL 60637, USA
First published on 20th April 2020
Abstract
Tumor hypoxia presents a major impediment to effective cancer therapy with ionizing radiation and immune checkpoint inhibitors. Here we report the design of a biomimetic nanoscale metal–organic-framework (nMOF), Hf-DBP-Fe, with catalase-like activity to decompose elevated levels of H2O2 in hypoxic tumors to generate oxygen and hydroxyl radical. The generated oxygen attenuates hypoxia to enable radiodynamic therapy upon X-ray irradiation and fixes DNA damage while hydroxyl radical inflicts direct damage to tumor cells to afford chemodynamic therapy. Hf-DBP-Fe thus mediates effective local therapy of hypoxic cancer with low-dose X-ray irradiation, leading to highly immunogenic tumor microenvironments for synergistic combination with anti-PD-L1 immune checkpoint blockade. This combination treatment not only eradicates primary tumors but also rejects distant tumors through systemic anti-tumor immunity. We have thus advanced an nMOF-based strategy to harness hypoxic tumor microenvironments for highly effective cancer therapy using a synergistic combination of low dose radiation and immune checkpoint blockade.
Introduction
Caused by rapid growth of tumor tissues and distorted blood vessels, tumor hypoxia leads to insufficient oxygen supply to solid tumors and promotes tumor progression and metastasis.1 Hypoxia also severely impairs the therapeutic efficacy of many cancer treatments, including chemotherapy,2 photodynamic therapy,3 radiation therapy (RT),4 and immunotherapy.5 In particular, it is well-established that low tumoral oxygen concentration prevents fixation of ionizing radiation-induced DNA damage and significantly compromises the therapeutic efficacy of RT.6 Tumor hypoxia is thus a promising target for improving the therapeutic efficacy of RT.7,8Over the past few decades, a number of strategies have been explored to overcome radioresistance of hypoxic tumors, including the use of nitroimidazole-based radiosensitizers,9,10 hypoxic cytotoxins such as tirapazamine,11,12 HIF-1 inhibitors,13–15 gene therapy,16–18 and innovative radiation dosing methods.19–22 Interestingly, a strategy targeting abnormal anaerobic respiration has recently been studied as an alternative to overcome hypoxia and enhance cancer radiation therapy.23,24 The anaerobic respiration in hypoxic tumors leads to higher hydrogen peroxide (H2O2) concentrations than healthy tissues. A number of catalase-mimicking nanomaterials such as MnFe2O4,25 and Pt26 have been shown to produce O2in situ by catalytically decomposing endogenous H2O2, thereby attenuating tumor hypoxia and leading to favorable cancer treatment outcomes.27 As we recently discovered the exceptional ability of nanoscale metal–organic frameworks (nMOFs) in enhancing the radiotherapeutic effects of ionizing radiations,28–30 we hypothesize that their synthetic tunability can be leveraged to design catalase-mimicking nMOFs which will decompose endogenous H2O2 to produce O2 and other species, thus enhancing the radiotherapy of hypoxic tumors.
As a class of hybrid nanomaterials, nMOFs have various advantages for biomedical applications due to their intrinsic porosity, good biocompatibility, and more importantly, the ease of imparting multifunctionality.31–35 Built from metal-based secondary building units (SBUs) and organic linkers, nMOFs can be readily functionalized by post-synthetic metalation on ligands36 and SBUs,37 ligand exchange,38 ion exchange,39 and cargo loading in the pores.40 Both Hf-based radiosensitizers41 and metalloporphyrins42 as therapeutics have been previously reported. We recently demonstrated that hafnium (Hf)-based nMOFs enhance radiosensitization via preferential X-ray absorption by electron-dense Hf-oxo SBUs over tissues28 and exert cytotoxic effects by enhancing the diffusion of reactive oxygen species (ROS) through porous channels.28,43,44 The incorporation of photosensitizing ligands into Hf-based nMOFs further enhanced the radiotherapeutic efficacy by eliciting radiotherapy-radiodynamic therapy (RT-RDT) mode of action29 rather than the established X-PDT process.45–47 Herein, we report compositional fine-tuning of nMOFs by incorporating catalase-mimicking porphyrin-Fe centers to extend the anticancer effect of RT-RDT to hypoxic tumors.48
The nMOF, Hf-DBP-Fe (DBP = 5,15-di(p-benzoato)porphyrin), was synthesized via postsynthetic metalation of the previously reported Hf-DBP nMOF to embed biomimetic porphyrin-FeIIICl centers as a functional mimic of the catalase active site. Test tube, in vitro and in vivo experiments revealed that porphyrin-FeIIICl decomposes elevated levels of H2O2 in hypoxic tumors to generate both O2 and hydroxyl radical (˙OH), which not only relieves tumor hypoxia but also causes direct damage to tumors. The generated O2 is used for RDT and fixing DNA damage caused by RT-RDT while ˙OH enables chemodynamic therapy (CDT), leading to greatly improved local tumor therapy with ionizing radiation by hijacking the abnormal metabolism of hypoxic tumors. Importantly, in conjunction with anti-PD-L1 antibody, an immune checkpoint inhibitor, Hf-DBP-Fe-mediated RT-RDT and CDT not only eradicate primary tumors but also reject distant tumors through systemic anti-tumor immunity on a hypoxic bilateral MC38 colorectal tumor model on immunocompetent C57BL/6 mice (Fig. 1). We further profiled the immune responses to investigate the fundamental mechanisms and provide insight into the exceptional levels of the abscopal effect and cure rate from a combination of Hf-DBP-Fe, low dose X-ray, and immune checkpoint inhibitor.
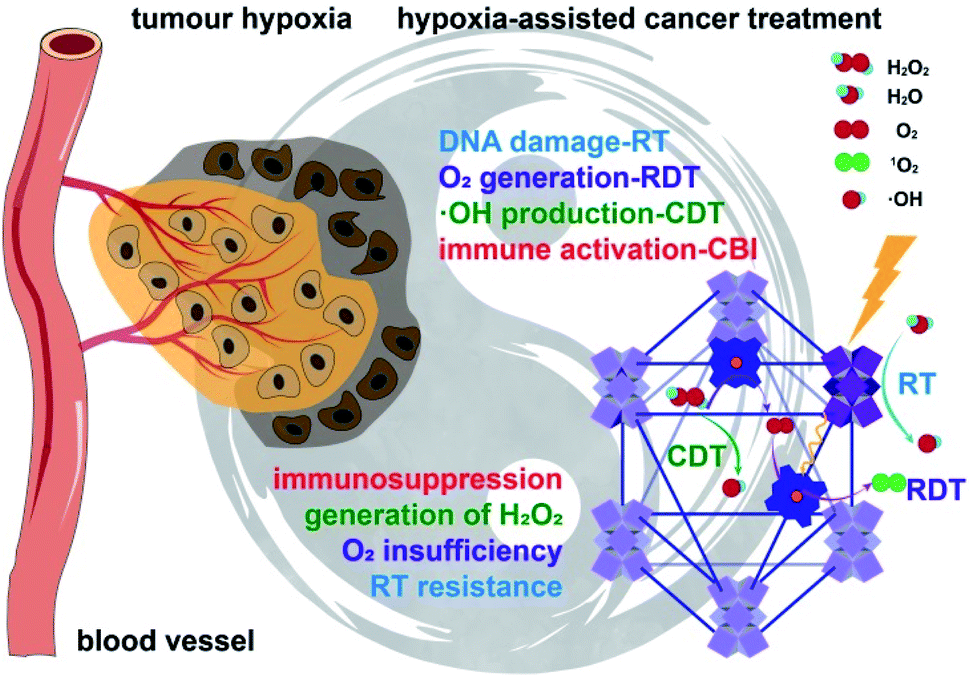 | ||
| Fig. 1 Hf-DBP-Fe harnesses tumor hypoxia for cancer treatment via RT-RDT and CDT as well as synergistic combination with CBI. Local injection of Hf-DBP-Fe in MC38 tumors followed by low-dose X-ray irradiation relieves tumor hypoxia by porphyrin-Fe(III)-mediated H2O2 decomposition to generate O2 for RDT and ˙OH for both CDT and RT. Immunogenic cell death caused by local RT-RDT and CDT synergizes with anti-PD-L1 CBI to lead to systemic antitumor immunity. | ||
Results and discussion
Synthesis and characterization of Hf-DBP-Fe
The hafnium-porphyrin nMOF, Hf-DBP, with a formula of Hf12(μ3-O)8(μ3-OH)8(μ3-H)6(DBP)6.8(AcO)3.5(OH)0.9(OH2)0.9, was synthesized through a solvothermal method by reaction between HfCl4 and 5,15-di(p-benzoato)porphyrin (H2DBP) with acetic acid and water as modulators in dimethylformamide (DMF) at 85 °C (Fig. S2†). Hf-DBP is constructed from Hf12 SBUs and DBP ligands with the hcp topological structure.44 Hf-DBP was then treated with FeCl2·4H2O in ethanol at 70 °C to afford Hf-DBP-Fe with [5,15-di(p-benzoato)porphyrin]-FeIIICl bridging ligands. Inductively coupled plasma-mass spectrometry (ICP-MS) studies gave a Hf to Fe ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7.1 for Hf-DBP-Fe, suggesting that all DBP ligands in Hf-DBP have been metalated with FeIII ions to afford Hf-DBP-Fe with a formula of Hf12(μ3-O)8(μ3-OH)8(μ3-OH)6(DBP-Fe)6.8(AcO)3.5(OH)0.9(OH2)0.9.
:7.1 for Hf-DBP-Fe, suggesting that all DBP ligands in Hf-DBP have been metalated with FeIII ions to afford Hf-DBP-Fe with a formula of Hf12(μ3-O)8(μ3-OH)8(μ3-OH)6(DBP-Fe)6.8(AcO)3.5(OH)0.9(OH2)0.9.
Hf-DBP-Fe maintained the same nanoplate shapes as Hf-DBP of ∼100 nm in dimensions as shown by transmission electron microscopy (TEM, Fig. 2a and S3†) and a thickness of ∼10 nm by atomic force microscopy (AFM, Fig. 2c). Dynamic light scattering (DLS) showed similar number-averaged sizes of 82.1 ± 2.1 for Hf-DBP-Fe and 81.6 ± 3.6 nm for Hf-DBP (Fig. 2d). Hf-DBP-Fe exhibited the same topological structure as Hf-DBP, as indicated by their similar powder X-ray diffraction (PXRD) patterns (Fig. 2e) and the same six-fold symmetry in the high resolution TEM (HRTEM) images as well as fast Fourier transformed (FFT) image (Fig. 2b).
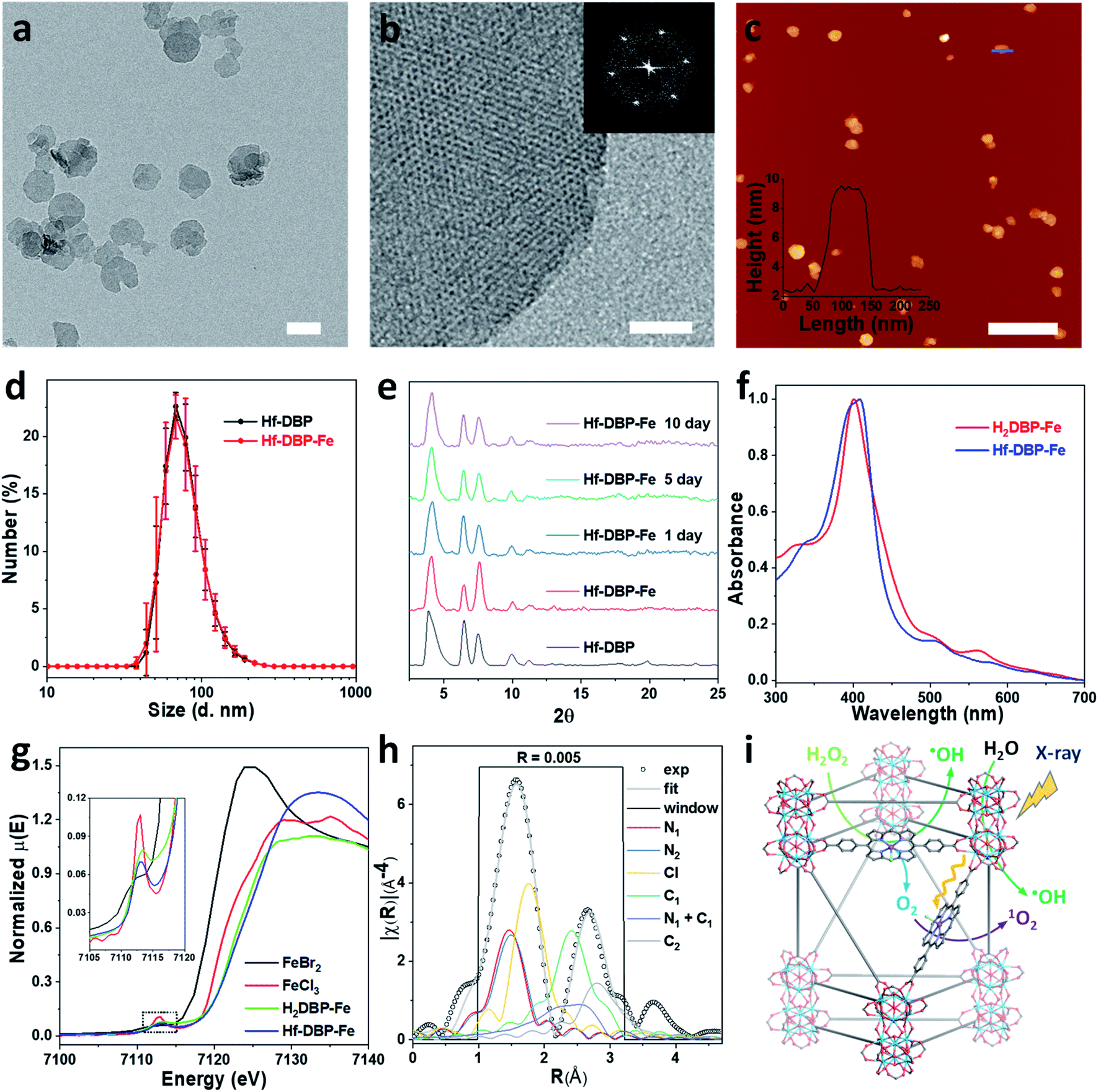 | ||
| Fig. 2 Characterization of Hf-DBP-Fe (a) TEM image and (b) HRTEM image with the FFT pattern (inset) of Hf-DBP-Fe. (c) AFM topography and its height profile (inset) along the blue line of Hf-DBP-Fe. (d) Number-averaged diameters of Hf-DBP-Fe and Hf-DBP in ethanol. (e) PXRD pattern and stability test of Hf-DBP-Fe. (f) UV-Vis spectra of Hf-DBP-Fe in comparison to H2DBP-Fe. (g) XANES analysis of Hf-DBP-Fe and H2DBP-Fe showing the pre-edge feature of FeIII in both Hf-DBP-Fe and H2DBP-Fe. (h) EXAFS fitting of Hf-DBP-Fe to five-coordinate porphyrin-FeIIICl as shown in Fig. S1.† (i) Schematic showing Hf-DBP-Fe-mediated CDT and RT-RDT processes. The DBP-Fe centers catalyze the generation of ˙OH (CDT) and O2 from H2O2. Upon X-ray irradiation, Hf-DBP-Fe enhances radiosensitization to produce additional ˙OH (RT) and convert the generated O2 into 1O2 (RDT). Scale bar = 100 nm in a, 20 nm in b, and 500 nm in c, respectively. | ||
To further investigate the chemical structure of the porphyrin-FeIIICl centers in Hf-DBP-Fe, H2DBP-FeIIICl (H2DBP-Fe) was synthesized as the molecular control. Hf-DBP-Fe displayed similar characteristic absorptions to H2DBP-Fe in the UV-vis spectra, supporting the complete FeIII metalation of all DBP ligands in Hf-DBP (Fig. 2f). X-ray absorption near edge structures (XANES) of Hf-DBP-Fe and H2DBP-Fe gave the same characteristic pre-edge features as FeIIICl3 (Fig. 2g), indicating the FeIII oxidation state in both Hf-DBP-Fe and H2DBP-Fe. Moreover, extended X-ray absorption fine structure (EXAFS) studies established the porphyrin-FeIIICl structure in Hf-DBP-Fe. Hf-DBP-Fe and H2DBP-Fe displayed similar EXAFS features, which were well fitted with the proposed five-coordinate porphyrin-FeIIICl molecular model (Fig. 2h, S1 and S4†). The stability of Hf-DBP-Fe in physiological environments was demonstrated by the identical PXRD patterns of Hf-DBP-Fe incubated in 6 mM phosphate buffered saline (PBS) solution for 10 days to the pristine sample (Fig. 2e), and was further validated by the TEM images of Hf-DBP-Fe in PBS (Fig. S3†).
Hf-DBP-Fe catalyzes H2O2 decomposition and ROS generation
As hypoxic tumors are shown to have high H2O2 concentrations, we hypothesize that with its porphyrin-FeIIICl centers as a mimic to the active site of catalase, Hf-DBP-Fe can catalyze the decomposition of H2O2 to generate O2 and ˙OH to overcome hypoxia. We first determined the ability of Hf-DBP-Fe in catalytically decomposing H2O2 using a H2O2 assay kit. 20 μM Hf-DBP-Fe or Hf-DBP was incubated in the aqueous solution of H2O2 with concentration ranging from 0 to 200 μM for 4 hours before the H2O2 assay kit was added to detect the remaining H2O2. Hf-DBP-Fe efficiently catalyzed H2O2 decomposition with no detectable H2O2 in the solution. In contrast, Hf-DBP did not decompose H2O2 in comparison to the H2O2 control solution (Fig. 3a and S6†).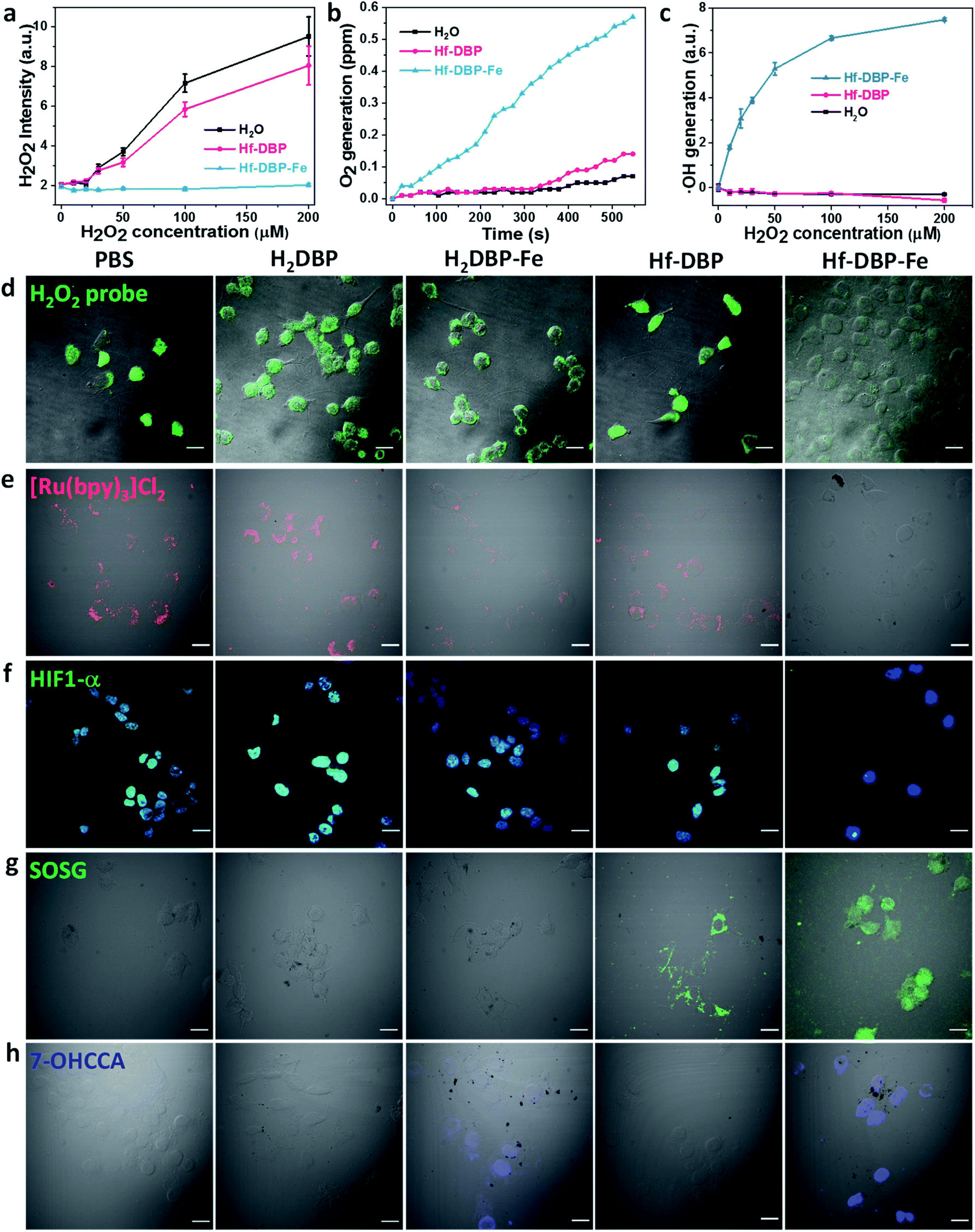 | ||
| Fig. 3 H2O2 decomposition and O2 and ˙OH generation by Hf-DBP-Fe. (a) H2O2 decomposition as probed with a H2O2 fluorescence kit, n = 6. (b) Time-dependent O2 generation as detected by an oxygen sensor, n = 6. (c) Quantification of ˙OH generation by the APF probe, n = 6. (d) Intracellular H2O2 decomposition by PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions as detected with a H2O2 kit. Green fluorescence indicates intracellular H2O2. (e) Intracellular O2 generation of cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP, or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions as detected with [Ru(bpy)3]Cl2. Intracellular red phosphorescence indicates O2-free area. (f) HIF1-α expression of cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions and immunostained with FITC-conjugated HIF1-α antibody. Green and blue fluorescence indicate intranuclear HIF1-α and nuclei, respectively. (g) 1O2 generation in live cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions and upon X-ray irradiation as detected using SOSG. Green fluorescence indicates the generated 1O2. (h) ˙OH generation in MC38 cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions as detected using CCA. Blue fluorescence indicates the presence of 7-OHCCA from the generated ˙OH. Scale bar = 20 μm. | ||
We then quantified the generation of O2 from H2O2 by Hf-DBP-Fe using an oxygen sensor to monitor the concentration of dissolved oxygen. The addition of 20 μM Hf-DBP-Fe to an oxygen-free aqueous solution with 150 μM H2O2 steadily generated O2 with a turnover frequency of 8.7 h−1 (Fig. 3b). No O2 was observed in the aqueous solution of 150 μM H2O2 or aqueous solution with 150 μM H2O2 and 20 μM Hf-DBP.
We next determined ˙OH generation from H2O2 by the aminophenyl fluorescein (APF) assay. 20 μM Hf-DBP-Fe or Hf-DBP was incubated in aqueous H2O2 solution (0–200 μM) in the presence of 5 μM APF for 4 hours. As shown in Fig. 3c and S7 (ESI†), Hf-DBP-Fe effectively generated ˙OH from H2O2 while Hf-DBP did not produce ˙OH. These results support the hypothesis that biomimetic porphyrin-Fe centers in Hf-DBP-Fe can decompose H2O2 to generate both O2 and ˙OH to enable the CDT of hypoxic tumors.
ICP-MS studies indicated that MC38 cells exhibited similarly efficient uptake of Hf-DBP-Fe and Hf-DBP after 4 h incubation (Fig. S8†). We then investigated whether Hf-DBP-Fe could overcome hypoxia in vitro. MC38 cells were seeded and cultured under either normoxic conditions or hypoxic conditions in an anaerobic incubator. We first demonstrated the ability of Hf-DBP-Fe to decompose H2O2in vitro. As shown in Fig. S9,† confocal laser scanning microscopy (CLSM) of MC38 cells cultured under normoxic conditions with the addition of 100 μM H2O2 and stained with a green fluorescent H2O2 kit showed strong H2O2 signals. The addition of Hf-DBP-Fe drastically reduced the green fluorescence, indicating the decomposition of H2O2 by Hf-DBP-Fe. In comparison, the addition of H2DBP and Hf-DBP to the same cell culture did not affect the H2O2 signals. We further showed that MC38 cells cultured under hypoxic conditions produced H2O2 which could be decomposed by Hf-DBP-Fe in vitro (Fig. 3d).
We then probed the generation of O2 from H2O2in vitro using cell-permeable [Ru(bpy)3]Cl2 whose red phosphorescence (at 621 nm) is inversely dependent on O2 concentration.49 Under normoxic conditions, the red phosphorescence of [Ru(bpy)3]Cl2 was completely quenched by dissolved O2 due to triplet energy transfer (Fig. S10†). Under hypoxic conditions, the red emission of [Ru(bpy)3]Cl2 was recovered in PBS, H2DBP, and Hf-DBP treated MC38 cells but remained completely quenched in Hf-DBP-Fe treated MC38 cells (Fig. 3e). These results indicate that Hf-DBP-Fe treatment under hypoxic conditions decomposes H2O2 to generate O2in vitro.
The reversal of hypoxia by Hf-DBP-Fe in MC38 cells was further demonstrated by HIF1-α staining. HIF1-α is an intranuclear protein that is upregulated in cells at low O2 concentrations. As shown in Fig. 3f and S11,† no HIF1-α signal was observed in all groups of MC38 cells under normoxic conditions. Under hypoxic conditions, PBS, H2DBP, and Hf-DBP treated MC38 cells showed high HIF1-α signals, but Hf-DBP-Fe treated MC-38 cells showed negligible HIF1-α signals because of the catalytic decomposition of H2O2 to generate O2 in a biomimetic fashion.
We then detected the generation of two different ROS, 1O2 and ˙OH, under either normoxic or hypoxic conditions using singlet oxygen sensor green (SOSG) and coumarin carboxylic acid (CCA), respectively. Under normoxic conditions, Hf-DBP and Hf-DBP-Fe generated similar amounts of 1O2 upon X-ray irradiation through the RDT process (Fig. S12†). However, under hypoxic conditions, Hf-DBP produced minimal 1O2 due to the low intracellular O2 concentration which shuts down to the O2-dependent RDT process. In contrast, the Hf-DBP-Fe treated group showed strong 1O2 signals as green fluorescence due to self-sufficient O2 generation from Hf-DBP-Fe catalyzed H2O2 decomposition (Fig. 3g).
We next determined if the porphyrin-Fe centers in Hf-DBP-Fe could biomimetically generate ˙OH from H2O2 under hypoxic conditions, which can be harnessed for CDT. The intracellular ˙OH was probed via hydroxylation of CCA to afford 7-hydroxy coumarin carboxylic acid (7-OHCCA) with blue fluorescence. As shown in Fig. 3h and S13,† no ˙OH signal was detected in all groups under normoxic conditions. In contrast, Hf-DBP-Fe treated MC38 cells showed a strong blue signal under hypoxic conditions, demonstrating the generation of ˙OH through Hf-DBP-Fe-mediated H2O2 decomposition. The ligand control, H2DBP-Fe, also decomposed H2O2 to generate both O2 and ˙OH, indicating the intrinsic catalytic properties of porphyrin-Fe centers.
Anticancer efficacy of Hf-DBP-Fe
We then carried out clonogenic assays to evaluate the radiosensitization properties of Hf-DBP-Fe or Hf-DBP on MC38 cells upon orthovoltage X-ray irradiation at 0–16 Gy. Cells were cultured for 12 days post irradiation, at which point cells were counted to calculate survival fractions. For convenience, we denote a sample plus X-ray irradiation as sample(+) and a sample without X-ray irradiation as sample(−) in this paper. For example, PBS(+) shows PBS plus X-ray irradiation while PBS(−) denotes PBS without X-ray radiation.Under normoxic conditions, Hf-DBP-Fe(+) showed similar radioenhancing effect to Hf-DBP(+) with the radiation enhancement factor at 10% survival (REF10) values of 1.86 ± 0.52 and 1.93 ± 0.27 (p value = 0.59), respectively (Fig. S14†). However, with hypoxia treatment, Hf-DBP-Fe(+) gave much stronger radioenhancing effect over Hf-DBP(+) with REF10 values of 3.30 ± 0.14 and 1.39 ± 0.03 (p value = 0.000017), respectively (Fig. 4a).
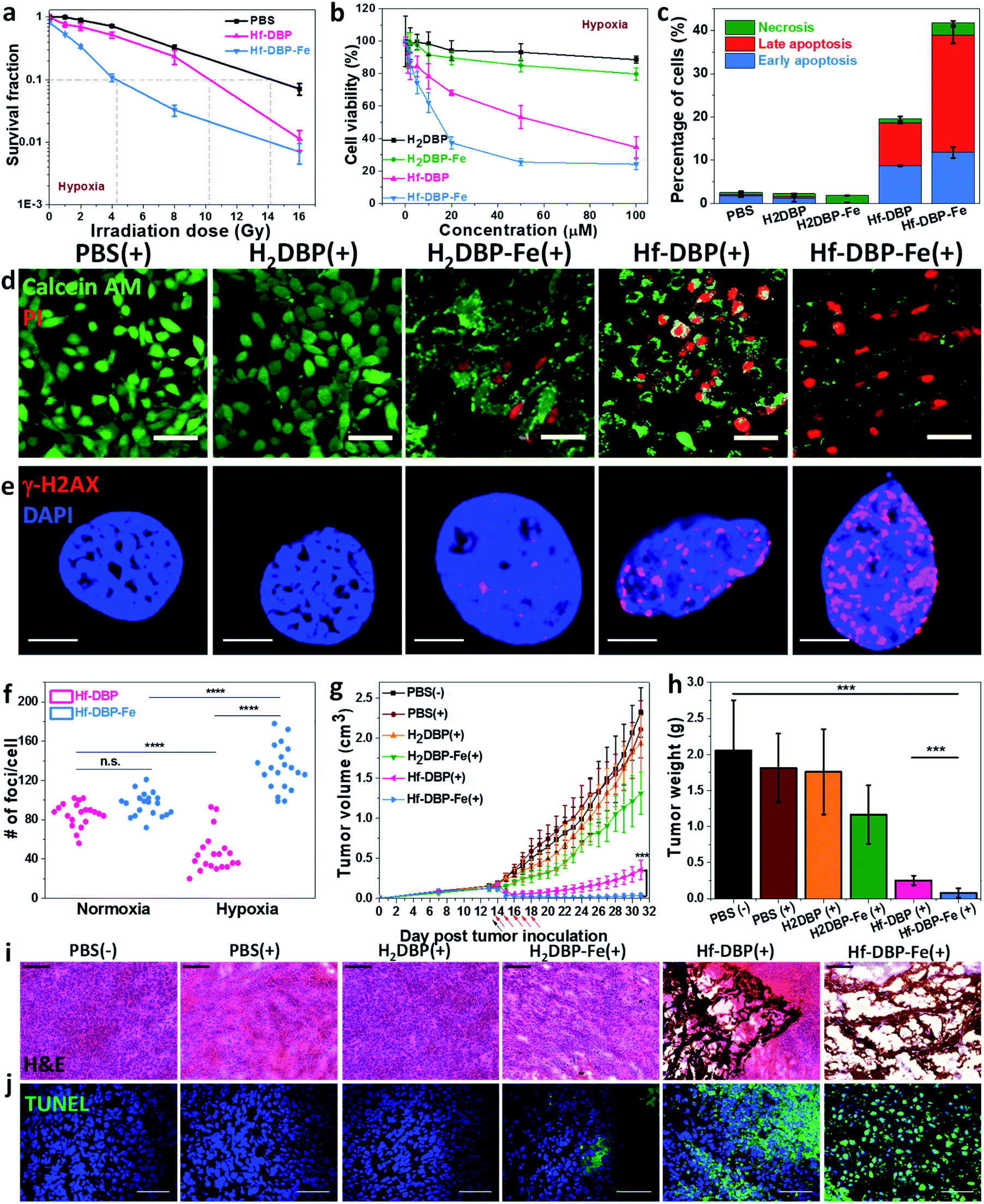 | ||
| Fig. 4 In vitro and in vivo anticancer efficacy of Hf-DBP-Fe. (a) Clonogenic assay to determine the radioenhancement of Hf-DBP-Fe, Hf-DBP or PBS upon X-ray irradiation under hypoxic conditions, n = 6. (b) Cytotoxicity of H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe upon 2 Gy X-ray irradiation on MC38 cells under hypoxic conditions, n = 6. (c) Statistical analysis of the percentages of early apoptotic cell (blue brick, annexin+PI−), late apoptotic cell (red brick, annexin+PI+), or necrotic cell (green brick, annexin−PI+) 48 hours after X-ray irradiation (2 Gy) of cells pre-treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose under hypoxic conditions, n = 3. (d) Live/dead cell assay to evaluate cell killing effect upon 2 Gy X-ray irradiation on MC38 cells under hypoxic conditions. Calcein AM (green) and PI (red) staining indicate live and dead cell, respectively. (e) Representative CLSM imaging of γ-H2AX assay to probe DNA DSBs on MC38 cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at a 20 μM equivalent dose upon X-ray irradiation at a dose of 2 Gy on MC38 cells under hypoxic conditions. (f) Statistical analysis of numbers of foci per nuclei on MC38 cells treated with Hf-DBP or Hf-DBP-Fe. 20 nuclei were randomly selected in each sample. (g) Tumor growth curves in MC38 tumor-bearing mice treated with PBS(−), PBS(+), H2DBP(+), H2DBP-Fe(+), Hf-DBP(+) or Hf-DBP-Fe(+) by intratumoral injection at an equivalent dose of 0.2 μmol per mouse, n = 6. Black arrows and red arrows refer to injection of different treatments and X-ray irradiation, respectively. (h) Excised tumor weights on day 31, n = 6. ***P < 0.001, ****P < 0.0001 from the control by the t-test. Representative (i) H&E histological staining and (j) TUNEL immunofluorescence staining of excised tumor slices for PBS(−), PBS(+), H2DBP(+), H2DBP-Fe(+), Hf-DBP(+) or Hf-DBP-Fe(+), respectively. Scale bar = 100 μm (i) or 50 μm (j). The error bars represent s.d. values. (+) and (−) refer to with and without X-ray irradiation, respectively. | ||
The much enhanced acute cytotoxicity of Hf-DBP-Fe(+) over Hf-DBP(+) under hypoxic conditions was validated by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. At 2 Gy, the IC50 values against MC38 cells under normoxic conditions were calculated to be 5.76 ± 3.54 and 4.22 ± 2.93 μM (p value = 0.067) for Hf-DBP-Fe(+) and Hf-DBP(+), respectively. Under hypoxic conditions, Hf-DBP-Fe(+) showed higher cytotoxicity than Hf-DBP(+) against MC38 cells, with IC50 values of 9.18 ± 3.72 and 42.59 ± 9.23 μM (p value = 6.5 × 10−5, Fig. 4b), respectively. No cytotoxicity was observed under normoxic conditions without irradiation, indicating the intrinsic non-toxicity of Hf-DBP-Fe and Hf-DBP (Fig. S15†). Clonogenic assays and MTS results indicate the higher acute cell killing effect and chronic cell killing effect of Hf-DBP-Fe(+) over Hf-DBP(+) under hypoxic conditions likely due to a combination of three factors: (1) sufficient O2 substrate for RDT, (2) generation of ˙OH by porphyrin-Fe centers and from radiosensitization by Hf12 SBUs, and (3) DNA damage fixation by the generated O2.
We further investigated cell death mechanism with an apoptosis kit. Under normoxic conditions, both Hf-DBP(+) and Hf-DBP-Fe(+) gave a large number of apoptotic/necrotic cells (Fig. S16†). Under hypoxic conditions, Hf-DBP-Fe(+) treatment led to cell apoptosis/necrosis with only 58.3% healthy cells, while Hf-DBP(+) and PBS(+) treatments were less effective, affording 80.4% and 97.4% healthy cells, respectively (Fig. 4c). These results were confirmed by CLSM imaging (Fig. S17†). The anticancer efficacy of different treatments was also determined by a live/dead cell assay. Under normoxic conditions, Hf-DBP(+) and Hf-DBP-Fe(+) exhibited similarly strong cell killing effect to give all propidium iodide (PI)-positive dead cells (Fig. S18†). Under hypoxic conditions, Hf-DBP-Fe(+) was equally effective with significant PI-positive cells while Hf-DBP(+) treatment led to fewer PI-positive dead cells but more live cells stained with green fluorescent calcein AM (Fig. 4d).
Phosphorylated γ-H2AX was used to qualitatively investigate DNA double-strand breaks (DSBs) caused by ˙OH in cells treated with PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe at an X-ray dose of 0 or 2 Gy under either normoxic or hypoxic conditions. 2 h after irradiation, cells pre-seeded on cover slides were fixed, permeabilized, and stained with Alexa-555 conjugated γ-H2AX antibodies. Under normoxic conditions, no fluorescence was observed 2 h post treatment in all treatment groups as dark control and X-ray irradiated groups without Hf-based nMOFs (Fig. S19†). In comparison, under hypoxic conditions, the Hf-DBP-Fe(−) treated group showed strong red fluorescence. The intensity of red fluorescence decreased in the Hf-DBP(+) treated group but increased in the Hf-DBP-Fe(+) treated group when compared to their respective signals under normoxic conditions, indicating the contribution from hypoxia-assisted DNA DSBs through Hf-DBP-Fe-mediated H2O2 decomposition to generate both O2 and ˙OH (Fig. S20†). This is supported by the number of DNA DSB foci shown in the zoomed-in CLSM images of cell nuclei (Fig. 4e and S21†). To quantify DNA DSB foci, 20 cell nuclei of each treatment group were randomly chosen and the numbers of red fluorescence-labeled γ-H2AX foci per cell nucleus were then counted (Fig. S22†). For the Hf-DBP(+) group, DNA DSB foci significantly decreased from normoxic (85.5 ± 12.1 per nucleus) to hypoxic conditions (46.2 ± 20.1 per nucleus, p value = 4.4 × 10−9, Fig. 4f). In contrast, DNA DSB foci significantly increased from normoxic (95.8 ± 11.9 per nucleus) to hypoxic conditions (132.5 ± 22.9 per nucleus, p value = 3.9 × 10−6) for the Hf-DBP-Fe(+) group, indicating that Hf-DBP-Fe-mediated ˙OH generation to cause DNA damage under hypoxic conditions.
The toxicity of Hf-DBP-Fe was examined on C57BL/6 mice. Subcutaneous injection of Hf-DBP-Fe at 0.8 μmol Hf per mouse did not alter body weight growth pattern from the PBS control (Fig. S23†). We then established a hypoxic murine colorectal tumor model to evaluate the anticancer efficacy of Hf-DBP-Fe. 5 × 105 MC38 cells were subcutaneously injected into the right flanks of C57BL/6 mice. On day 14 post tumor inoculation, PBS, H2DBP, H2DBP-Fe, Hf-DBP or Hf-DBP-Fe was intratumorally injected to the tumors at an equivalent dose of 0.2 μmol per mouse. 12 h post injection, tumors were irradiated with X-rays at a dose of 1 Gy per fraction for a total of 5 fractions. Tumor growth inhibition indices (TGIs) were calculated as [1 − (mean volume of treated tumors/mean volume of control tumors)] × 100% to evaluate the efficacy of Hf-DBP-Fe mediated hypoxic RT-RDT. As shown in Fig. 4g, the H2DBP-Fe(+) treated group showed minor tumor growth inhibition with a TGI of 37.9%, which was likely due to DNA damage from ˙OH generated by H2DBP-Fe. Hf-DBP(+) showed moderate tumor inhibition with a TGI of 84.7%, indicating the radiosensitization effect of Hf12 SBUs. In contrast, Hf-DBP-Fe(+) effectively regressed tumors to afford an impressive TGI of 98.3%. On day 31 post tumor inoculation, the tumors were excised (Fig. S24†), weighted, and sectioned for histology and immunostaining. The weights of excised tumors were 0.08 ± 0.06, 0.25 ± 0.07, 1.17 ± 0.40, 1.76 ± 0.59, 1.81 ± 0.48, and 2.06 ± 0.70 g for mice treated with Hf-DBP-Fe(+), Hf-DBP(+), H2DBP-Fe(+), H2DBP (+), PBS(+), and PBS(−), respectively (Fig. 4h). H&E histological staining indicated severe necrosis of tumor slices from Hf-DBP-Fe(+) treatment (Fig. 4i). A TdT-mediated dUTP nick end labeling assay was performed to further evaluate the in vivo apoptosis. As shown in Fig. 4j, Hf-DBP-Fe(+) treated tumors presented strong green fluorescence, indicating strong apoptosis in treated tumors. In comparison, negligible green fluorescence was found in other groups. Finally, both stable body weights and normal histological images of frozen major organ slices showed that Hf-DBP-Fe-mediated, hypoxia-tolerant RT-RDT was not systemically toxic (Fig. S25 and S26†).
Hf-DBP-Fe(+) treatment overcomes hypoxia and induces immunogenicity
H2O2 levels and hypoxia biomarker HIF1-α expressions in the excised tumor sections were quantitatively determined to assess the hypoxia-overcoming effect of Hf-DBP-Fe(+) treatment. As shown in Fig. 5a, S27 and 5b, S28,† Hf-DBP-Fe(+) significantly reduced H2O2 levels and HIF1-α signals in tumor slices when compared to all other treatment groups. These qualitative immunofluorescence results indicate that MC38 tumors in this model are highly hypoxic and Hf-DBP-Fe(+) treatment decomposes H2O2 to overcome tumor hypoxia and downregulate the expression of intranuclear HIF1-α, which are confirmed by the quantitative determination of intratumoral H2O2 levels and statistical analysis of H2O2 signal levels and HIF1-α positive areas in immunofluorescence images (Fig. 5g and h). After PBS(+), Hf-DBP(+) or Hf-DBP-Fe(+) treatment, intratumoral H2O2 was extracted with acetone and then titrated with TiOSO4 to form the yellow TiIV peroxide complex which is quantified by UV-vis spectroscopy. The intratumoral H2O2 concentrations 12 h post injection were found to be 0.43 ± 0.24, 2.41 ± 0.16, and 2.61 ± 0.43 μmol g−1 for Hf-DBP-Fe(+), Hf-DBP(+), and PBS(+) treatment groups, respectively (Fig. 5f). The decrease of intratumoral H2O2 concentration and relief of hypoxia after Hf-DBP-Fe injection was further supported by quantitative determination of H2O2 and HIF1-α after immunostaining. As shown in Fig. 5g and h, both H2DBP-Fe and Hf-DBP-Fe injected groups showed significant decrease of fluorescence intensities, indicating the capability of porphyrin-Fe to decompose H2O2 and relieve hypoxia in vivo.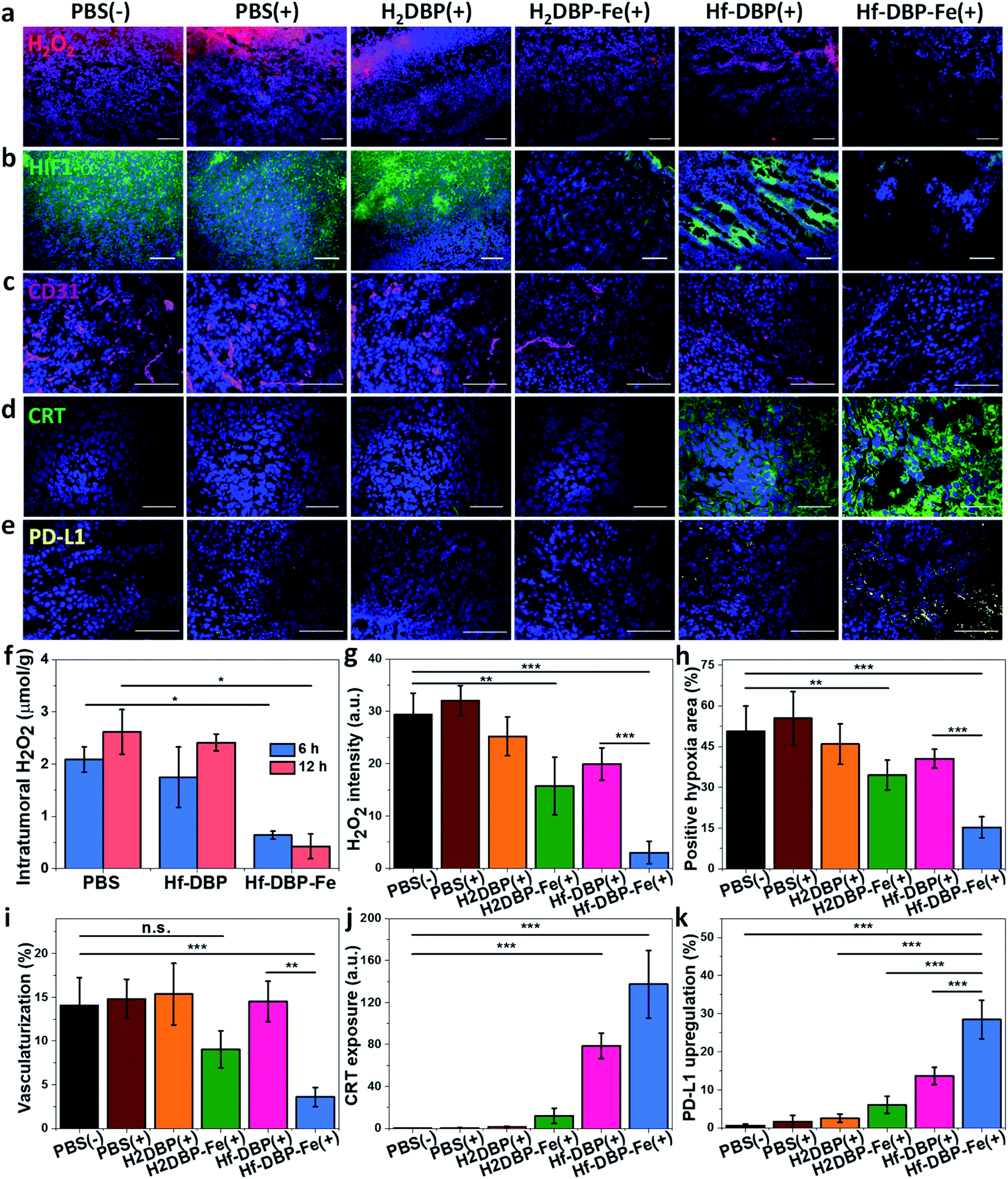 | ||
| Fig. 5 Hf-DBP-Fe overcomes hypoxia and causes immunogenicity in vivo. Representative CLSM images of (a) intratumoral H2O2 levels detected using a H2O2 fluorescence kit, (b) intratumoral HIF1-α expression levels stained with FITC-conjugated HIF1-α antibody, (c) CD31 expression for tumoral angiogenesis level probed by Cy3-conjugated CD31 antibody, (d) CRT expression probed by FITC-conjugated CRT antibody and (e) PD-L1 upregulation probed by allophycocyanin-conjugated PD-L1 antibody after treatment with PBS(−), PBS(+), H2DBP(+), H2DBP-Fe(+), Hf-DBP(+) or Hf-DBP-Fe(+) at an equivalent dose of 0.2 μmol per mouse. (f) H2O2 concentration of tumors treated with PBS, Hf-DBP or Hf-DBP-Fe at an equivalent dose of 0.2 μmol per mouse 6 or 12 hours after injection quantified with TiOSO4, n = 3. Quantitative analysis of (g) the total fluorescence intensity of the H2O2 kit, (h) the positive area of hypoxia, (i) tumoral vascularization, (j) CRT exposure, and (k) PD-L1 upregulation based on CLSM images of (a), (b), (c), (d) and (e), respectively. Scale bar = 100 μm. | ||
We further examined tumor angiogenesis caused by insufficient O2 supply from hypoxia using the immunofluorescence of CD31, a biomarker for tumor blood vessels. As shown in Fig. 5c, S29,† and 5i, the vasculaturization of Hf-DBP-Fe(+) or H2DBP-Fe(+) treated tumors significantly decreased in comparison to other treatment groups, suggesting increased O2 supply in Hf-DBP-Fe(+) or H2DBP-Fe(+) treatment groups owing to porphyrin-Fe mediated biomimetic O2 generation to reduce tumor angiogenesis. Taken together, detailed comparisons of intratumoral H2O2 levels, HIF1-α expressions, and tumor angiogenesis levels among different treatment groups indicate that Hf-DBP-Fe(+) treatment effectively overcomes tumor hypoxia via porphyrin-Fe mediated H2O2 decomposition to generate O2 and ˙OH.
We next determined the immunogenicity of different treatments by detecting in vitro calreticulin (CRT) levels. As a chaperone protein, CRT is transposed to the cell surface when encountering ER stress, acting as an immunogenic cell death (ICD) biomarker. MC38 cells were cultured under hypoxic conditions and treated with Hf-DBP-Fe, Hf-DBP, H2DBP-Fe, H2DBP, and PBS with or without irradiation. Their CRT exposure levels were determined by CLSM and flow cytometry. Without X-ray irradiation, green fluorescence was observed only in the Hf-DBP-Fe(−) treated group, suggesting the immunogenicity of porphyrin-Fe mediated CDT under hypoxic conditions. Upon X-ray irradiation, Hf-DBP-Fe(+) group possessed a much stronger CRT signal than Hf-DBP(+), indicating the higher immunogenicity induced by Hf-DBP-Fe(+) treatment under hypoxic conditions (Fig. S30†). Flow cytometry quantification studies confirmed higher CRT exposure by Hf-DBP-Fe(+) treatment (Fig. S31†).
We also performed CRT immunostaining on tumor section slides with different treatments as shown in Fig. 5d, S32,† and 5j. Quantitative green CRT fluorescence analysis showed that Hf-DBP-Fe(+) treatment induced significantly stronger ICD in hypoxic tumor tissues than all other groups. Finally, immunostaining of PD-L1 expression levels on the same tumor samples showed slightly increased PD-L1 level in the Hf-DBP(+) treatment group but significantly enhanced PD-L1 expression in the Hf-DBP-Fe(+) treatment group. Increased ICD and elevated PD-L1 level in Hf-DBP-Fe(+) treated tumors suggest that the addition of anti-PD-L1 CBI will likely enhance the anticancer efficacy of Hf-DBP-Fe mediated RT-RDT and extend this local treatment into a systemic therapy for hypoxic tumors (Fig. 5e and k).
Hf-DBP-Fe(+) plus α-PD-L1 leads to strong abscopal effects
We combined Hf-DBP-Fe(+) local treatment and intraperitoneal (i.p.) administration of an anti-PD-L1 antibody (α-PD-L1) for systemic cancer rejection. A bilateral model of hypoxic MC38 tumor was established by subcutaneously inoculating 5 × 105 and 2.5 × 105 MC38 cells onto two flanks of C57BL/6 mice as primary and secondary tumors. 14 days post inoculation, Hf-DBP-Fe was injected to the primary tumors at a dose of 0.2 μmol per mouse. 12 h later, the primary tumor was irradiated at 1 Gy X-ray (225 kVp, 13 mA, 0.3 mm Cu filter) and followed by four more daily fractions of X-ray at a dose of 1 Gy/fraction. 75 μg of anti-PD-L1 antibody (α-PD-L1) was administered by intraperitoneal injection every three days for a total of 3 doses. Fig. 6a and b show that PBS(+) and PBS(−) groups showed minimal difference in tumor growth, suggesting that there was no radiotherapeutic effect from low dose X-rays alone on such a hypoxic tumor model. Anti-PD-L1 plus X-ray radiation [α-PD-L1(+)] slightly delayed MC38 tumor progression on primary tumors but showed a weak effect on distant tumors. Hf-DBP-Fe(+) alone effectively eliminated local tumors, but had only a modest effect in delaying distant tumor growth. The Hf-DBP-Fe(−) and anti-PD-L1 combination group [Hf-DBP-Fe(−)/α-PD-L1] demonstrated similar moderate inhibition of primary tumors from the local CDT effect but little effect on distant tumors. In stark contrast, the Hf-DBP-Fe(+) and anti-PD-L1 combination group [Hf-DBP-Fe(+)/α-PD-L1] significantly regressed both primary and distant MC38 tumors, supporting the synergy between Hf-DBP-Fe(+) and α-PD-L1. The Hf-DBP-Fe(+)/α-PD-L1 treatment completely eradicated both primary and distant tumors in five out of six mice, affording a cure rate of 83.33%. Kaplan–Meier survival curves showed that Hf-DBP-Fe(+)/α-PD-L1 significantly extended the survival time with five out of six mice alive and tumor free on day 150 post Hf-DBP-Fe(+)/α-PD-L1 treatment when the mice were used for a tumor re-challenge study by injecting B16F10 melanoma cells. In comparison, the mice from other groups all have to be sacrificed by day 59 post treatment with median survival time of 30.5, 29.5, 38, 51, and 42.5 days for PBS(−), PBS(+), α-PD-L1(+), Hf-DBP-Fe(+) and Hf-DBP-Fe(−)/α-PD-L1 treatment groups, respectively (Fig. 6c). In addition, the absence of weight loss and other histopathological changes in the main organs in all treated groups supports the lack of general toxicity (Fig. S33†).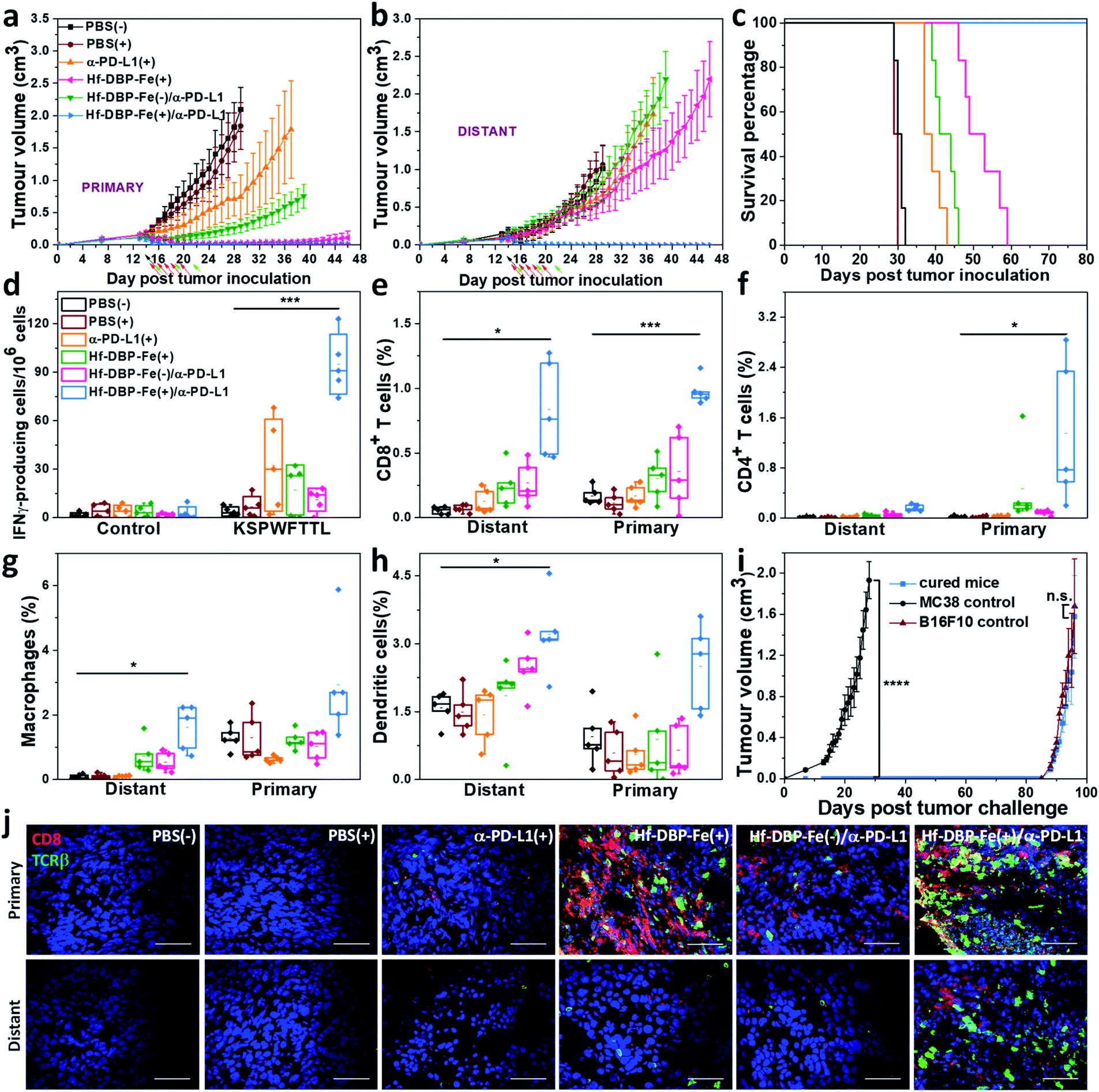 | ||
| Fig. 6 Abscopal effect of Hf-DBP-Fe(+)/α-PD-L1 on an MC38 bilateral model. Primary (a) and distant (b) tumor growth curves of MC38 bilateral tumor-bearing mice treated with PBS(−), PBS(+), α-PD-L1 (+), Hf-DBP-Fe(+), or Hf-DBP-Fe/α-PD-L1(−), and Hf-DBP-Fe/α-PD-L1(+). Black, red, and green arrows correspond to PBS or Hf-DBP-Fe intratumoral injections at equivalent doses of 0.2 μmol per mouse, X-ray irradiation, and intraperitoneal antibody administration at a dose of 75 μg per mouse, respectively. n = 6. (c) Survival curves of MC38 bilateral tumor-bearing mice with the aforementioned treatments. (d) ELISPOT assay for the detection of tumor-specific IFN-γ producing T-cells. Splenocytes were harvested 12 days after the first treatment and were co-cultured with the stimulating KSPWFTTL peptide for 42 h. Percentages of tumor-infiltrating (e) CD8+ T cells, (f) CD4+ T cells, (g) macrophages, and (h) dendritic cells with respect to the total tumor cells, calculated by flow cytometric analysis. Data are shown as means ± s.d. (n = 6). *P < 0.05, **P < 0.01 and ***P < 0.001 by the t-test. Central lines, bounds of box and whiskers correspond to mean values, 25% to 75% of the range of data and 1.5 fold of interquartile range away from outliers, respectively. (i) Tumor growth curves after the treated mice challenged with MC38 cells and re-challenged with B16F10 cells. (j) CLSM imaging of immunostained tumor section slides of mice with the aforementioned treatment to show CD8+ T cells infiltration. Red and green fluorescence indicate CD8+ cells and TCRβ+ cells, respectively. Scale bar = 50 μm. | ||
Anti-tumor immunity
The rejection of untreated distant tumors suggests that systemic anti-tumor immunity was effectively induced. Using an IFN-γ Enzyme-Linked ImmunoSpot (ELISPOT) assay, the presence of tumor-antigen specific cytotoxic T cells was determined. 12 days following the first treatment, harvested splenocytes from MC38-bearing mice were stimulated with peptide epitope KSPWFTTL for 42 hours to detect the activation of tumor-specific CD8+ T cells after treatment. The number of antigen-specific IFN-γ producing T cells per 106 splenocytes significantly increased in the Hf-DBP-Fe(+)/α-PD-L1 treatment group (94.8 ± 14.6) compared to the PBS(−) (1.6 ± 0.8) treatment group (Fig. 6d), suggesting that Hf-DBP-Fe(+)/α-PD-L1 effectively generates a tumor-specific T cell response. Next, we profiled infiltrating leukocytes in both primary and distant tumors. The Hf-DBP-Fe(+)/α-PD-L1 treatment group demonstrated a significant increase in tumor-infiltrating CD45+ T cells (Fig. S34†) and CD8+ T cells for both primary and distant tumors (Fig. S35†). Following Hf-DBP-Fe(+)/α-PD-L1 treatment, the percentages of CD8+ T cells in total primary and distant tumor cells significantly increased from 0.17 ± 0.07% and 0.06 ± 0.02% in the PBS(−) group to 0.98 ± 0.20% and 0.85 ± 0.30%, respectively (Fig. 6e). The Hf-DBP-Fe(+)/α-PD-L1 group also demonstrated a significant increase in tumor-infiltrating CD4+ T cells in the primary tumors compared to PBS(−) (0.02 ± 0.02 to 1.25 ± 0.83, Fig. 6f). The increase of CD8+ T cell infiltration in both primary and distant tumors after treatment with Hf-DBP-Fe(+)/α-PD-L1 was further confirmed with CLSM imaging (Fig. 6j). In addition, the percentages of tumor-infiltrated neutrophils, macrophages and dendritic cells in the Hf-DBP-Fe(+)/α-PD-L1 group were also enhanced compared to the PBS(−) group (Fig. 6g, h and S36–S38†). In summary, our results suggest that the Hf-DBP-Fe(+) and α-PD-L1 combination treatment bolsters both innate immune response and systemic adaptive immune responses.Tumor challenge studies
4 weeks after tumor eradication, the treated mice were injected with 5 × 105 MC38 cells subcutaneously on the contralateral flank. Naïve healthy mice inoculated with the same amount of cells served as the control. Within 27 days post MC38 challenge inoculation, no treated mice had developed tumors, while the tumors of the control mice reached 2 cm3 (Fig. 6i). On day 136 after the first treatment, the surviving mice were re-challenged by subcutaneous injection of 1 × 106 B16F10 cells into the right flanks with naive mice inoculated serving as a control. All mice developed B16F10 tumors, and no significant difference was found between the re-challenged group and the control group at the end point. The tumor challenge study suggests that tumor specific immune memory was produced in mice following tumor eradication by the Hf-DBP-Fe(+)/α-PD-L1 combination treatment.Discussion
A major challenge in RT is maximizing the anticancer effect while minimizing side effects on the adjacent normal tissues. Although many innovative irradiation techniques have been developed, significant amounts of ionizing radiations are deposited in normal tissues surrounding tumors to cause severe side effects. In the past several decades, significant efforts have been devoted to searching for radioenhancers that can increase the therapeutic index of RT. One strategy relies on increasing the difference of radiation absorption between healthy and tumor tissues. To this end, nanoparticles (NPs) of high-Z elements, such as HfO2 and Au NPs, have been extensively studied, but to date no radioenhancer has been approved by the Food and Drug Administration for clinical use. We previously showed that porous nMOFs built from Hf-oxo SBUs significantly outperform ultrasmall HfO2 nanoparticles via both enhanced radiosensitization and facile ROS diffusion through MOF channels.28 More importantly, we discovered that nMOFs based on Hf-oxo SBUs and photosensitizing ligands further enhanced radiotherapeutic effects of ionizing radiations via the RT-RDT mode of action.29,30,50 Hf-oxo SBUs absorb X-ray to produce ˙OH (RT) and transfer energy to photosensitizers for 1O2 generation (RDT).Another major obstacle for improving the anticancer efficacy of RT is the hypoxic microenvironment found in many advanced tumors.51,52 Tumor hypoxia results from insufficient and heterogeneous blood supply to deeply seated tumors, promoting local tumor progression and systemic metastasis as well as compromising the efficacy of many anticancer treatments, particularly RT.6 In the absence of oxygen, radiation-induced radicals can abstract hydrogen atom from non-protein sulfhydryls and lose the ability to cause DNA damage, leading to hypoxia-induced radiation resistance. Herein we incorporate porphyrin-Fe bridging ligands as a biomimetic oxygen generator into nMOFs to overcome hypoxia and afford highly effective RT-RDT. The porphyrin-Fe centers catalyze H2O2 decomposition to generate O2 and ˙OH while the nMOF efficiently generates 1O2 as well as sensitizing the production of additional ˙OH upon X-ray irradiation. Hf-DBP-Fe thus hijacks abnormal anaerobic metabolism of hypoxic tumors to enable CDT in the absence of X-ray irradiation. The ROSs generated from Hf-DBP-Fe mediated RT-RDT and CDT processes are also shown to be immunogenic, providing a strong rationale for combination with ICB.
Tumor hypoxia has also been shown to significantly contribute to the establishment of an immunosuppressive tumor microenvironment.53 Hypoxia renders cancer progression and drug resistance by upregulating the HIF family of transcription factors. In particular, HIF1-α has been shown to drive the expression of PD-L1, helping the tumors to resist cytotoxic T-cell therapy.51 The upregulation of HIF1-α signaling also promotes the enzymatic activity of CD73, an ectonucleotidase that converts extracellular adenosine triphosphate to adenosine.54 The accumulation of adenosine hampers the activation of antitumor immunity through many different pathways.55 Separately, cytotoxic CD4+ and CD8+ T cells were found to be more potent with the knockout of HIF1-α, providing another support for the negative impact of hypoxia on T-cell based immunotherapy. More importantly, we found that hypoxic MC38 tumors have PD-L1 expression which is further upregulated with Hf-DBP-Fe(+) treatment. We thus rationalized that Hf-DBP-Fe mediated RT-RDT and CDT can overcome hypoxia and have strong synergy with CBI to promote systemic antitumor immunity. In conjunction with α-PD-L1, Hf-DBP-Fe-mediated RT-RDT and CDT not only eradicate local tumors but also reject distant tumors on a hypoxic syngeneic bilateral MC38 colorectal tumor model.
Conclusions
We have synthesized a novel nMOF with heavy metal-based SBUs and photosensitizing linkers not only for RT-RDT but also CDT of hypoxic tumors. The porphyrin-Fe bridging ligands overcome hypoxia to enable CDT and RT-RDT via biomimetic decomposition of elevated levels of H2O2 to generate both O2 and ˙OH. The synergistic combination of Hf-DBP-Fe-mediated CDT and RT-RDT with α-PD-L1 led to superior anti-tumor outcome on bilateral syngeneic models of colon cancer, expanding the local efficacy of CDT and RT-RDT to untreated tumors via systemic anticancer immunity. We have thus developed a strategy to harness hypoxic tumor microenvironments for highly effective cancer therapy by combining an nMOF with low dose X-ray irradiation and immune checkpoint blockade.Conflicts of interest
The authors declare the following competing financial interest(s): W. L. is founder and chairman of Coordination Pharmaceuticals Inc. which licensed the RT-RDT technology from the University of Chicago.Acknowledgements
All animal experiments were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985). The Institutional Animal Care and Use Committee at the University of Chicago reviewed and approved the study protocol ACUP 72408. We thank Dr Jacqueline Ramírez of the Pharmacology Core Facility at the University of Chicago for experimental help and Mr August Culbert for editing. We acknowledge National Cancer Institute (U01-CA198989), Department of Defense (PC170934P2), the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599) and the Ludwig Institute for Metastasis Research for funding support.References
- M. Hockel and P. Vaupel, Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Targeting Hypoxia in Cancer Therapy, Nat. Rev. Cancer, 2011, 11, 393 CAS.
- T. M. Busch, S. M. Hahn, S. M. Evans and C. J. Koch, Depletion of Tumor Oxygenation During Photodynamic Therapy: Detection by the Hypoxia Marker EF3 [2-(2-Nitroimidazol-1 [H]-yl)-N-(3, 3, 3-Trifluoropropyl) Acetamide], Cancer Res., 2000, 60, 2636–2642 CAS.
- R. G. Bristow and R. P. Hill, Hypoxia and Metabolism: Hypoxia, DNA Repair and Genetic Instability, Nat. Rev. Cancer, 2008, 8, 180 CAS.
- J. Galon and D. Bruni, Approaches to Treat Immune Hot, Altered and Cold tumours with Combination Immunotherapies, Nat. Rev. Drug Discovery, 2019, 18, 197–218 CrossRef CAS PubMed.
- J. M. Brown and W. R. Wilson, Exploiting Tumour Hypoxia in Cancer Treatment, Nat. Rev. Cancer, 2004, 4, 437 CrossRef CAS PubMed.
- G. L. Semenza, Targeting HIF-1 for Cancer Therapy, Nat. Rev. Cancer, 2003, 3, 721 CrossRef CAS PubMed.
- H. Harada, How Can We Overcome Tumor Hypoxia in Radiation Therapy?, J. Radiat. Res., 2011, 52, 545–556 CAS.
- G. Adams, I. Flockhart, C. Smithen, I. Stratford, P. Wardman and M. Watts, Electron-Affinic Sensitization: VII. A Correlation between Structures, One-electron Reduction Potentials, and Efficiencies of Nitroimidazoles as Hypoxic Cell Radiosensitizers, Radiat. Res., 1976, 67, 9–20 CAS.
- C. N. Coleman, Hypoxic Cell Radiosensitizers: Expectations and Progress in Drug Development, Int. J. Radiat. Oncol., Biol., Phys., 1985, 11, 323–329 CrossRef CAS.
- J. M. Brown, SR 4233 (Tirapazamine): A New Anticancer Drug Exploiting Hypoxia in Solid Tumours, Br. J. Cancer, 1993, 67, 1163 CrossRef CAS PubMed.
- K. B. Peters and J. M. Brown, Tirapazamine: A Hypoxia-Activated Topoisomerase II Poison, Cancer Res., 2002, 62, 5248–5253 CAS.
- G. L. Semenza, Evaluation of HIF-1 Inhibitors as AntiCancer Agents, Drug Discovery Today, 2007, 12, 853–859 CAS.
- P. K. Majumder, P. G. Febbo, R. Bikoff, R. Berger, Q. Xue, L. M. McMahon, J. Manola, J. Brugarolas, T. J. McDonnell and T. R. Golub, mTOR Inhibition Reverses Akt-Dependent Prostate Intraepithelial Neoplasia through Regulation of Apoptotic and HIF-1-Dependent Pathways, Nat. Med., 2004, 10, 594 CrossRef CAS PubMed.
- E.-J. Yeo, Y.-S. Chun, Y.-S. Cho, J. Kim, J.-C. Lee, M.-S. Kim and J.-W. Park, YC-1: A Potential Anticancer Drug Targeting Hypoxia-Inducible Factor 1, J. Natl. Cancer Inst., 2003, 95, 516–525 CrossRef CAS PubMed.
- N. M. Mazure, M. C. Brahimi-Horn, M. A. Berta, E. Benizri, R. L. Bilton, F. Dayan, A. Ginouvès, E. Berra and J. Pouysségur, HIF-1: Master and Commander of the Hypoxic World: A Pharmacological Approach to Its Regulation by siRNAs, Biochem. Pharmacol., 2004, 68, 971–980 CrossRef CAS PubMed.
- R. L. Cowen, K. J. Williams, E. C. Chinje, M. Jaffar, F. C. Sheppard, B. A. Telfer, N. S. Wind and I. J. Stratford, Hypoxia Targeted Gene Therapy to Increase the Efficacy of Tirapazamine as an Adjuvant to Radiotherapy: Reversing Tumor Radioresistance and Effecting Cure, Cancer Res., 2004, 64, 1396–1402 CrossRef CAS PubMed.
- G. U. Dachs, A. V. Patterson, J. D. Firth, P. J. Ratcliffe, K. S. Townsend, I. J. Stratford and A. L. Harris, Targeting Gene Expression to Hypoxic Tumor Cells, Nat. Med., 1997, 3, 515 CrossRef CAS PubMed.
- R. Hill, Sensitizers and Radiation Dose Fractionation: Results and Interpretations, Int. J. Radiat. Oncol., Biol., Phys., 1986, 12, 1049–1054 CrossRef CAS.
- A. Prasanna, M. M. Ahmed, M. Mohiuddin and C. N. Coleman, Exploiting Sensitization Windows of Opportunity in Hyper and Hypo-Fractionated Radiation Therapy, J. Thorac. Dis., 2014, 6, 287 Search PubMed.
- R. Mohan, Q. Wu, M. Manning and R. Schmidt-Ullrich, Radiobiological Considerations in the Design of Fractionation Strategies for Intensity-Modulated Radiation Therapy of Head and Neck Cancers, Int. J. Radiat. Oncol., Biol., Phys., 2000, 46, 619–630 CrossRef CAS.
- H. Harada, S. Itasaka, Y. Zhu, L. Zeng, X. Xie, A. Morinibu, K. Shinomiya and M. Hiraoka, Treatment Regimen Determines Whether an HIF-1 Inhibitor Enhances or Inhibits the Effect of Radiation Therapy, Brit. J. Cancer, 2009, 100, 747 CrossRef CAS PubMed.
- W. Fan, W. Bu, Z. Zhang, B. Shen, H. Zhang, Q. He, D. Ni, Z. Cui, K. Zhao and J. Bu, X-ray Radiation-Controlled NO-Release for On-Demand Depth-Independent Hypoxic Radiosensitization, Angew. Chem., Int. Ed., 2015, 54, 14026–14030 CrossRef CAS PubMed.
- N. Lu, W. Fan, X. Yi, S. Wang, Z. Wang, R. Tian, O. Jacobson, Y. Liu, B. C. Yung and G. Zhang, Biodegradable Hollow Mesoporous Organosilica Nanotheranostics for Mild Hyperthermia-Induced Bubble-Enhanced Oxygen-Sensitized Radiotherapy, ACS Nano, 2018, 12, 1580–1591 CrossRef CAS PubMed.
- J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, Continuous O2-Evolving MnFe2O4 Nanoparticle-Anchored Mesoporous Silica Nanoparticles for Efficient Photodynamic Therapy in Hypoxic Cancer, J. Am. Chem. Soc., 2017, 139, 10992–10995 CrossRef CAS PubMed.
- Y. Zhang, F. Wang, C. Liu, Z. Wang, L. Kang, Y. Huang, K. Dong, J. Ren and X. Qu, Nanozyme Decorated Metal–Organic Frameworks for Enhanced Photodynamic Therapy, ACS Nano, 2018, 12, 651–661 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, H2O2-Activatable and O2-Evolving Nanoparticles for Highly Efficient and Selective Photodynamic Therapy Against Hypoxic Tumor cells, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS PubMed.
- K. Ni, G. Lan, C. Chan, B. Quigley, K. Lu, T. Aung, N. Guo, P. La Riviere, R. R. Weichselbaum and W. Lin, Nanoscale Metal-Organic Frameworks Enhance Radiotherapy to Potentiate Checkpoint Blockade Immunotherapy, Nat. Commun., 2018, 9, 2351 CrossRef PubMed.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, G. Lan, H. Tang, C. Pelizzari, Y.-X. Fu, M. T. Spiotto, R. R. Weichselbaum and W. Lin, Low-Dose X-Ray Radiotherapy-Radiodynamic Therapy via Nanoscale Metal–organic frameworks Enhances Checkpoint Blockade Immunotherapy, Nat. Biomed. Eng., 2018, 2, 600 CrossRef CAS PubMed.
- K. Ni, G. Lan, S. S. Veroneau, X. Duan, Y. Song and W. Lin, Nanoscale Metal-Organic Frameworks for Mitochondria-Targeted Radiotherapy-Radiodynamic Therapy, Nat. Commun., 2018, 9, 4321 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, Zinc-Adeninate Metal-Organic Framework for Aqueous Encapsulation and Sensitization of Near-Infrared and Visible Emitting Lanthanide Cations, J. Am. Chem. Soc., 2011, 133, 1220–1223 CrossRef CAS PubMed.
- W. Morris, W. E. Briley, E. Auyeung, M. D. Cabezas and C. A. Mirkin, Nucleic Acid-Metal Organic Framework (MOF) Nanoparticle Conjugates, J. Am. Chem. Soc., 2014, 136, 7261–7264 CrossRef CAS PubMed.
- J. Park, Q. Jiang, D. Feng, L. Mao and H. C. Zhou, Size-Controlled Synthesis of Porphyrinic Metal–Organic Framework and Functionalization for Targeted Photodynamic Therapy, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS PubMed.
- L. Wang, M. Zheng and Z. Xie, Nanoscale Metal-Organic Frameworks for Drug Delivery: A Conventional Platform With New Promise, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- J. M. Falkowski, T. Sawano, T. Zhang, G. Tsun, Y. Chen, J. V. Lockard and W. Lin, Privileged Phosphine-Based Metal-Organic Frameworks for Broad-Scope Asymmetric Catalysis, J. Am. Chem. Soc., 2014, 136, 5213–5216 CrossRef CAS PubMed.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Chemoselective Single-Site Earth-Abundant Metal Catalysts at Metal-Organic Framework Nodes, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, Postsynthetic Modification of Metal-Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- C. Brozek and M. Dincă, Cation Exchange at the Secondary Building Units of Metal-Organic Frameworks, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- O. T. Wilcox, A. Fateeva, A. P. Katsoulidis, M. W. Smith, C. A. Stone and M. J. Rosseinsky, Acid Loaded Porphyrin-Based Metal–Organic Framework for Ammonia Uptake, Chem. Commun., 2015, 51, 14989–14991 RSC.
- Y. Liu, P. Zhang, F. Li, X. Jin, J. Li, W. Chen and Q. Li, Metal-Based Nanoenhancers for Future Radiotherapy: Radiosensitizing and Synergistic Effects on Tumor Cells, Theranostics, 2018, 8, 1824 CrossRef CAS PubMed.
- S. Shao, V. Rajendiran and J. F. Lovell, Metalloporphyrin Nanoparticles: Coordinating Diverse Theranostic Functions, Coord. Chem. Rev., 2019, 379, 99–120 CrossRef CAS PubMed.
- G. Lan, K. Ni and W. Lin, Nanoscale Metal-Organic Frameworks for Phototherapy of Cancer, Coord. Chem. Rev., 2019, 379, 65–81 CrossRef CAS PubMed.
- K. Lu, C. He and W. Lin, Nanoscale Metal-Organic Framework for Highly Effective Photodynamic Therapy of Resistant Head and Neck Cancer, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- H. Chen, G. D. Wang, Y. J. Chuang, Z. Zhen, X. Chen, P. Biddinger, P. Biddinger, H. Liu, B. Shen, Z. Pan and J. Xie, Nanoscintillator-Mediated X-Ray Inducible Photodynamic Therapy for in Vivo Cancer Treatment, Nano Lett., 2015, 15, 2249–2256 CrossRef CAS PubMed.
- S. Shrestha, J. Wu, B. Sah, A. Vanasse, L. N. Cooper, L. Ma, G. Li, H. Zheng, W. Chen and M. P. Antosh, X-Ray Induced Photodynamic Therapy with Copper-Cysteamine Nanoparticles in Mice Tumors, Proc. Natl. Acad. Sci., 2019, 116, 16823–16828 CrossRef CAS PubMed.
- W. Chen and J. Zhang, Using Nanoparticles to Enable Simultaneous Radiation and Photodynamic Therapies for Cancer Treatment, J. Nanosci. Nanotechnol., 2006, 6, 1159–1166 CrossRef CAS PubMed.
- M. C. DeRosa, D. J. Hodgson, G. D. Enright, B. Dawson, C. E. Evans and R. J. Crutchley, Iridium Luminophore Complexes for Unimolecular Oxygen Sensors, J. Am. Chem. Soc., 2004, 126, 7619–7626 CrossRef CAS PubMed.
- A. C. Kent, D. Liu, A. Ito, T. Zhang, K. M. Brennaman, J. T. Meyer and W. Lin, Rapid Energy Transfer in Non-porous Metal–Organic Frameworks with Caged Ru (bpy)32+ Chromophores: Oxygen Trapping and Luminescence Quenching, J. Mater. Chem. A, 2013, 1, 14982–14989 RSC.
- K. Ni, G. Lan, C. Chan, X. Duan, N. Guo, S. S. Veroneau, R. R. Weichselbaum and W. Lin, Ultrathin Metal-Organic-Layer Mediated Radiotherapy-Radiodynamic Therapy, Matter, 2019, 1, 1331–1353 CrossRef.
- Y. Chen, H. Zhong, J. Wang, X. Wan, Y. Li, W. Pan, N. Li and B. Tang, Catalase-like Metal–Organic Framework Nanoparticles to Enhance Radiotherapy in Hypoxic Cancer and Prevent Cancer Recurrence, Chem. Sci., 2019, 10, 5773–5778 RSC.
- Z. Yu, P. Zhou, W. Pan, N. Li and B. Tang, A Biomimetic Nanoreactor for Synergistic Chemiexcited Photodynamic Therapy and Starvation Therapy Against Tumor Metastasis, Nat. Commun., 2018, 9, 1–9 Search PubMed.
- I. B. Barsoum, C. A. Smallwood, D. R. Siemens and C. H. Graham, A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells, Cancer Res., 2014, 74, 665–674 CrossRef CAS PubMed.
- C. A. Corzo, T. Condamine, L. Lu, M. J. Cotter, J.-I. Youn, P. Cheng, H.-I. Cho, E. Celis, D. G. Quiceno and T. Padhya, HIF-1α Regulates Function and Differentiation of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment, J. Exp. Med., 2010, 207, 2439–2453 CrossRef CAS PubMed.
- D. Vijayan, A. Young, M. W. Teng and M. J. Smyth, Targeting Immunosuppressive Adenosine in Cancer, Nat. Rev. Cancer, 2017, 17, 709 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis of H2DBP-Fe and Hf-DBP-Fe, X-ray absorption spectroscopy, biomimetic properties of Hf-DBP-Fe in test tubes, in vitro biomimetic properties of Hf-DBP-Fe, in vitro anti-cancer efficacy, in vivo anti-cancer efficacy, in vivo biomimetic properties of Hf-DBP-Fe, abscopal effect and anti-cancer immunity. See DOI: 10.1039/d0sc01949f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |