
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputer-generated “synthetic contingency” plans at times of logistics and supply problems: scenarios for hydroxychloroquine and remdesivir†
Sara
Szymkuć‡
a,
Ewa P.
Gajewska‡
a,
Karol
Molga‡
a,
Agnieszka
Wołos
a,
Rafał
Roszak
a,
Wiktor
Beker
a,
Martyna
Moskal
a,
Piotr
Dittwald
a and
Bartosz A.
Grzybowski
 *abc
*abc
aInstitute of Organic Chemistry, Polish Academy of Sciences, ul. Kasprzaka 44/52, Warsaw 02-224, Poland. E-mail: nanogrzybowski@gmail.com
bIBS Center for Soft and Living Matter, 50, UNIST-gil, Eonyang-eup, Ulju-gun, Ulsan, 689-798, South Korea
cDepartment of Chemistry, UNIST, 50, UNIST-gil, Eonyang-eup, Ulju-gun, Ulsan, 689-798, South Korea
First published on 10th June 2020
Abstract
A computer program for retrosynthetic planning helps develop multiple “synthetic contingency” plans for hydroxychloroquine and also routes leading to remdesivir, both promising but yet unproven medications against COVID-19. These plans are designed to navigate, as much as possible, around known and patented routes and to commence from inexpensive and diverse starting materials, so as to ensure supply in case of anticipated market shortages of commonly used substrates. Looking beyond the current COVID-19 pandemic, development of similar contingency syntheses is advocated for other already-approved medications, in case such medications become urgently needed in mass quantities to face other public-health emergencies.
Introduction
Faced with the eruption of the coronavirus pandemic, individual academic and clinical laboratories, funding agencies, and entire governments are intensifying efforts to develop and deploy safe and effective vaccines and/or antiviral medications. While vaccines may become available within a year or so, development and approval of a brand new drug will, most likely, require a significantly longer time, not relevant to the current exigency. Accordingly, much of the ongoing effort has been focused on drugs that are already approved and could be re-purposed against COVID-19. For example, reports have been emerging in the scientific literature1,2 that chloroquine (CQ) and hydroxychloroquine (HCQ) – vintage drugs to treat malaria as well as some autoimmune diseases – seem to inhibit SARS-CoV-2 infection in vitro by slowing down entry of viruses into the cell and by blocking their transport from early endosomes to endolysosomes,2,3 causing noticeable enlargement of the former and affecting the pH levels4 within the endolysosomal tract. Since HCQ is less toxic than CQ5,6 and given the current scarcity of viable alternatives, the use of this drug against the COVID-19 pandemic has been sanctioned by the FDA (but only temporarily and in clinical settings), even in the absence of comprehensive clinical data. In another example, Gilead's remdesivir – originally developed to treat hepatitis C and then tested against Ebola and Marburg disease – has shown promise7,8 and recently gained the FDA's authorization for emergency use in the U.S. Still, should either HCQ, remdesivir or any other repurposed drug prove effective – and, to reiterate, this is still uncertain – the demand might soon surpass supply. Moreover, the key synthetic methods involved in their production are very often protected by patents (including some very recent ones, even for the off-label HCQ, see Fig. 1), and we cannot exclude the possibility that monetary, corporate interests might interfere with humanitarian inspirations. In addition, the failure of the worldwide logistics and supply chains that has accompanied the COVID-19 pandemic might render some key substrates temporarily unavailable, in effect delaying the execution of the proven synthetic routes and calling for alternative synthetic solutions. Anticipating such complications, we harnessed the power of Chematica9–18 – an experimentally tested10,11 platform for computer-assisted retrosynthesis of both known and unknown target molecules – to design syntheses of HCQ and remdesivir. We were most interested in synthetic plans that would (1) commence from various inexpensive and popular starting materials (so that the syntheses minimize the abovementioned supply problems); (2) circumvent patented methodologies whenever possible;16 and (3) minimize the use of expensive methodologies and/or reagents. As described below, these analyses had different outcomes for HCQ and remdesivir. For the structurally simpler HCQ, Chematica was able to rapidly identify a large number of alternative syntheses differing in the key disconnections and meeting criteria (1)–(3). For the structurally more complex remdesivir, the program also found chemically correct routes but very similar to the current and patented approaches (with minor differences in the choices of low-cost substrates and protecting groups). These results illustrate the prowess of modern computer-assisted retrosynthesis in cases when multiple methodologies can be applied to a target of interest (here, the HCQ scenario), but also their limitations when challenged with scaffolds that can be made by only one known methodology (here, remdesivir's case). It should be noted, however, that the first of these scenarios is by far more common (see ref. 10,11 and 16 describing novel syntheses designed by Chematica and validated by experiments) and computers can be of tangible help in suggesting alternative and, as we demonstrate here, cost-effective syntheses on timescales commensurate with emergency situations such as the one presented by the COVID-19 pandemic. In this context, we remain open to performing – on a pro bono basis – similar synthetic analyses for organizations considering production and unrestricted (both geographically and economically) distribution of other potential anti-COVID-19 agents, should such agents become available in the near future.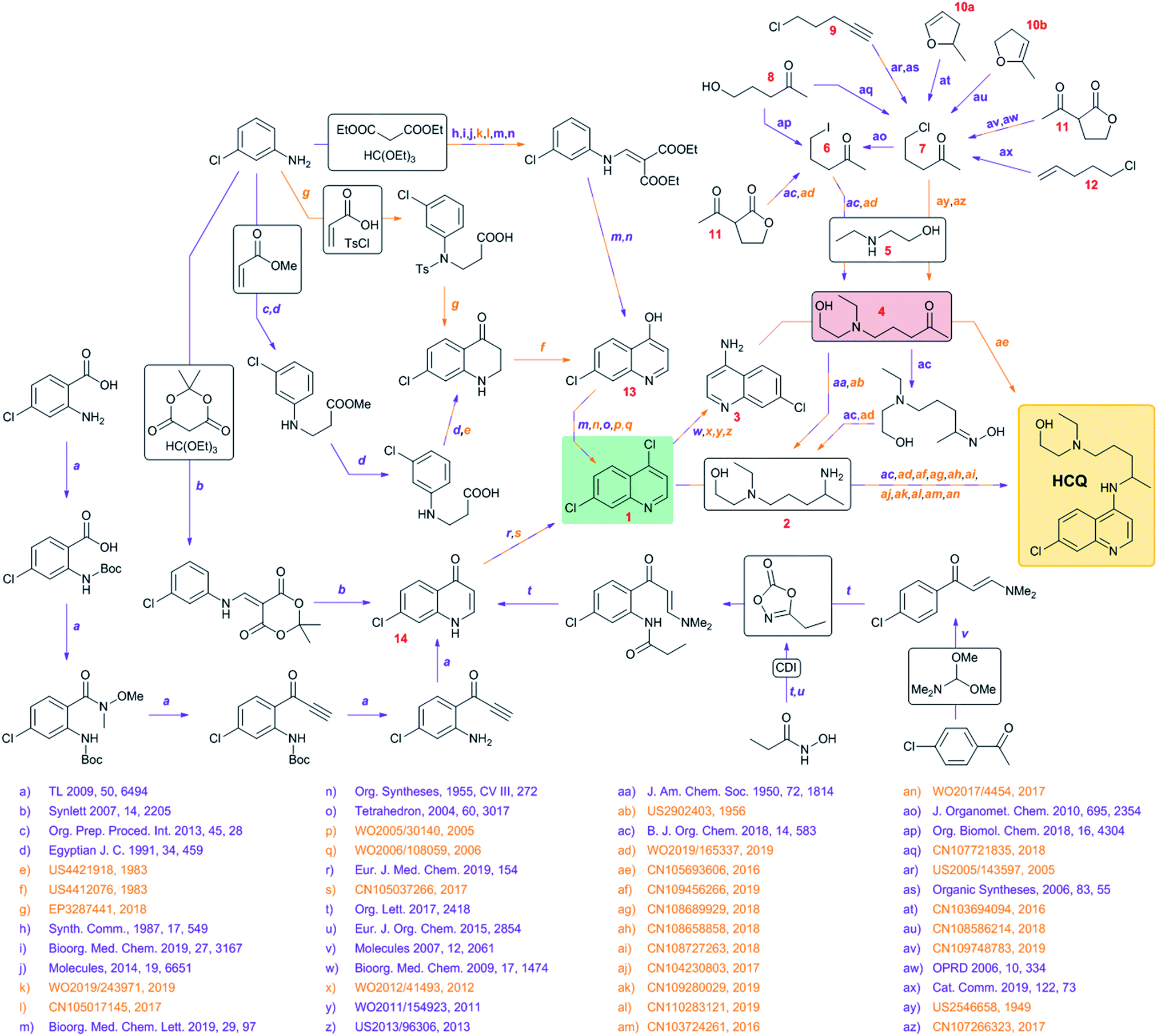 | ||
| Fig. 1 Synthetic network summarizing known syntheses of hydroxychloroquine, HCQ, along with the pertinent literature (patents in orange, scientific publications in violet). The two key intermediates are highlighted by colored boxes: in terms of availability and price, 1 (green) is significantly less problematic than 4 (red). For the full literature references, see the ESI, Section S3.† | ||
Computational methods
Chematica is a sophisticated platform for fully automated design of pathways leading to arbitrary (i.e., both known and new) targets. The software combines elements of network theory17,18 with an expert knowledge-base of synthetic transformations as well as multiple reaction-evaluation routines (based on machine learning,12,13 quantum mechanics,9,10 and molecular dynamics10,14) to search over vast trees of synthetic possibilities. The reaction transforms (currently, ∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) are expert-coded based on the underlying reaction mechanisms and are broader than any specific literature precedents (for comparison with machine extraction of rules from reaction repositories, see ref. 14). Each rule specifies the scope of admissible substituents, accounts for stereo- and regio-chemistry requirements, recognizes groups that must be protected under given reaction conditions, and identifies functionalities that are outright incompatible. The searches are guided by combinations of functions (either heuristic9,10 or best-in-class AI-based13) that score both synthetic positions as well as costs of individual reactions. The pathways identified by the program terminate in either commercially available chemicals (here, more than 200000 molecules from Sigma-Aldrich catalogs, each with the price per unit quantity; also see below for price re-scaling) or those already known in the literature (ca. 6 million substances, each accompanied by a measure of synthetic popularity,9,17i.e., how many times a given substance was used in prior syntheses). Since the program typically identifies a large number of possible routes, the network of viable syntheses already found is queried by dynamic linear programming algorithms to select pathways with the lowest cost (propagated recursively from substrates to products with the consideration of estimated yields) and highest diversity of the proposed retrosynthetic strategies.15 In setting up a particular search, the user can specify parameters influencing the economy of the solutions, notably, the upper price threshold and/or the minimal synthetic popularity of the starting materials, the relative cost of performing a reaction operation, or the desired estimated yield. The user can also eliminate certain types of transformations or unwanted reagents (e.g., expensive catalysts). He/she is also able to “lock” certain bonds or fragments in the target such that they are not disconnected along the synthetic plan – as described in detail in ref. 16; this functionality is useful in navigating around patented routes. Depending on the number of imposed constraints, a typical search for a drug-like molecule takes from few to tens of minutes and within this time inspects tens to hundreds of thousands of reaction candidates. Ultimately, a user-specified number of top-scoring pathways (typically 50–100) are returned and displayed as bipartite graphs with nodes that are expandable to display molecular structures, suggested reaction conditions typical to a given reaction class, and more.9,10
000) are expert-coded based on the underlying reaction mechanisms and are broader than any specific literature precedents (for comparison with machine extraction of rules from reaction repositories, see ref. 14). Each rule specifies the scope of admissible substituents, accounts for stereo- and regio-chemistry requirements, recognizes groups that must be protected under given reaction conditions, and identifies functionalities that are outright incompatible. The searches are guided by combinations of functions (either heuristic9,10 or best-in-class AI-based13) that score both synthetic positions as well as costs of individual reactions. The pathways identified by the program terminate in either commercially available chemicals (here, more than 200000 molecules from Sigma-Aldrich catalogs, each with the price per unit quantity; also see below for price re-scaling) or those already known in the literature (ca. 6 million substances, each accompanied by a measure of synthetic popularity,9,17i.e., how many times a given substance was used in prior syntheses). Since the program typically identifies a large number of possible routes, the network of viable syntheses already found is queried by dynamic linear programming algorithms to select pathways with the lowest cost (propagated recursively from substrates to products with the consideration of estimated yields) and highest diversity of the proposed retrosynthetic strategies.15 In setting up a particular search, the user can specify parameters influencing the economy of the solutions, notably, the upper price threshold and/or the minimal synthetic popularity of the starting materials, the relative cost of performing a reaction operation, or the desired estimated yield. The user can also eliminate certain types of transformations or unwanted reagents (e.g., expensive catalysts). He/she is also able to “lock” certain bonds or fragments in the target such that they are not disconnected along the synthetic plan – as described in detail in ref. 16; this functionality is useful in navigating around patented routes. Depending on the number of imposed constraints, a typical search for a drug-like molecule takes from few to tens of minutes and within this time inspects tens to hundreds of thousands of reaction candidates. Ultimately, a user-specified number of top-scoring pathways (typically 50–100) are returned and displayed as bipartite graphs with nodes that are expandable to display molecular structures, suggested reaction conditions typical to a given reaction class, and more.9,10
Results and discussion
The results described in the following come from various searches executed by our team over the course of two days and using three machines, each with 64 cores. Multiple searches were performed on the newest version of the program that is currently being transitioned onto the commercial Synthia™ platform owned and distributed by Sigma-Aldrich/Merck KGaA. These searches used various parameters (summarized in the ESI, Section S2†) to reflect different economic scenarios of the desired syntheses and with different types of the abovementioned constraints. In all, the searches considered on the order of millions of potential intermediates and synthetic plans. The common feature of the searches was the desire to offer alternatives to existing syntheses and to suggest multiple synthetic plans using diverse but inexpensive starting materials. In considering the prices of the starting materials, we realized that catalog prices from a specialty-chemicals retailer such as Sigma-Aldrich, S-A, are understandably higher than those from whole-sale producers. What is important for the validity of our analysis, however, is that the S-A prices correlate with the wholesale ones, as evidenced by the data plotted in Fig. 2 and spanning substrates of the new syntheses of HCQ we identified. We will discuss these issues in more detail along with specific routes.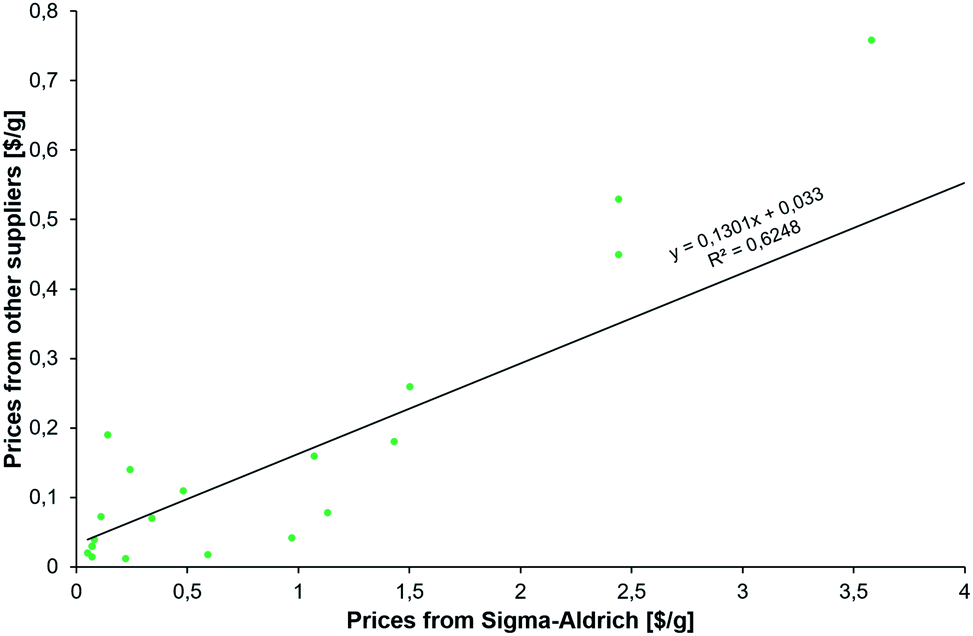 | ||
| Fig. 2 Correlation between prices (scaled per gram) of the substrates for new syntheses of HCQ (see Fig. 3) from (x-axis) Sigma-Aldrich' catalog interfaced with Chematica and (y-axis) the least inexpensive options we were able to identify from larger-scale manufacturers. Not surprisingly, the latter are, on average, twelve times less expensive (median = 4.67). | ||
Known syntheses of hydroxychloroquine (HCQ)
To begin with, we surveyed the available literature to construct a synthetic network summarizing currently known syntheses of HCQ (Fig. 1). Somewhat remarkably, although HCQ has been off-patent for decades, a large proportion of methods involved have been patented, some quite recently, substantiating our concern of potential IP complications in case of emergency production by independent agents. These solutions hinge on the late stage attachment of the side chain performed via either (i) nucleophilic aromatic substitution of dichloroquinoline 1 and amine 2 or (ii) reductive amination of aminochloroquinoline 3 (itself derived from 1) and ketone 4, the latter being the starting material for the preparation of 2. The two “hubs” of the network are, obviously, 1 or 4 though they are quite different from the economic and logistic points of view. The heterocyclic part of HCQ, 1, is rather inexpensive (1.50 $ per g from S-A, 0.26 $ per g from Biosynth Carbosynth) and in case of supply problems, can be sourced (in 94% yield, via chlorination using POCl3) from hydroxychloroquinoline 13 which, in turn, can be made in ∼40% yields in two steps either from 3-chloroaniline, diethyl malonate and ethyl orthoformate (respectively, 9.51 $ per g from S-A, 0.05 $ per g from Oakwood Chemical, OC; 0.04 $ per g from S-A, 0.015 $ per g from OC; 0.12 $ per g from S-A, 0.03 $ per g from OC) or from 3-chloroaniline, acrylic acid (or methyl acrylate) and tosyl chloride (respectively, 9.51 $ per g from S-A, 0.05 $ per g from OC; 1.48 $ per g from S-A, 0.02 $ per g from Gelest Inc.; 1.43 $ per g from S-A, 0.03 $ per g from Alfa Aesar; 0.02 $ per g from S-A, 0.04 $ per g from Alfa Aesar). Alternatively, 1 can be obtained from chloroquinolinone 14, available via a similar two-step sequence starting from 3-chloroaniline, Meldrum's acid (1.75 $ per g from S-A, 0.07 $ per g from AbaChemScene) and ethyl orthoformate. Some more recent approaches for the preparation of 14 rely on different starting materials (4-chloroacetophenone or 2-amino-4-chlorobenzoic acid) but require at least four steps. Additionally, C–H activation of enaminone derived from chloroacetophenone requires an expensive bimetallic catalyst (Cp*Co(CO)I2/AgSbF6).In contrast, ketone 4 is not easily sourced (no prices listed on eMolecules) and is likely the production bottleneck. This intermediate can be prepared via alkylation of aminoalcohol 5 (0.11 $ per g from S-A, 0.04 $ per g from Acros Organics) with haloketones 6/7, which in turn can be derived from hydroxyketone 8, chloroalkyne 9, enol ethers 10a/10b, lactone 11 or chloroalkene 12. These substrates, except for 8 and 11 (both available for less than 0.5 $ per g from suppliers like Combi-Blocks or ChemScene), are relatively expensive (from 4 $ per g to even 585 $ per g) so these methodologies are probably unsuitable for industrial up-scaling.
Syntheses of hydroxychloroquine (HCQ) designed automatically by Chematica
Without any search constraints, Chematica generally identified, among its top choices, many of these known solutions (or their very close analogs, differing in insignificant details). The program began to find substantially different pathways especially upon application of restrictive thresholds for the prices/popularities of the starting materials. Fig. 3 summarizes 17 routes we found most economically viable, concise, and diverse (see the ESI, Section 1† for enlarged views). In addition to routes relying on nucleophilic aromatic substitution of dichloroquinoline and reductive amination of aminochloroquinoline, the software was able to avoid these steps, replacing them with methodologies such as A3-coupling (path 15), Cu-cat. coupling between a heteroaryl iodide and an amine (paths 2 and 3), three-component reaction between an amine, an aldehyde and a halide under Barbier-type conditions (path 14), or alkylation of an aromatic amine with an alkyl iodide (path 11). Other innovative aspects of Chematica's plans are manifested in the routes to prepare the side-chain of HCQ which, as we saw before, is the major factor driving availability/cost of the overall synthesis. The machine's proposals include, for example, opening of a lactam with a Grignard reagent (paths 1 and 16) or alkylation of a lactone followed by ring-opening to install a primary iodide functionality – which is a very convenient group for subsequent alkylation (path 13). Other interesting approaches use the multicomponent Mannich reaction. In pathway 2, this reaction is combined with a subsequent Henry reaction, and in pathway 10 it follows a Curtius rearrangement. Both the Henry reaction and Curtius rearrangement are interesting alternatives to reductive amination or reduction of an oxime used for the introduction of the nitrogen atom. As already mentioned, all of these proposed routes avoid expensive catalysts and commence from inexpensive starting materials, readily available in large quantities (e.g., ethylamine at 0.018 $ per g, 2-bromoethyl acetate at 0.22 $ per g, 5-chloro-2-pentanone at 0.08 $ per g, or ethanolamine at 0.012 $ per g). Only a few of these substrates were used in previously published/patented syntheses. In Fig. 3, their prices are indicated in red font. We observe that several pathways start from materials available in ton quantities or from biofeedstocks, making these routes especially appealing for scale-up. For example, path 12 commences from hydroxyketone (which can be produced from pentoses via a furfural intermediate) and uses ethylene oxide to install the required hydroxyethyl side chain. Path 3 navigates the HCQ's side chain to levulinic acid, preparable on ton-scales from carbohydrates.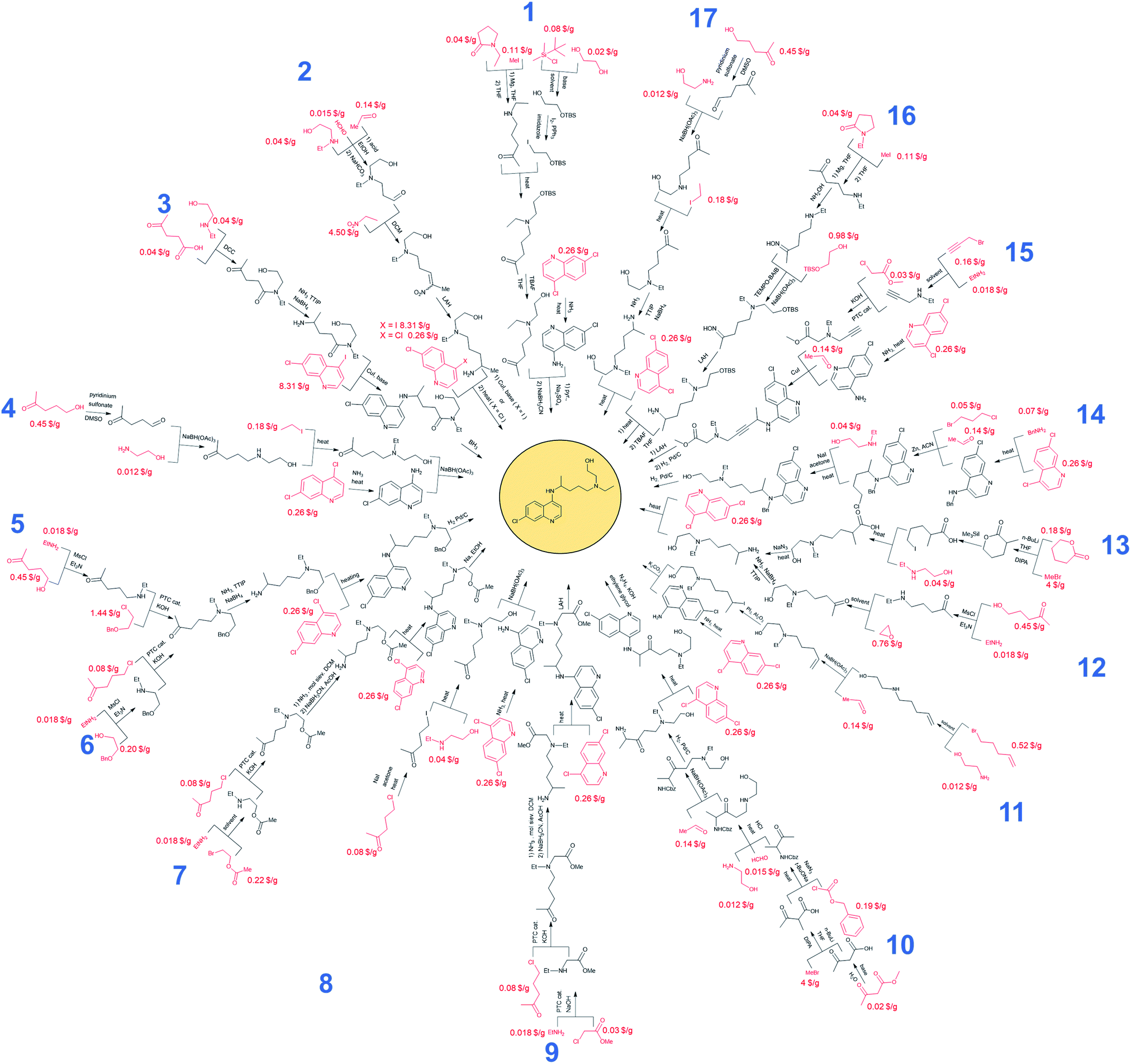 | ||
| Fig. 3 Novel and economically appealing syntheses of hydroxychloroquine (HCQ) designed automatically by Chematica. Commercially available substrates and their prices (the lowest ones we were able to identify) scaled to $ per g are colored in red. | ||
In addition to the above analyses, it is also prudent to consider syntheses of the dichloroquinoline starting material which, as already mentioned, is currently inexpensive but may become less so if demand surpasses supply. As shown in Fig. 4, the program rediscovered the patented approach (pathway marked as 18) based on Meldrum's acid and also generated a similar path, 19, starting from a commercially available dimethylamino Meldrum's acid adduct not used in the patented synthesis. Moreover, Chematica proposed three alternative routes, 20–22, which rely on copper-catalyzed cyclization forming the quinolone ring. These pathways involve different methodologies for the preparation of the acetophenone derivative, which is commercially available but expensive (29.4 $ per g from S-A, 11.07 $ per g from Chemenu). Specifically, this compound can be synthesized from a methyl benzoate derivative (6.56 $ per g from S-A, 1.16 $ per g from PharmaBlock) either via Tebbe olefination followed by ketone synthesis (pathway 20) or via the Grignard reaction (pathway 21). Chematica also proposed the synthesis of the acetophenone derivative from 2-bromo-4-chlorotoluene (0.96 $ per g from S-A, 0.35 $ per g from BLD Pharmatech) via benzylic oxidation followed by the Grignard reaction and oxidation of an alcohol (pathway 22).
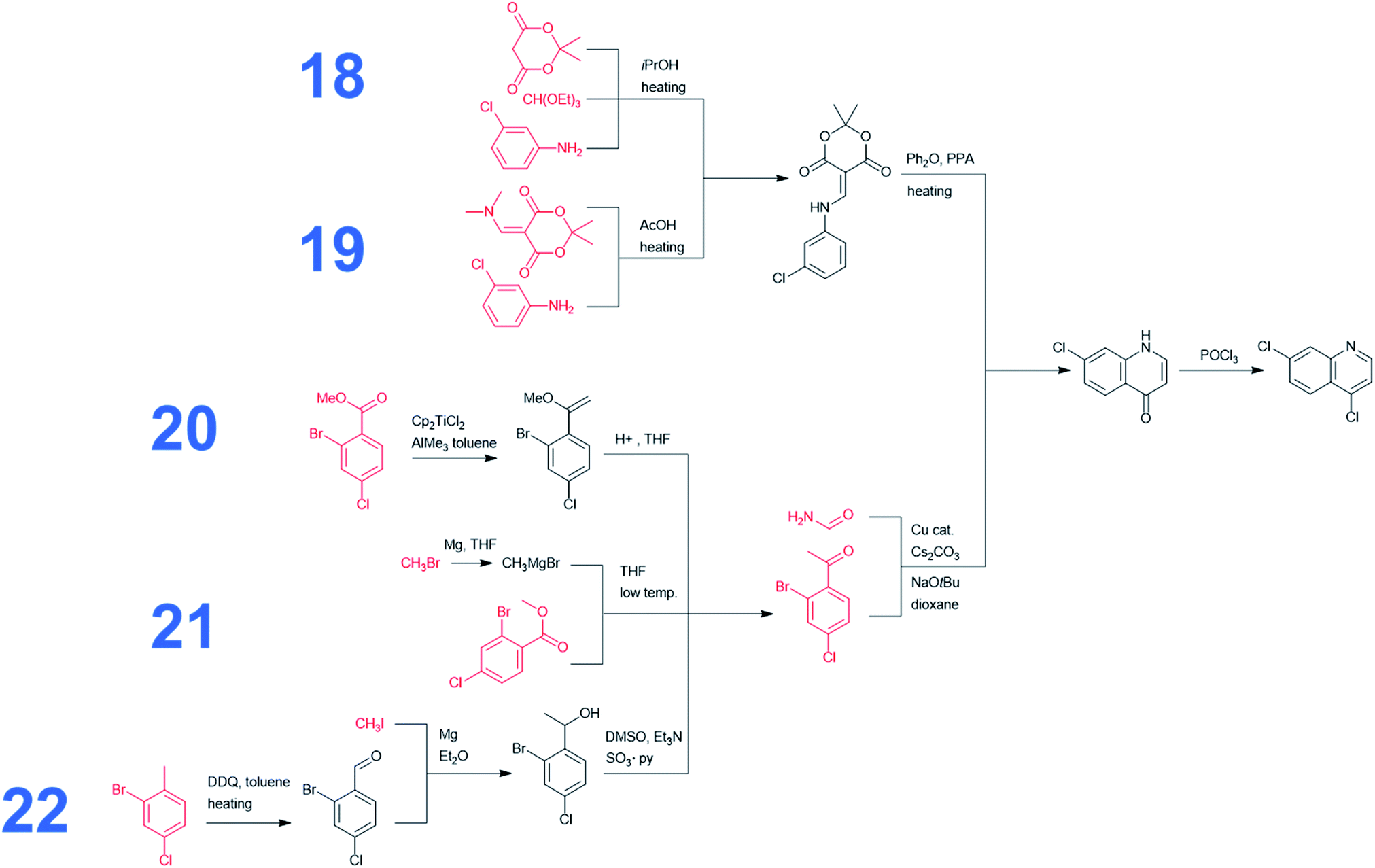 | ||
| Fig. 4 Chematica-designed syntheses of the dichloroquinoline intermediate. Commercially available substrates and their prices (the lowest ones we were able to identify) scaled to $ per g are colored in red. | ||
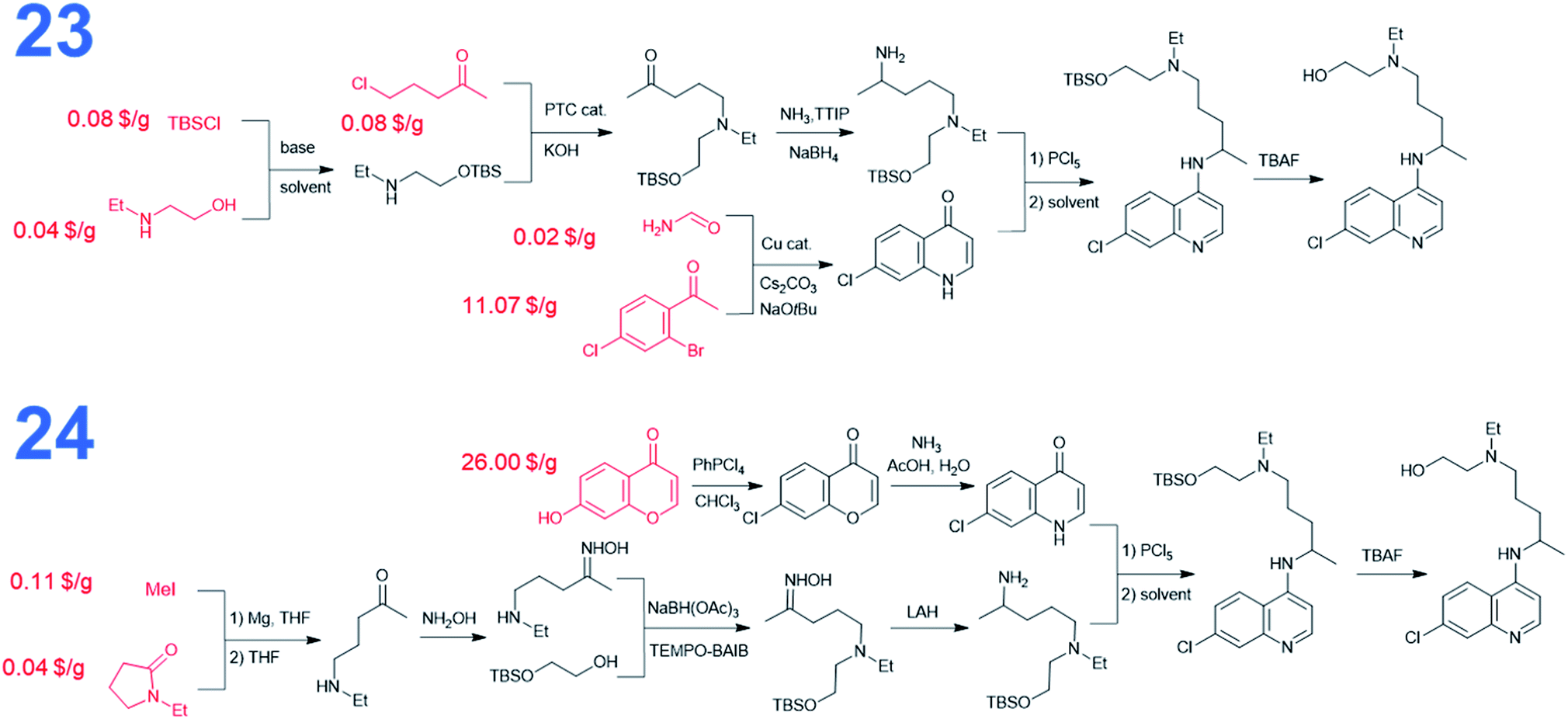
We also performed searches to synthesize HCQ taking advantage of Chematica's functionality to avoid certain user-specified substructures. Here, we excluded from the searches both 8-chloro-4-chloroquinoline and 8-chloro-4-iodoquinoline scaffolds. The top-scoring routes avoiding these motifs share a common key intermediate, 7-chloro-1H-quinolin-4-one, also identified as a precursor in some of the pathways (3–5) leading to dichloroquinoline (see Fig. 5). However, “downstream” syntheses of this heterocycle differ in both synthetic plans. In the first pathway marked as 23, the quinolone scaffold is assembled from a bromoacetophenone derivative and formamide via copper-catalyzed cyclization. These starting materials were also used to synthesize dichloroquinoline and both their pricing and accessibility from less expensive sources were described in the previous paragraph. The synthetic strategy for the alkyl part of hydroxychloroquine is the same as in path 16 from Fig. 3. The second synthetic route, numbered 24 in Fig. 5, uses a heterocyclic starting material, benzopyrone, which is further transformed into the quinolinone precursor in a two-step sequence. The cost of this substrate is 46.50 $ per g at S-A and 26 $ per g at CombiBlocks, making this solution less economical. The approach used to construct the alkyl part of the molecule resembles the strategy from path 5 in Fig. 3.
 | ||
| Fig. 5 Chematica-designed syntheses of HCQ constrained to avoid dichloro- or iodo,chloro-quinolone scaffolds. Commercially available substrates and their prices (the lowest ones we were able to identify) scaled to $ per g are colored in red. | ||
Finally, we comment on the conditions proposed by the software, especially on the usability of certain reagents at large scales. The reader will, no doubt, recognize many conditions used widely on industrial scales – for instance, the use of H2/Pd19,20 in pathways 5, 6, 10, 14, and 15 or PTC21 in pathways 5, 6, 7, 9, 15, and 23. On the other hand, the uses of reagents such as TEMPO, n-BuLi or LAH merit further discussion.
TEMPO, used in pathways 16 and 24, is relatively expensive, which might be a deterrent in using it on large scales as a stoichiometric reagent. However, it is widely used in industrial organic synthesis, although usually in small amounts (0.1–10% mol).22 In this context, we note that the N-alkylation methodology (via a one-pot oxidationreduction amination sequence)23 proposed by the software also uses reduced amounts of TEMPO (0.2 mmol of TEMPO to 1 mmol of the substrate), suggesting some potential for scale-up. Alternatively, one could consider its replacement with 4-hydroxy derivatives (e.g., 4-hydroxy-TEMPO or 4-acetamido-TEMPO) obtained from an inexpensive (3 $ per kg) triacetoneamine precursor.22 Moreover, if the cost of the one-pot methodology still proves impractical, it could readily be replaced with a “classic” two-step approach involving oxidation of a primary alcohol followed by reductive amination. Such a strategy was, in fact, used in pathway 17 for the synthesis of 5-[ethyl(2-hydroxyethyl)amino]pentan-2-one. Therein, Chematica's suggestion for the oxidation of a primary alcohol was the sulphur trioxide/pryridine complex, an oxidant used in industry, e.g., in the 190 kg scale synthesis of an HIV protease inhibitor.24 For the reductive amination step, the software proposed NaBH(OAc)3 (the same reductant was proposed in the one-pot procedure) for which multi-kilogram-scale syntheses were described in the literature.25,26 In addition, NaBH(OAc)3 is easily obtainable in situ from NaBH4 and AcOH.27
Regarding n-butyllithium – used in pathways 10 and 13 – the use of this reagent requires safety precautions, but there are examples of its usage in the pharmaceutical industry.28 For instance, n-BuLi has been used in kilogram-scale syntheses of 2,2-dimethyl-1-(4-methylthio-5-pyrimidinyl)indane29 and 1-[4-(2-chloroethoxy)phenyl]-1-(4-iodophenyl)-2-phenyl-1-butanol30 as well as in the synthesis of AZD6906 performed in a flow-reactor.31 In Chematica's synthetic plans, both reactions employing n-butyllithium are alkylations of esters and the reagent is used to generate LDA in situ; this approach is also used in industry owing to its cost efficiency.29
Regarding LAH – used in pathways 2, 9 and 16 – and according to the review by Magano and Dunetz on large-scale reductions,32 it is the most common reagent for the reduction of acyclic esters. This assertion is supported by numerous kilogram-scale syntheses using this reducing agent (e.g., Glaxo's synthesis of Sodelglitazar,33 Eli Lilly's route to DPP IV Inhibitor LY2497282,34 or Sanochemia's nine-step synthesis leading to (−)-galanthamine35).
Chematica's syntheses of remdesivir
Since HCQ is a synthetically simple target, we were also interested in Chematica's performance on a more complex target such as remdesivir. In this case, the program was able to construct chemically correct pathways though the key steps in these routes – marked as A, B and C in Fig. 6 – were identical (save minor differences in the protecting groups used) to the known approach. The choice of these key retrosynthetic disconnections is perhaps not surprising given that the presence of the C-arylated ribose, phosphorylated alanine and pyrrolotriazine fragments leaves relatively little room for other strategies. For instance, assuming that these building blocks are kept intact during synthesis, there does not seem to be a more straightforward means to construct the cyanated C-aryl nucleoside than via addition of an appropriate organometallic reagent to a protected lactone and subsequent cyanation of the hemiacetal obtained. In light of these considerations, the major virtue of computational analysis is the ability of the machine to rapidly navigate the searches to inexpensive starting materials. For instance, in Chematica's synthetic plan in Fig. 6, the heterocyclic part is navigated to commercially available aminopyrrolotriazine (5.5 $ per g from PharmaBlock), while the necessary C-aryl nucleoside is constructed from ribonolactone (5.7 $ per g from Biosynth Carbosynth). Finally, the aminoacid part of remdesivir can be sourced from three different, commercially available and inexpensive (0.25–0.31 $ per g from CoolPharm) protected (with Cbz, Boc, or Fmoc) derivatives of alanine.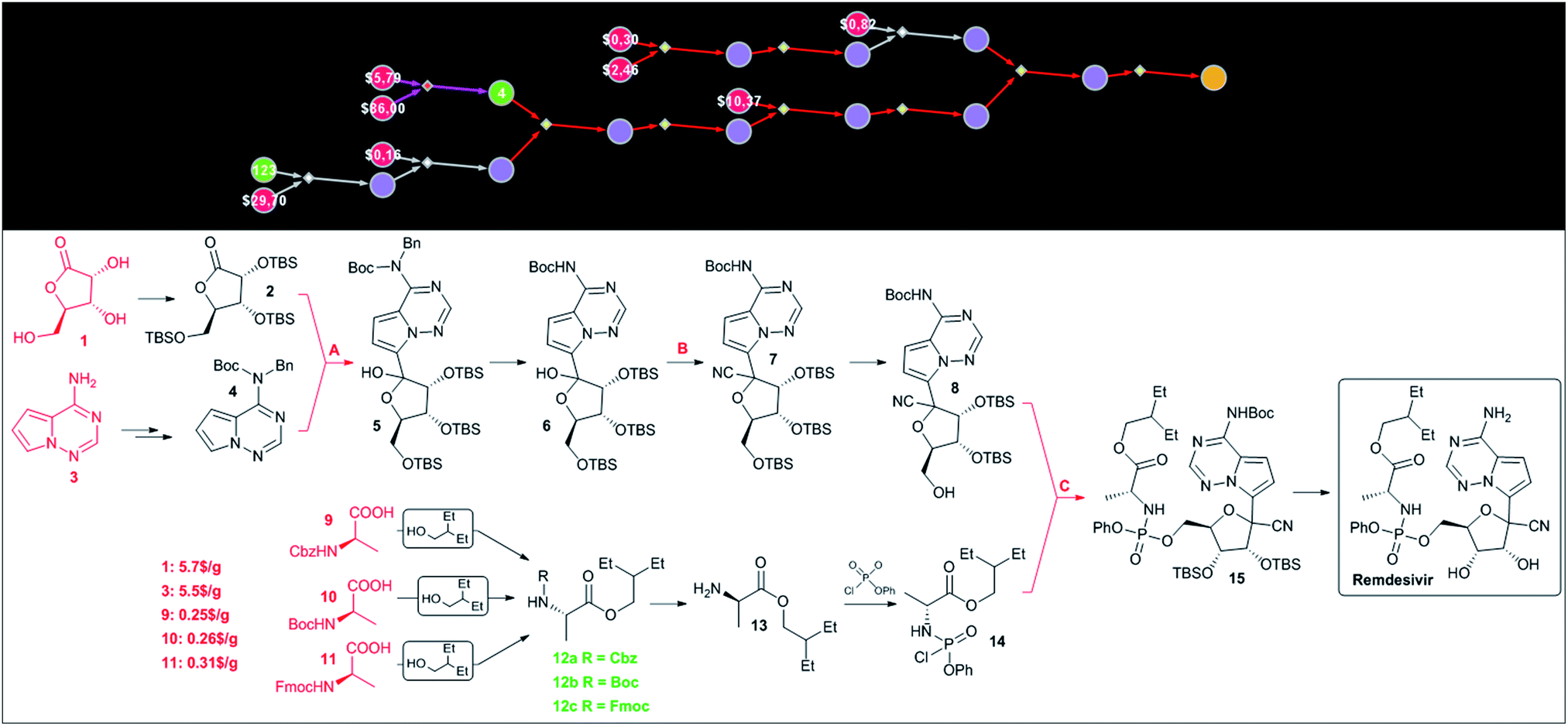 | ||
| Fig. 6 Chematica-designed syntheses of remdesivir. The top panel shows the screenshot of the pathway in a graphical representation. The colors of the nodes are: yellow = target; violet = molecules not known in Chematica's literature collection; green = known molecules with numbers indicating their synthetic popularity (i.e., how many times these molecules were used in prior syntheses); red = commercially available molecules with prices in $ per g from the S-A catalog. Red- and violet-colored reaction arrows indicate reaction sequences in which the machine was strategizing over multiple steps (using, respectively, functional group interconversion and global protection/deprotection routines). Details of the pathway are shown in the bottom panel. The key steps are marked with capital letters: A – addition of an organometallic reagent to lactone, B – cyanation, and C – phosphorylation. Commercially available substrates and their prices (the lowest ones we were able to identify) scaled to $ per g are colored in red. For Chematica's raw screenshot of these routes, see the ESI, Section S4.† | ||
Conclusions
In summary, we capitalized on the speed and chemical accuracy of modern computer-assisted synthetic planning to develop alternative and economical “contingency” plans for the synthesis of HCQ and remdesivir. Although these syntheses could, without doubt, also be identified by human experts alone, tracing them to inexpensive substrates while minimizing the use of previously described methodologies might be a rather tedious and time-consuming enterprise, incompatible with the COVID-19 emergency at hand. In a broader context, this exercise made us realize that the current system of chemical/pharmaceutical production is heavily reliant on efficient but few-and-far-between synthetic approaches – while this approach works at “peacetime,” it might be very vulnerable to the disruption of global supply chains of key starting materials, effectively leaving us without alternative means of production. Consequently, we advocate development of similar contingency plans for other approved drugs, in case they are needed in large quantities on short notice.Author contributions
K. M., E. P. G, S. S., P. D., W. B., and M. M. were the key developers of Chematica. K. M., E. P. G, and S. S. performed the synthetic analyses described in the paper. A. W. and R. R. helped with the pricing and synthetic data. B. A. G. conceived Chematica in graduate school and has directed its development ever since. All authors contributed to the writing of the manuscript.Conflicts of interest
The authors declare no financial interest in this work. While Chematica was originally developed and owned by B.A.G.‘s Grzybowski Scientific Inventions, LLC, neither he nor the co-authors currently hold any stock in this company, which is now the property of Merck KGaA, Darmstadt, Germany. Most of the authors have, until recently, collaborated with Merck KGaA, Darmstadt, on Chematica's development within the DARPA “Make-It” award. All queries about access options to Chematica (now rebranded as SynthiaTM), including academic collaborations, should be directed to Dr Sarah Trice at E-mail: sarah.trice@sial.com.Acknowledgements
This work was performed on a volunteer basis in the authors' free time (over the weekend of March 21/22, 2020). This being said, we are grateful to the U.S. DARPA who, for many years, sustained the development of Chematica under the “Make-It” award, 69461-CH-DRP #W911NF1610384. We also thank MilliporeSigma/Merck KGaA for the use of their computer resources. B. A. G. also gratefully acknowledges the personal support from the Institute for Basic Science Korea, Project Code IBS-R020-D1. A.W. gratefully acknowledges the personal support from the National Science Center, NCN, Poland under the Symfonia Award (#2014/12/W/ST5/00592).References
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res, 2020, 30, 269–271 CrossRef CAS PubMed.
- J. Liu, R. Cao, M. Xu, X. Wang, H. Zhang, H. Hu, Y. Li, Z. Hu, W. Zhong and M. Wang, Cell Discov, 2020, 6, 16 CrossRef CAS PubMed.
- M. Mauthe, I. Orhon, C. Rocchi, X. Zhou, M. Luhr, K.-J. Hijlkema, R. P. Coppes, N. Engedal, M. Mari and F. Reggiori, Autophagy, 2018, 14, 1435–1455 CrossRef CAS PubMed.
- M. Borkowska, M. Siek, D. V. Kolygina, Y. I. Sobolev, S. Lach, S. Kumar, Y.-K. Cho, K. Kandere-Grzybowska and B. A. Grzybowski, Nat. Nanotechnol., 2020, 15, 331–341 CrossRef CAS PubMed.
- H. Weniger, Review of side effects and toxicity of chloroquine, Bull. World Health, 1979, 79, 906 Search PubMed.
- E. W. McChesney, Am. J. Med., 1983, 75, 11–18 CrossRef CAS PubMed.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer, S. Kline, D. Lopez de Castilla, R. W. Finberg, K. Dierberg, V. Tapson, L. Hsieh, T. F. Patterson, R. Paredes, D. A. Sweeney, W. R. Short, G. Touloumi, D. C. Lye, N. Ohmagari, M. Oh, G. M. Ruiz-Palacios, T. Benfield, G. Fätkenheuer, M. G. Kortepeter, R. L. Atmar, C. B. Creech, J. Lundgren, A. G. Babiker, S. Pett, J. D. Neaton, T. H. Burgess, T. Bonnett, M. Green, M. Makowski, A. Osinusi, S. Nayak and H. C. Lane, N. Engl. J. Med., 2020 DOI:10.1056/NEJMoa2007764.
- W. Yin, C. Mao, X. Luan, D.-D. Shen, Q. Shen, H. Su, X. Wang, F. Zhou, W. Zhao, M. Gao, S. Chang, Y.-C. Xie, G. Tian, H.-W. Jiang, S.-C. Tao, J. Shen, Y. Jiang, H. Jiang, Y. Xu, S. Zhang, Y. Zhang and H. E. Xu, Science, 2020 DOI:10.1126/science.abc1560.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chemie Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- E. P. Gajewska, S. Szymkuć, P. Dittwald, M. Startek, O. Popik, J. Młynarski and B. A. Grzybowski, Chem, 2020, 6, 280–293 Search PubMed.
- W. Beker, E. P. Gajewska, T. Badowski and B. A. Grzybowski, Angew. Chemie Int. Ed., 2019, 58, 4515–4519 CrossRef CAS PubMed.
- T. Badowski, E. P. Gajewska, K. Molga and B. A. Grzybowski, Angew. Chemie Int. Ed., 2020, 59, 725–730 CrossRef CAS PubMed.
- K. Molga, E. P. Gajewska, S. Szymkuć and B. A. Grzybowski, React. Chem. Eng., 2019, 4, 1506–1521 RSC.
- T. Badowski, K. Molga and B. A. Grzybowski, Chem. Sci., 2019, 10, 4640–4651 RSC.
- K. Molga, P. Dittwald and B. A. Grzybowski, Chem, 2019, 5, 460–473 CAS.
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, Nat. Chem., 2009, 1, 31–36 CrossRef CAS PubMed.
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chemie Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed.
- C. Hardouin, F. Pin, J.-F. Giffard, Y. Hervouet, J. Hublet, S. Janvier, C. Penloup, J. Picard, N. Pinault, B. Schiavi, P. Zhang, W. Zhao and X. Zhu, Org. Process Res. Dev., 2019, 23, 1932–1947 CrossRef CAS.
- P. Lienard, P. Gradoz, H. Greciet, S. Jegham and D. Legroux, Org. Process Res. Dev., 2017, 21, 18–22 CrossRef CAS.
- J. Tan and N. Yasuda, Org. Process Res. Dev., 2015, 19, 1731–1746 CrossRef CAS.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS.
- C. Guérin, V. Bellosta, G. Guillamot and J. Cossy, Org. Lett., 2011, 13, 3534–3537 CrossRef PubMed.
- C. Liu, J. S. Ng, J. R. Behling, C. H. Yen, A. L. Campbell, K. S. Fuzail, E. E. Yonan and D. V. Mehrotra, Org. Process Res. Dev., 1997, 1, 45–54 CrossRef CAS.
- D. H. B. Ripin, S. Abele, W. Cai, T. Blumenkopf, J. M. Casavant, J. L. Doty, M. Flanagan, C. Koecher, K. W. Laue, K. McCarthy, C. Meltz, M. Munchhoff, K. Pouwer, B. Shah, J. Sun, J. Teixeira, T. Vries, D. A. Whipple and G. Wilcox, Org. Process Res. Dev., 2003, 7, 115–120 CrossRef CAS.
- E. J. Kiser, J. Magano, R. J. Shine and M. H. Chen, Org. Process Res. Dev., 2012, 16, 255–259 CrossRef CAS.
- A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 CrossRef CAS.
- T. L. Rathman and W. F. Bailey, Org. Process Res. Dev., 2009, 13, 144–151 CrossRef CAS.
- T. J. Dietsche, D. B. Gorman, J. A. Orvik, G. A. Roth and W. R. Shiang, Org. Process Res. Dev., 2000, 4, 275–285 CrossRef CAS.
- D. S. Ennis, D. C. Lathbury, A. Wanders and D. Watts, Org. Process Res. Dev., 1998, 2, 287–289 CrossRef CAS.
- T. Gustafsson, H. Sörensen and F. Pontén, Org. Process Res. Dev., 2012, 16, 925–929 CrossRef CAS.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- A. D. Brown, R. D. Davis, R. N. Fitzgerald, B. N. Glover, K. A. Harvey, L. A. Jones, B. Liu, D. E. Patterson and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 297–302 CrossRef CAS.
- H. Yu, R. N. Richey, J. R. Stout, M. A. LaPack, R. Gu, V. V. Khau, S. A. Frank, J. P. Ott, R. D. Miller, M. A. Carr and T. Y. Zhang, Org. Process Res. Dev., 2008, 12, 218–225 CrossRef CAS.
- B. Küenburg, L. Czollner, J. Fröhlich and U. Jordis, Org. Process Res. Dev., 1999, 3, 425–431 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01799j |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |