
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereo-controlled anti-hydromagnesiation of aryl alkynes by magnesium hydrides†
Bin
Wang
a,
Derek Yiren
Ong
a,
Yihang
Li
a,
Jia Hao
Pang
a,
Kohei
Watanabe
 b,
Ryo
Takita
*b and
Shunsuke
Chiba
*a
b,
Ryo
Takita
*b and
Shunsuke
Chiba
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore. E-mail: shunsuke@ntu.edu.sg
bGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: takita@mol.f.u-tokyo.ac.jp
First published on 4th May 2020
Abstract
A concise protocol for anti-hydromagnesiation of aryl alkynes was established using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar combination of sodium hydride (NaH) and magnesium iodide (MgI2) without the aid of any transition metal catalysts. The resulting alkenylmagnesium intermediates could be trapped with a series of electrophiles, thus providing facile accesses to stereochemically well-defined functionalized alkenes. Mechanistic studies by experimental and theoretical approaches imply that polar hydride addition from magnesium hydride (MgH2) is responsible for the process.
:1 molar combination of sodium hydride (NaH) and magnesium iodide (MgI2) without the aid of any transition metal catalysts. The resulting alkenylmagnesium intermediates could be trapped with a series of electrophiles, thus providing facile accesses to stereochemically well-defined functionalized alkenes. Mechanistic studies by experimental and theoretical approaches imply that polar hydride addition from magnesium hydride (MgH2) is responsible for the process.
Introduction
Stereo-controlled construction of substituted alkenes is one of the most fundamental yet important processes in synthetic chemistry.1 For this purpose, hydrometallation of readily available alkynes2 has been investigated as a powerful and promising tactic of choice (Scheme 1A). Among various metals involved, use of a magnesium element for hydrometallation of alkynes (i.e. hydromagnesiation) allows for direct preparation of alkenylmagnesium species, which is very useful for a sequential bond-forming process with various electrophiles,3 thus providing facile access to multi-substituted alkenes. For this purpose, alkyl Grignard reagents having β-hydrogen atom(s) have been used as a hydrogen source in the presence of a catalytic amount of titanocene dichloride (Cp2TiCl2)4 or iron(II) chloride (FeCl2),5 in which metal hydride species (H–M: M = Ti or Fe), generated via β-hydride elimination from the corresponding alkylmetal intermediates, undergo syn-hydrometallation onto alkynes (Scheme 1B-i). Ashby demonstrated syn-hydromagnesiation of alkynes using magnesium hydride (MgH2) in the presence of Cp2TiCl2 (ref. 6) or CuI7 as a catalyst (Scheme 1B-ii). Very recently, Cavallo and Rueping revealed in their magnesium-catalysed hydroboration of alkynes that butylmagnesium hydride generated in situ from dibutylmagnesium and pinacolborane induces syn-hydromagnesiation (Scheme 1B-iii).8 Moreover, Mahon and Hill showed that structurally well-defined dimeric β-diketoiminato magnesium hydrides undergo syn-hydromagnesiation onto diphenylacetylene.9 As such, the stereochemical mode taking place in all the cases above is syn-hydromagnesiation. Herein, we report anti-hydromagnesiation of aryl alkynes using 1:1 molar combination of sodium hydride (NaH) and magnesium iodide (MgI2), which does not need the aid of any transition metal catalyst (Scheme 1C). The resulting alkenylmagnesium species could be functionalized with a series of electrophiles, to provide stereochemically well-defined substituted alkenes. Discovery, optimization, and substrate scope as well as preliminary mechanistic proposals are described.
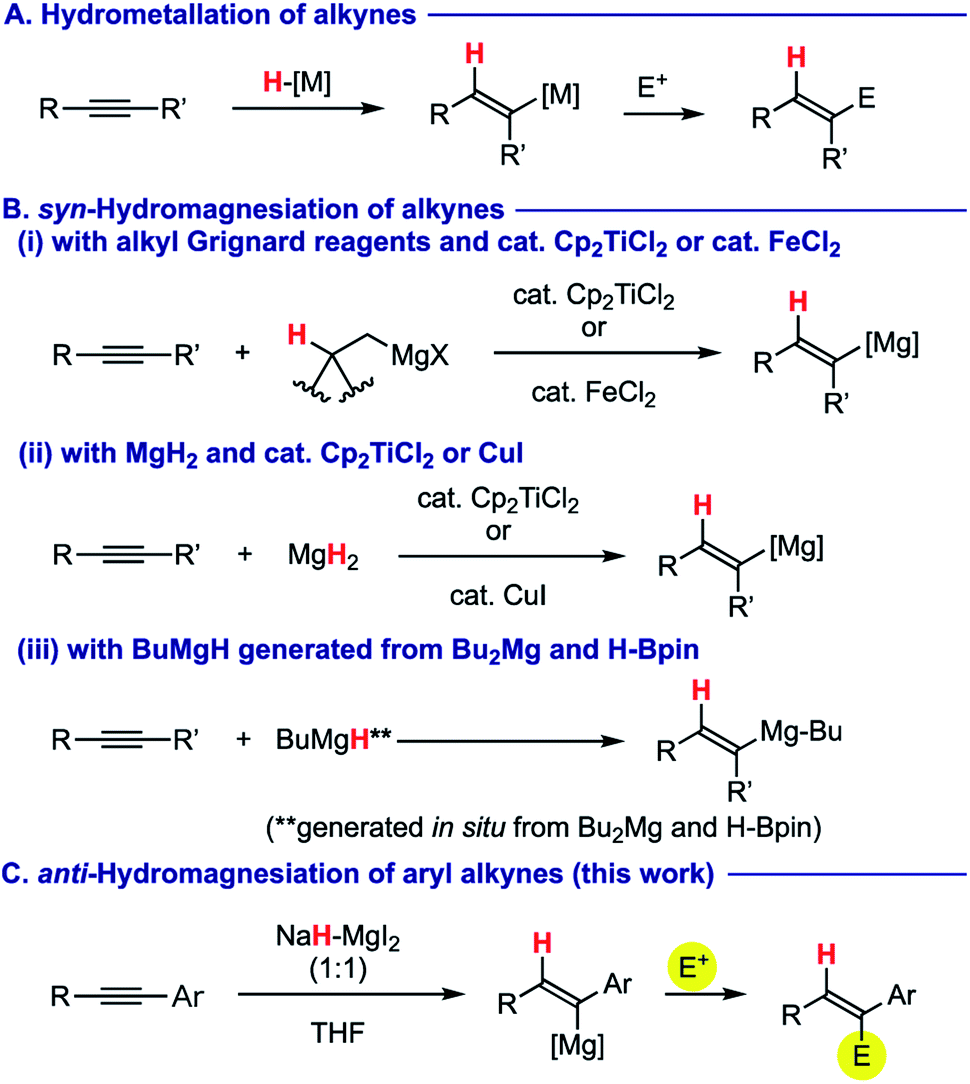 | ||
| Scheme 1 Hydrometallation of alkynes. | ||
Results and discussion
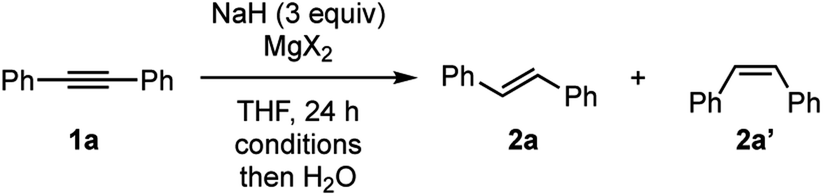
Our group has recently uncovered unprecedented hydride reduction of polar π-electrophiles such as nitriles and amides by NaH in the presence of dissolving iodide salts such as sodium iodide (NaI),10 where counter ion metathesis between bulk NaH and NaI is supposed to be a key to activate NaH.11,12 We also found NaH could be used as a hydride source for facile generation of main group metal hydrides. For example, we demonstrated controlled reduction of carboxamides into alcohols or amines by a combination of NaH with ZnI2 or ZnCl2.13,14 Based on these findings, our current attention is directed to seek for reductive molecular transformations of non-polar π-systems such as alkynes by main group metal hydrides.We embarked on our investigation of chemical reactivity of various main group metal hydrides, derived from NaH and the corresponding metal halides, toward reduction diphenylacetylene (1a). Among main group metal iodides examined for the optimization (see the ESI† for details), we found that a combination of NaH and MgI2 shows a promising reactivity toward semi-hydrogenation of 1a to trans-stilbene (2a) (Table 1).15 Use of NaH and MgI2 in 1:1 molar ratio resulted in the best outcome for the formation of 2a (entries 1–3). Full conversion of 1a was attained at 100 °C as the reaction temperature, providing stilbene 2a in 96% yield with high trans-selectivity (trans:cis = 94:6) (entry 4). Interestingly, the reactions with MgBr2 and MgCl2 gave comparable results (entries 5 and 6),16 implying that a common reactive magnesium hydride species is generated and responsible for the present reduction of 1a (vide infra). It should be noted that the reactions with other alkaline earth metal iodides based on Ca, Sr, and Ba were not optimal for this transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | MgX2 (equiv.) | Temp [°C] | Conv.b [%] | Yieldb [%] | |
| 2a | 2a′ | ||||
|
a All the reactions were conducted using 0.5 mmol of 1a in THF (2.5 mL).
b GC yields with n-dodecane as an internal standard.
c Isolated yield was 96% as a 94:6 trans/cis-mixture.
d Isolated yield was 93% as a 94:6 trans/cis-mixture.
|
|||||
| 1 | MgI2 (1.5) | 80 | 60 | 38 | 1 |
| 2 | MgI2 (2) | 80 | 62 | 40 | 2 |
| 3 | MgI2 (3) | 80 | 73 | 68 | 4 |
| 4 | MgI2 (3) | 100 | >99 | 93c | 6 |
| 5 | MgBr2 (3) | 100 | 99 | 93d | 5 |
| 6 | MgCl2 (3) | 100 | 94 | 89 | 4 |
Having optimized the reaction conditions in hands (Table 1, entry 4), we next investigated the substrate scope of alkynes for their trans-semi-reduction (Scheme 2A). Various diarylalkynes 1b–1g could be converted selectively into the corresponding trans-alkenes 2b–2g in good yields in general. Stereoselectivity was slightly dropped when a sterically bulky aryl group is installed onto the substrates (for 2c and 2d), while the electronic nature of the aryl substituents does not affect much onto the stereoselectivity (2e–2gvs.2h–2i). Alkynes having heteroaryl motifs such as furan, thiophene, and indole could also be reduced in efficient manners (for 2j–2l). Reduction of 1-phenyl-2-t-butylacetylene (1m) afforded the corresponding trans-alkene 2m in good yield, while that of 1-phenyl-2-phenyldimethylsilylacetylene (1n) resulted in formation of alkenyl silane 2n in only moderate yield. However, this method was found not applicable for reduction of dialkyl alkynes such as 5-decyne (1o). Deuterium labeling experiments using D2O (for quenching) and NaD unambiguously suggested the presence of alkenylmagnesium species A as an intermediate, that is formed via anti-hydromagnesiation (Scheme 2B).
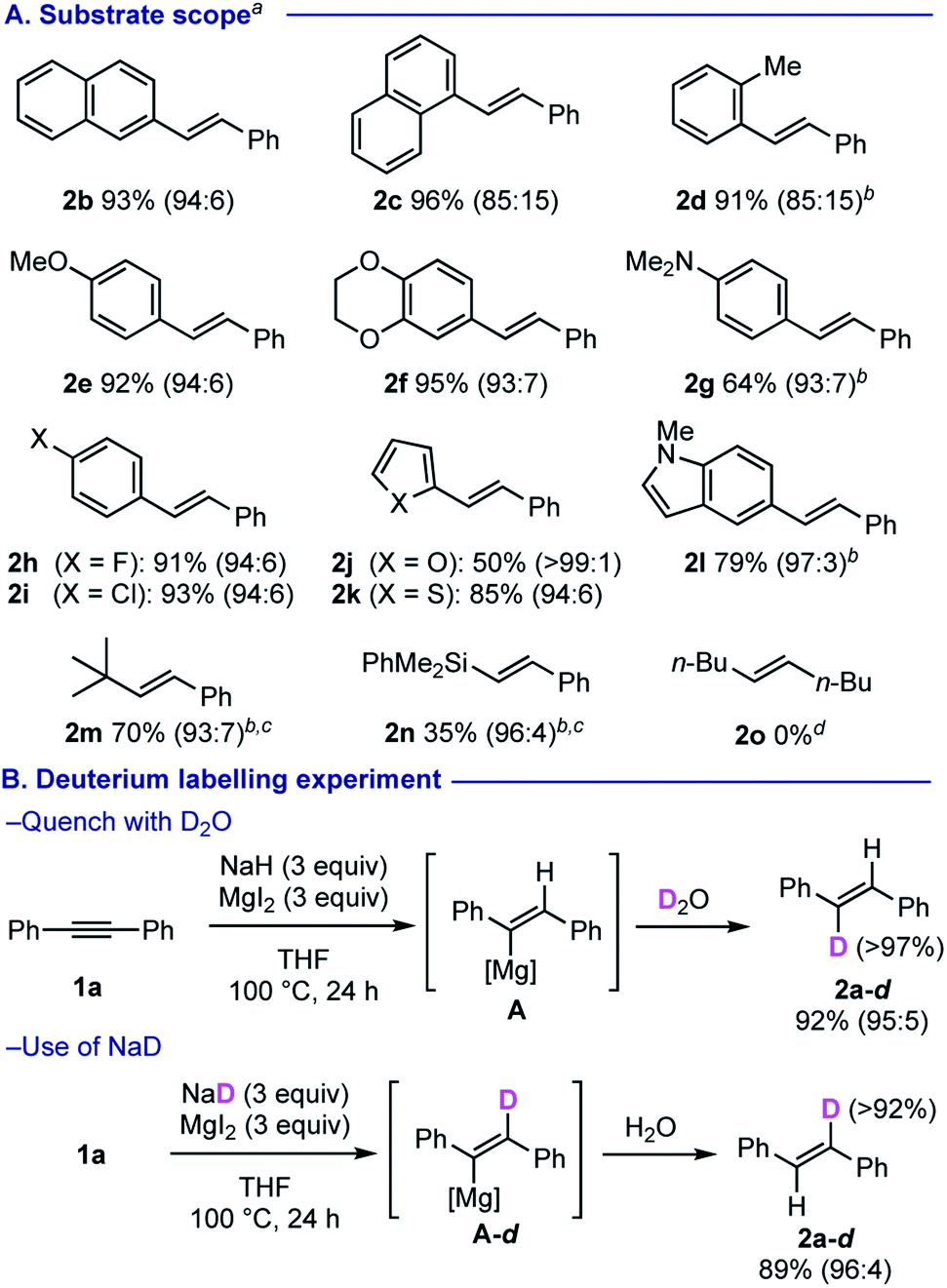 | ||
| Scheme 2 Trans-selective reduction of aryl alkynes 1. a Unless otherwise stated, the reactions were conducted using 0.5 mmol of alkynes 1 with NaH (3 equiv.) and MgI2 (3 equiv.) THF (2.5 mL) at 100 °C. The trans/cis ratio was indicated in the parentheses. b The reactions were performed using NaH (5 equiv.) and MgI2 (5 equiv.). c The reaction was performed at 120 °C. d Recovery of 5-decyne (1o) in >90% yield. | ||
The alkenylmagnesium A could be further functionalized by subsequent treatment with various electrophiles (Scheme 3A).17 Formylation with N,N-dimethylformamide (DMF) proceeded smoothly to form 2,3-diphenylacrylaldehyde (3a) in 70% yield. Addition of 1.2 equiv. of 4-anisaldehyde afforded allylalcohol 4a in 87% yield, whereas use of 2.1 equiv. of 4-anisaldehyde resulted in Oppenauer-type oxidation18 to form α,β-unsaturated ketone 5a in 76% yield. Allylation with allylbromide was facilitated by a catalytic amount (10 mol%) of CuCN·2LiCl19 to afford skipped diene 6a in 94% yield. Similarly, installation of a benzyloxymethyl (BOM) unit was achieved for synthesis of 7a using BOMCl. Use of 1,2-dibromo-1,1,2,2-tetrachloroethane allowed for smooth bromination of A to form alkenyl bromide 8a. Borylation of A could be achieved with pinacolborane by following the Singaram's protocol,20 affording alkenylboronic ester 9a in 91% yield.
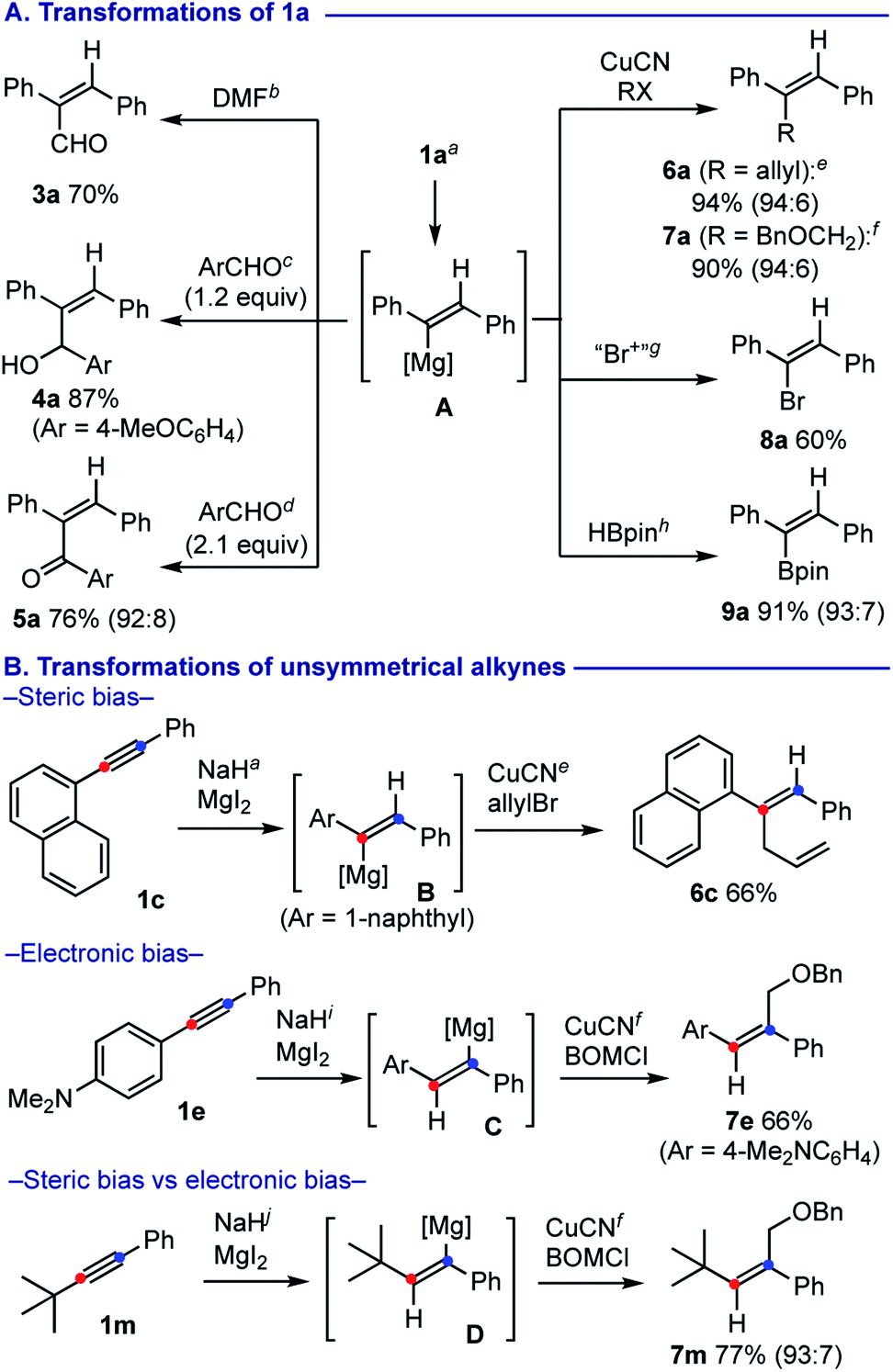 | ||
| Scheme 3 Functionalization with various electrophiles. a The reactions were conducted using 0.5 mmol of 1 with NaH (3 equiv.) and MgI2 (3 equiv.) in THF (2.5 mL) at 100 °C. b DMF (4 equiv.), 0–24 °C, 6 h. Minor isomer 3a′ was isolated in 9% yield. c 4-MeOC6H4CHO (1.2 equiv.), 0 °C, 3.5 h. Minor isomer 4a′ was isolated in 7% yield. d 4-MeOC6H4CHO (2.1 equiv.), 0–24 °C, 15 h. e CuCN·2LiCl (10 mol%), allylbromide (4 equiv.), 0 °C, 3 h. f CuCN·2LiCl (10 mol%), BOMCl (1.2 equiv.), 0 °C, 2 h. g BrCl2CCCl2Br (2.2 equiv.), 0 °C, 30 min. h HBpin (1.1 equiv.), 0–24 °C, 2.5 h. i The reaction was conducted using 0.5 mmol of 1e with NaH (5 equiv.) and MgI2 (5 equiv.) in THF (2.5 mL) at 100 °C. j The reaction was conducted using 0.5 mmol of 1m with NaH (5 equiv.) and MgI2 (5 equiv.) in THF (2.5 mL) at 120 °C. BOM = benzyloxymethyl. | ||
We next questioned how the steric and electronic bias could affect the regioselectivity on the hydromagnesiation of unsymmetrical aryl alkynes (Scheme 3B). The reaction of 1c having a sterically bulkier 1-naphthyl group underwent selective hydromagnesiation to form alkenylmagnesium B with installation of a hydride at the less hindered distal carbon (marked in blue) as a major form, that could be trapped by subsequent allylation to provide 6c in 66% yield.21 On the other hand, hydromagnesiation of 1e having an electron-rich 4-dimethylaminophenyl group resulted in installation of hydride on the proximal carbon (marked in red) to generate alkenylmagnesium C, that was functionalized with BOMCl to provide 7e in 66% yield. Hydromagnesiation of 1-phenyl-2-t-butylacetylene (1m) occurred in regioselective manner, where hydride attack was observed at the β-carbon (marked in red) to the phenyl group, while the side was sterically shielded by the t-Bu group. The resulting alkenylmagnesium D could be further transformed into BOM adduct 7m in 77% yield.
To elucidate the active hydride species responsible for the present anti-hydromagnesiation of alkynes, we conducted several control experiments (Scheme 4 and see the ESI†). Ashby reported preparation of magnesium hydride MgH2 in bulk state by treatment of MgBr2 with 2 equiv. of NaH in THF at room temperature (in quantitative yield together with the formation of inert sodium bromide).22,23 We observed that the reaction of alkyne 1a with bulk MgH2, prepared from 3 equiv. of NaH and 1.5 equiv. of MgBr2 by following the Ashby's protocol, resulted in selective formation of trans-stilbene (2a) despite poor conversion of 1a (Scheme 4A). On the other hand, treatment of 1a with bulk MgH2 in the presence of MgI2 (1.5 equiv.) greatly enhanced the conversion of 1a, providing almost the same outcome with that in the optimized reaction conditions (Scheme 4B). The IR spectrum of the mixture obtained from the reaction of NaH and MgI2 in 1:1 molar ratio showed the presence of only MgH2 and MgI2 (NaI is transparent in the IR window) (Scheme 4C). We reasoned that while bulk MgH2 itself is less hydridic due to its polymerized structure with high lattice energy,24 synergistic cooperation between bulk MgH2 and MgI2 through counter ion metathesis allows for freshly generating more hydridic MgH2 probably of smaller units (Scheme 4D), that is the key for the success in use of NaH and MgI2 in 1:1 molar ratio for the efficient anti-hydromagnesiation.
 | ||
| Scheme 4 Elucidation of active hydride species. | ||
The DFT calculations for the reactions of diphenylacetylene (1a) with a MgH2 dimer as the model of activated (MgH2)n species were thus carried out at the ωB97XD/6-311++G**/SMD(THF)//ωB97XD/6-31++G** level of theory (Scheme 5). From INT1anti, the reduction of diphenylacetylene smoothly proceeded viaTSanti (ΔG‡ +23.6 kcal mol−1), in the manner of anti-hydromagnesiation to afford alkenylmagnesium species INT2anti. In this event, the hydride in the same plane as one of the benzene rings attacks on the proximal alkyne carbon center and, the magnesium cation successively shifts to the distal alkyne carbon, indicating the polar hydride transfer mechanism. Thus, the steric and electronic bias of the aromatic ring should result in profound effects on the regioselectivity (i.e.Scheme 3B). Further investigations led to find the syn-reduction pathway. In TSsyn, diphenylacetylene is distorted, where two phenyl rings are almost perpendicular (the dihedral angle is ca. 84°). Accordingly, TSsyn was located 2.6 kcal mol−1 higher than TSanti. These computed results well corroborated the reducing ability of the NaH–MgI2 system for the anti-selective hydromagnesiation.25
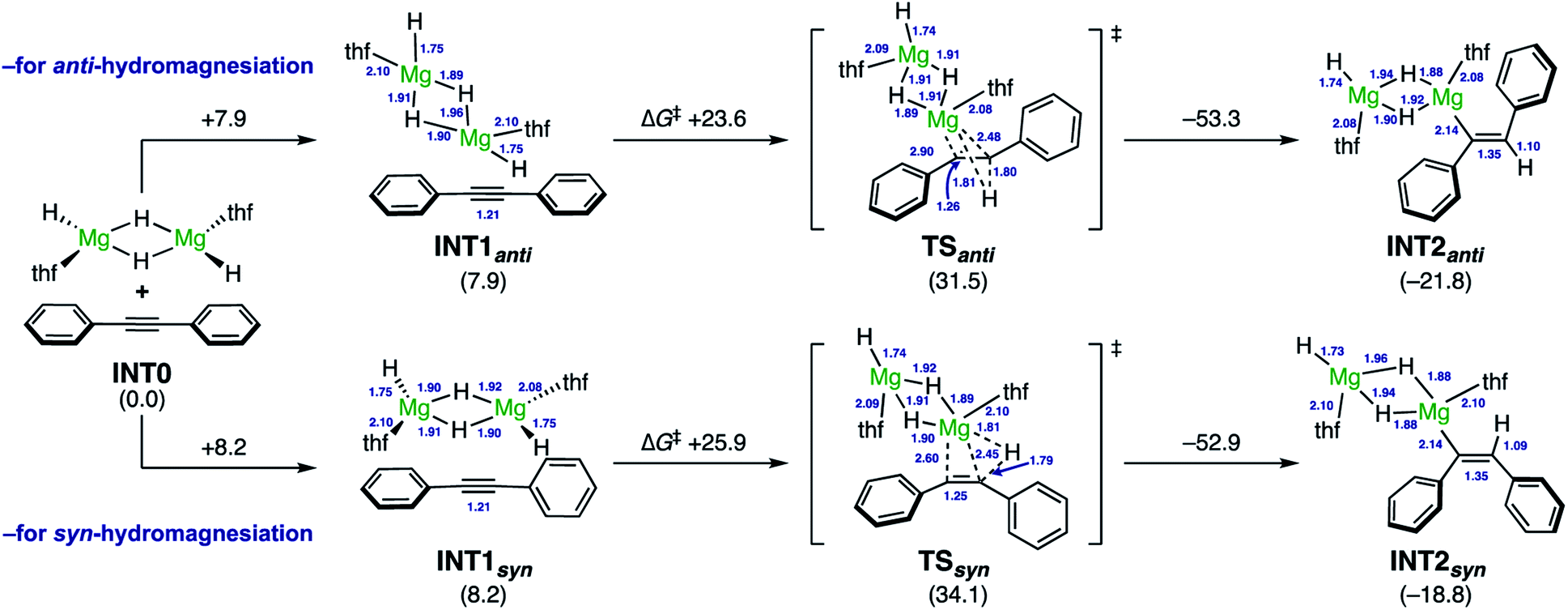 | ||
| Scheme 5 DFT calculations for model reactions of diphenylacetylene with MgH2·thf dimeric species. Energy changes and bond lengths at the ωB97XD/6-311++G**/SMD(THF)//ωB97XD/6-31++G** level of theory are shown in kcal mol−1 and Å, respectively. thf = tetrahydrofuran. | ||
We observed that the reaction of propargyl alcohol 10a with 3 equiv. of NaH26 and 2 equiv. of MgI2 proceeded smoothly at 60 °C, affording trans-alkene 11a as a single isomer in 87% yield (Scheme 6A). Based on the deuterium labelling experiments using NaD and D2O (Scheme 6A for use of NaD in the reduction of 10a. See the ESI† for details), we proposed that the process is triggered by the formation of alkoxymagnesium hydride E, that mediates hydromagnesiation to afford 5-membered magnesiocycle F. This hydroxy-guided approach27 allowed for trans-semi-reduction of various propargylic alcohols 10b–10g into the corresponding allylic alcohols 11b–11g (Scheme 6B). Heteroaromatic motifs such as 2-thienyl and 2-pyridyl groups were compatible with the process (for 11b and 11c). Sterically more hindered substrates based on adamantane (for 10d) and derived from (R)-carvone (for 10e) generated homoallylic alcohols 11d and 11e in 56% and 77% yields, respectively. Secondary propargylic alcohols 10f and 10g could also be smoothly reduced. Formation of 11e and 11g kept alkenyl moieties intact, suggesting that the present protocol is selective in the hydromagnesiation of alkynes. Phenol and aniline moieties were also capable in guiding trans-semi-reduction of alkynes for providing the corresponding 11h–11k in good to moderate yields.
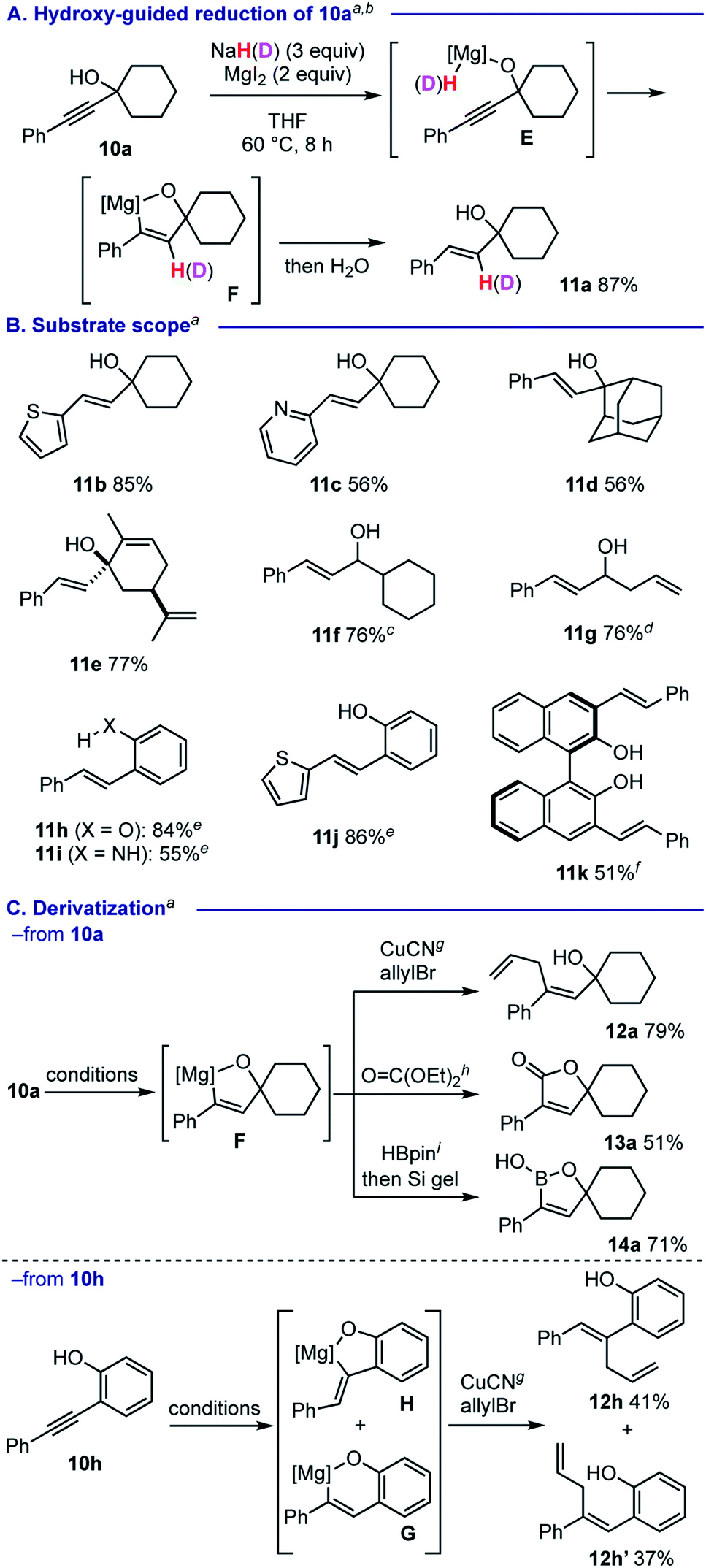 | ||
| Scheme 6 Guided reduction. a The reactions were conducted using 0.5 mmol of 10 with NaH (3 equiv.) and MgI2 (2 equiv.) THF (2.5 mL) at 60 °C. b The reaction with NaD installed a deuterium on the β-styryl moiety in 93% incorporation rate (see the ESI† for details). cTrans:cis = 99:1. d11g was isolated as its TBS ether. See the ESI† for details. e The alkyne reduction was conducted at 80 °C. f The alkyne reduction was conducted using NaH (6 equiv.) and MgI2 (4 equiv.) at 100 °C. g CuCN·2LiCl (10 mol%), allylbromide (4 equiv.), 0 °C, 2 h. h Diethyl carbonate (4 equiv.), 40 °C, 15 h. i HBpin (1.1 equiv.), 0–24 °C, 0.5 h then treatment with Si gel. | ||
The 5-membered magnesiocycle intermediate F could be further functionalized by CuCN-catalyzed allylation to form skipped diene 12a in good yield (Scheme 6C). Treatment of F with diethyl carbonate allowed for construction of lactone 13a. Furthermore, the reaction with pinacolborane followed by workup with Si gel resulted in formation of 1,5-dihydro-1,2-oxaborole 14a. On the other hand, we found that the reaction of 2-(phenylethynyl)phenol (10h) likely involves a mixture of 6-membered magnesiocycle G and 5-membered one H, which could be trapped under CuCN-catalyzed allylation reaction conditions to afford skipped dienes 12h (41% yield) and 12h′ (37% yield), respectively.
Conclusions
In conclusion, we have developed a concise protocol for anti-hydromagnesiation of aryl alkynes that operates with the magnesium hydride species derived from NaH and MgI2 under transition-metal free conditions. Subsequent treatment of the resulting alkenylmagnesium intermediates with various electrophiles allowed for the synthesis of stereochemically defined tri-substituted alkenes. Efforts are currently underway to apply the NaH–MgI2 system for reductive functionalization of other π-conjugate systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from Nanyang Technological University (NTU) and the Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2019-T2-1-089) (for S. C.) as well as JSPS (Grant-in-Aid for Scientific Research (C) (19K0662)), Uehara Memorial Foundation, and the Naito Foundation (for R. T.). The computations were performed using Research Center for Computational Science at Okazaki, Japan.Notes and references
- For selected reviews, see: (a) A. Fürstner, J. Am. Chem. Soc., 2019, 141, 11 CrossRef PubMed; (b) C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313 CrossRef PubMed; (c) E. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef PubMed; (d) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef PubMed; (e) J. Wang, Stereoselective Alkene Synthesis, Topics in Current Chemistry, Springer, 2012 CrossRef.
- For reviews, see: (a) J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075 CrossRef; (b) T. G. Frihed and A. Fürstner, Bull. Chem. Soc. Jpn., 2016, 89, 135 CrossRef; (c) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190 CrossRef.
- G. S. Silverman and P. E. Rakita, Handbook of Grignard Reagents, Marcel Decker, New York, 1996 Search PubMed.
- (a) Y. Gao and F. Sato, J. Chem. Soc., Chem. Commun., 1995, 659 RSC; (b) F. Sato, H. Ishikawa, H. Watanabe, T. Miyake and M. Sato, J. Chem. Soc., Chem. Commun., 1981, 718 RSC; (c) F. Sato, H. Ishikawa and M. Sato, Tetrahedron Lett., 1981, 22, 85 CrossRef CAS.
- L. Ilies, T. Yoshida and E. Nakamura, J. Am. Chem. Soc., 2012, 134, 16951 CrossRef CAS PubMed.
- E. C. Ashby and T. Smith, J. Chem. Soc., Chem. Commun., 1978, 30b RSC.
- E. C. Ashby, J. J. Lin and A. B. Goel, J. Org. Chem., 1978, 43, 757 CrossRef CAS.
- M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 7025 CrossRef CAS PubMed.
- L. Garcia, M. F. Mahon and M. S. Hill, Organometallics, 2019, 38, 3778 CrossRef CAS.
- (a) G. H. Chan, D. Y. Ong, Z. Yen and S. Chiba, Helv. Chim. Acta, 2018, 101, e1800049 CrossRef; (b) G. H. Chan, D. Y. Ong and S. Chiba, Org. Synth., 2018, 95, 240 CrossRef CAS; (c) Y. Huang, G. H. Chan and S. Chiba, Angew. Chem., Int. Ed., 2017, 56, 6544 CrossRef CAS; (d) P. C. Too, G. H. Chan, Y. L. Tnay, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2016, 55, 3719 CrossRef CAS PubMed.
- Z. Hong, D. Y. Ong, S. K. Muduli, P. C. Too, G. H. Chan, Y. L. Tnay, S. Chiba, Y. Nishiyama, H. Hirao and H. S. Soo, Chem.–Eur. J., 2016, 22, 7108 CrossRef CAS PubMed.
- For a review on synergistic cooperation of main group metal elements, see: S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332 CrossRef CAS PubMed.
- D. Y. Ong, Z. Yen, A. Yoshii, J. Revillo Imbernon, R. Takita and S. Chiba, Angew. Chem., Int. Ed., 2019, 58, 4992 CrossRef CAS PubMed.
- For controlled reduction of nitriles to aldehydes by NaH–ZnCl2 system, see: D. Y. Ong and S. Chiba, Synthesis, 2020, 52, 1369 CrossRef.
- For a review on semi-hydrogenation of alkynes, see: K. C. K. Swamy, A. S. Reddy and S. A. Kalyani, Tetrahedron Lett., 2018, 59, 419 CrossRef.
- MgI2 (98%, powder, Sigma-Aldrich, 394599), MgBr2 (>99%, powder, anhydrous, Sigma-Aldrich, 495093), and MgCl2 (>99%, beads, -10 mesh, anhydrous, Sigma-Aldrich, 449164) were used. See the ESI† for details.
- For reports on functionalization of alkenylmagnesium species generated through Fe-catalyzed syn-carbomagnesiation of alkynes, see: (a) E. Shirakawa, D. Ikeda, S. Masui, M. Yoshida and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 272 CrossRef PubMed; (b) T. Yamagami, R. Shintani, E. Shirakawa and T. Hayashi, Org. Lett., 2007, 9, 1045 CrossRef PubMed.
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Huskens, Synthesis, 1994, 1007 CrossRef.
- I. Klement, M. Rottländer, C. E. Tucker, T. N. Majid and P. Knochel, Tetrahedron, 1996, 52, 7201 CrossRef CAS.
- J. W. Clary, T. J. Rettenmaier, R. Snelling, W. Bryks, J. Banwell, W. T. Wipke and B. Singaram, J. Org. Chem., 2011, 76, 9602 CrossRef CAS.
- The structures of 6c and 7e were confirmed by the X-ray crystallographic analyses.
- E. C. Ashby and R. D. Schwartz, Inorg. Chem., 1971, 10, 355 CrossRef CAS.
- For a review on molecular magnesium hydrides, see: D. Mukherjee and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 1458 CrossRef CAS.
- (a) R. W. P. Wagemans, J. H. van Lenthe, P. E. de Jongh, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2005, 127, 16675 CrossRef CAS PubMed; (b) C. M. Stander and R. A. Pacey, J. Phys. Chem. Solids, 1978, 39, 829 CrossRef CAS.
- The reaction profile on the conversion of 1a to 2a generated using GC analysis clearly showed anti-hydromagnesiation is a predominant pathway over syn-one. See the ESI† for details.
- 1 equiv. of NaH is used for deprotonation of alcohols.
- A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including procedures, syntheses and characterization of new compounds; 1H and 13C NMR spectra. CCDC 1987693 and 1987694. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01773f |
| This journal is © The Royal Society of Chemistry 2020 |