
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced transition-metal- and external-photosensitizer-free intramolecular aryl rearrangement via C(Ar)–O bond cleavage†
Qian
Dou
a,
Chao-Jun
Li
 b and
Huiying
Zeng
*a
b and
Huiying
Zeng
*a
aThe State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 Tianshui Road, Lanzhou, 730000, P. R. China. E-mail: zenghy@lzu.edu.cn
bDepartment of Chemistry and FQRNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. West, Montreal, Quebec H3A 0B8, Canada
First published on 26th May 2020
Abstract
The use of photochemical reactions that do not require expensive photocatalysts or transition metals is an environmentally friendly strategy for accomplishing a variety of structural transformations. Herein, we report a protocol for photoinduced transition-metal- and external-photocatalyst-free intramolecular heteroaryl/aryl rearrangement reactions of 2-heteroaryl/aryloxybenzaldehydes. The protocol was compatible with a variety of functionalities, including methyl, methoxy, cyano, ester, trifluoromethyl, halogen, and heteroaromatic rings. Control experiments suggested that the reaction proceeded via a photoinduced intramolecular heteroaryl/aryl rearrangement process involving photoexcitation of the aldehyde carbonyl group, radical addition, C–C bond formation and C(Ar)–O bond cleavage.
Introduction
Catalytic cleavage of C(Ar)–O bonds is among the most promising methods for degradation and transformation of renewable naturally occurring aromatic resources such as lignins,1 which can thus serve as sustainable alternatives to aromatic halides. For example, transition-metal-catalyzed cross-coupling reactions involving C(Ar)–O bond cleavage have become a powerful tool for modification of the functional groups of aromatic compounds.2 In particular, reactions involving transition-metal-catalyzed activation of the C(Ar)–O bonds of phenol derivatives (e.g., sulfonates,3 esters,4 and carbamates5) have been extensively explored. However, compared with these phenol derivatives, aryl ethers are more readily available (owing to their presence in naturally occurring, renewable lignins), and their use allows for more step-economical cross-coupling reactions.6 Considerable progress has been made in developing methods for the coupling of aromatic alkyl ethers with various nucleophiles by means of direct transition-metal-catalyzed activation of C(Ar)–O bonds. Notably, ortho functional groups with chelating ability can facilitate C(Ar)–OMe bond cleavage and eliminate the need for an external ligand bearing a directing group.7 This strategy was recently used to accomplish the challenging task of cleaving the 4-O-5 linkage of a lignin model compound, despite the fact that this diaryl ether C(Ar)–O bond has a high dissociation energy and good stability.8 In addition, we reported the Pd/C-catalyzed conversion of diaryl ethers to amines via a process involving C(Ar)–O bond cleavage and C–N bond formation.9Recently, ortho-functional group assisted cleavages of the C(Ar)–O bond of diaryl ethers via an intramolecular rearrangement have been reported. For example, Rao and Li developed a rhodium-catalyzed rearrangement of 2-aryloxybenzaldehydes involving cleavage of the C(Ar)–O bond of a diaryl ether, aldehyde C–H bond insertion, and an intramolecular chelation-assisted SNAr process in the presence of a stoichiometric oxidant at high temperature10 (Scheme 1a). Recently, the Glorius's group reported a N-heterocyclic carbene-catalyzed intramolecular arylation reactions of aldehyde C–H bonds involving cleavage of the C–O bonds of diaryl ethers (Scheme 1b).11 However, in these reactions, the migrating groups are limited to heteroaryl or aryl rings bearing electron-withdrawing groups.
 | ||
| Scheme 1 Strategies for the cleavage of the C(Ar)–O bonds of diaryl ethers. | ||
Progress in photochemistry research has led to the development of methods for photoinduced transition-metal- and photosensitizer-free functionalization of C(Ar)–O bonds,12 which results in the formation of new C–C, C–N, C–P, and C–B bonds. Herein, we report that photoexcitation of the aldehyde carbonyl group of 2-aryl/heteroaryloxybenzaldehydes under transition-metal- and external-photosensitizer-free conditions generates C–O diradicals that undergo C(Ar)–O bond cleavage via an intramolecular Minisci13-rearrangement process (Scheme 1c).
Results and discussion
For our preliminary studies, we selected 2-(pyridin-2-yloxy)benzaldehyde (1a) as a model substrate. Its UV-vis absorption spectrum indicated that the wavelength of its absorption maximum was approximately 254 nm (see ESI†), which was thus chosen as the excitation wavelength. To our delight, when an acetonitrile solution of 1a containing trifluoroacetic acid (TFA) as a proton source was UV-irradiated (254 nm) at room temperature, C(Ar)–O bond cleavage and rearrangement product 2a was detected (23% yield; Table 1, entry 1). The effects of varying the solvent were subsequently investigated (entries 1–4), and EtOAc was found to give the best yield of the rearrangement product (76%, entry 4). We also explored different proton sources (TfOH, HCl, and H2SO4), all of which gave lower yields than TFA (entries 5–7). Decreasing the concentration of 1a to 0.1 M increased the yield to 90% (entry 8), but further dilution slightly lowered the yield (85%, entry 9). When TFA was replaced with one equivalent of a Lewis acid (Sc(OTf)3, Zn(OTf)2, In(OTf)3, or Yb(OTf)3), 2a was still obtained, albeit in a lower yield (6–78%, entries 10–13). Using a different irradiation wavelength (405 or 385 nm) gave an inferior yield (entries 14 and 15), and control experiments revealed that both light and the additive were essential for the rearrangement (entries 16 and 17).|
|
|||
|---|---|---|---|
| Entry | Additive | Solvent | Yield of 2ab/[%] |
| a General conditions: 1a (0.2 mmol) and the additive (1.0 equiv.) in the solvent (1.0 mL) was irradiated at 254 nm for 24 h at room temperature under argon. b Yields were determined by 1H NMR spectroscopy using nitromethane as an internal standard; n.r. = no reaction. c EtOAc (2.0 mL). d Isolated yield. e EtOAc (4.0 mL). f 405 nm. g 385 nm. h In the dark. i No additive. | |||
| 1 | TFA | CH3CN | 23 |
| 2 | TFA | DCE | 29 |
| 3 | TFA | H2O | Trace |
| 4 | TFA | EtOAc | 76 |
| 5 | TfOH | EtOAc | n.p. |
| 6 | HCl | EtOAc | 10 |
| 7 | H2SO4 | EtOAc | 14 |
| 8c | TFA | EtOAc | 90 (83d) |
| 9e | TFA | EtOAc | 85 |
| 10c | Zn(OTf)2 | EtOAc | 28 |
| 11c | Sc(OTf)3 | EtOAc | 9 |
| 12c | In(OTf)3 | EtOAc | 6 |
| 13c | Yb(OTf)3 | EtOAc | 78 |
| 14c,f | TFA | EtOAc | 13 |
| 15c,g | TFA | EtOAc | 19 |
| 16c,h | TFA | EtOAc | n.r. |
| 17c,i | — | EtOAc | 15 |

We explored the substrate scope of this rearrangement (Table 2) under the optimized reaction conditions (Table 1, entry 8). Firstly, various substituents on the migrating heteroaryl ring were investigated. Substrates with pyridine rings bearing an electron-donating group such as methoxy or methyl provided up to 96% yields of the desired rearrangement products (2b–2f). Interestingly, substrates containing a halogen atom (F, Cl, or Br) successfully underwent the reaction upon UV irradiation, giving moderate to high yields of products with their photosensitive C–halogen bonds14 intact (2g–2i). The rearrangement also proceeded smoothly when the pyridine ring bore a strongly electron-withdrawing trifluoromethyl group, giving 2j in moderate yield. In addition to pyridine, we tested substrates with a benzothiazole and a pyrazine ring, which smoothly afforded products 2k and 2l, respectively, even in the absence of TFA.

Next we elucidated the effects of substituents on the salicylaldehyde portion of the substrate. Both electron-donating and electron-withdrawing substituents (methyl, methoxy, and halide) were tolerated; and the products 2m–2r were obtained in 51–85% yields. In addition, we assessed substrates with a substituent on both the heteroaryl ring and the salicylaldehyde moiety and found that such compounds were suitable for the rearrangement, providing the products (2s–2w) with good yields (54–95%). However, no products (2x and 2y) were detected and the starting materials were recovered when 1-isoquinolinyl and 2-quinolinyl were used as the migrating groups, possibly due to the fact that the fused-ring has a stronger absorption of the light than the carbonyl group and inhibits the reaction.
We also explored the use of substrates bearing various substituted aryl groups (Table 3). To our delight, we found that in the absence of TFA, the substrates underwent the desired C(Ar)–O bond cleavage/rearrangement in EtOAc upon irradiation at 254 nm. A variety of functional groups were tolerated: including electron-withdrawing ester, cyano, trifluoromethyl, fluoro, chloro, and bromo, and electron-donating methoxy and methyl groups (2z–2ag). Interestingly, even substrates that had substituents at both of the ortho positions of the migrating ring afforded the desired rearranged products (2ae and 2ag), despite such substrates being sterically hindered.
| a Reaction conditions: 1 (0.2 mmol) in EtOAc (2.0 mL) was irradiated at 254 nm for 24 h at room temperature under argon; isolated yields are provided. |
|---|

|
To gain insight into the mechanism of this photoinduced intramolecular heteroaryl/aryl rearrangement, we carried out a series of control experiments. First, we determined the effect of periodically turning the light on and off on the time course of the yield of product 2a (Fig. 1a). We observed that the reaction proceeded smoothly when the light was on and that there was no increase in the product yield during periods when light was off. These results demonstrated that the UV light was necessary. In addition, we monitored the time course of the amounts of 1a and 2a (Fig. 1b) and found that the yield of 2a reached 80% after only 4 h of reaction time, after which point the reaction rate decreased. Kinetic isotope effect experiments (KH/KD = 1.2) carried out under the standard conditions (Scheme 2a) suggested that the C–H cleavage of the aldehyde is not involved in the rate-limiting step. This finding also indicated that an aryl formyl free radical of aldehyde 1 was unlikely to have been generated. Moreover, we found that the model reaction was suppressed only slightly when a radical inhibitor (2,2,6,6-tetramethyl-1-piperidinyloxy) was added, implying that the rearrangement did not involve an intermolecular free-radical process (Scheme 2b). Finally, no cross-rearrangement products were detected when a mixture of 1a and 1t was subjected to the standard conditions (Scheme 2c), confirming that the photoinduced rearrangement was an intramolecular process.
 | ||
| Fig. 1 (a) Time course of the yield of 2a for a reaction of 1a during which the light was periodically turned on and off. (b) Time course of the yield of 2a and consumption of 1a during a reaction under the standard conditions. | ||
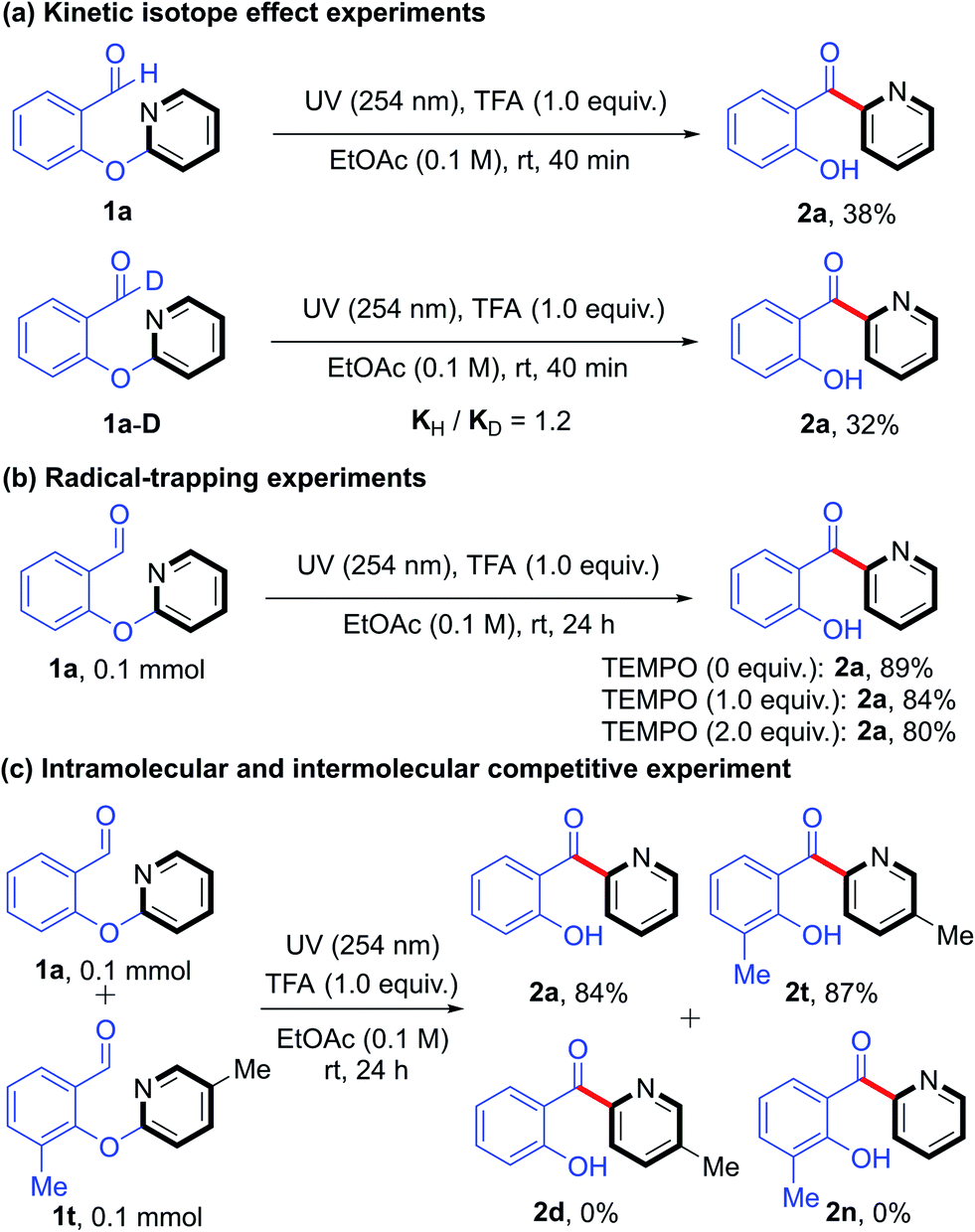 | ||
| Scheme 2 Mechanistic studies. | ||
On the basis of the above-described control experiments, a plausible mechanism for this photoinduced rearrangement is proposed in Scheme 3. Substrate A reacts with acid to form organic salt B. Photoirradiation generates diradical C, and subsequent intramolecular attack of the carbon radical on the pyridine ring generates cation radical Dvia a Minisci-type reaction. Rearomatization of D accompanied by homolytic cleavage of the C–O bond forms diradical E. Further transformation can proceed by one of two paths. In path a, abstraction of a hydrogen radical from the benzyl position by one of the oxygen radicals generates diradical F, which tautomerizes to form ortho-quinone G. Tautomerization of G generates intermediate H, which affords neutral product 2a upon workup. Alternatively, in path b, a hydrogen radical is abstracted from the benzyl position of E by the other oxygen radical to generate diradical G, which then forms intermediate H.
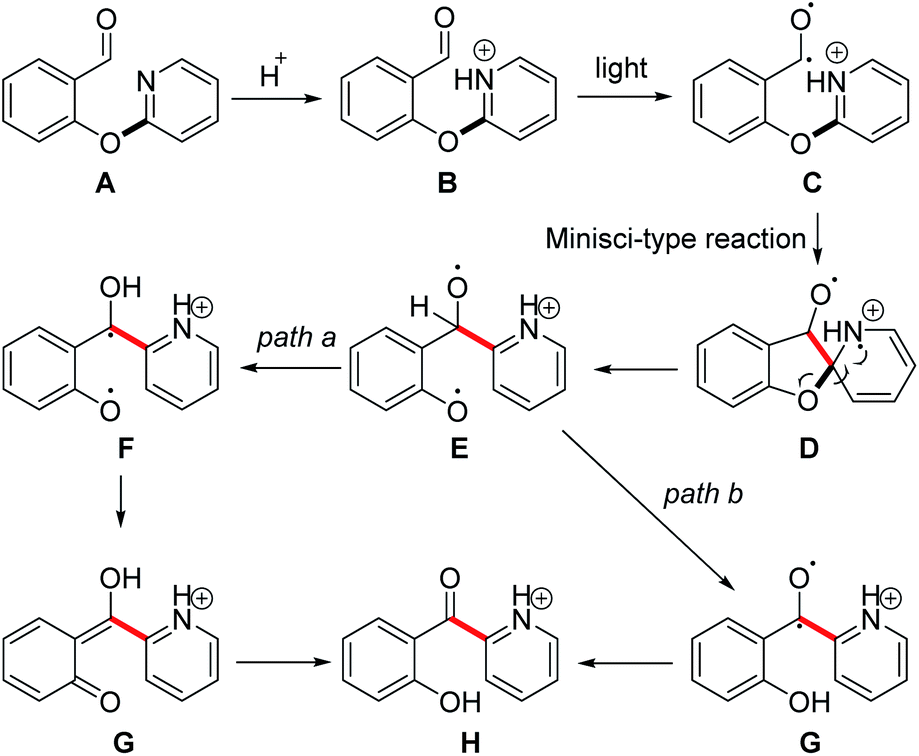 | ||
| Scheme 3 The plausible mechanism. | ||
Conclusions
In summary, we have developed a photoinduced aldehyde-mediated rearrangement reaction that involves C(Ar)–O bond cleavage and C–C bond formation. This efficient protocol affords rapid access to 2-hydroxybenzophenone derivatives under transition-metal- and external-photosensitizer-free conditions at room temperature and is thus environmentally friendly. The protocol is compatible with a variety of functional groups and is suitable for substrates bearing heteroaryl and electron-deficient or -rich aryl migrating groups. Mechanistic studies confirmed that the reaction proceeds by means of an intramolecular rearrangement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21971093), the International Joint Research Centre for Green Catalysis and Synthesis (no. 18JR3RA284 and 2016B01017), and the 111 project for support of our research. We also thank the Canada Research Chair (Tier I) foundation, the E. B. Eddy Endowment Fund, and the FQRNT for support provided to C.-J. Li.Notes and references
- (a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed; (b) W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852 RSC.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (b) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed; (c) H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen and C.-J. Li, ACS Catal., 2017, 7, 510 CrossRef CAS; (d) Z. Wang, J. Niu, H. Zeng and C.-J. Li, Org. Lett., 2019, 21, 7033 CrossRef CAS PubMed; (e) H. Zeng, Z. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2019, 58, 2859 CrossRef CAS PubMed; (f) H. Zeng, J. Yu and C.-J. Li, Chem. Commun., 2020, 56, 1239 RSC; (g) D. Cao, J. Yu, H. Zeng and C.-J. Li, J. Agric. Food Chem., 2020 DOI:10.1021/acs.jafc.0c00644.
- (a) L. Lv, D. Zhu, J. Tang, Z. Qiu, C.-C. Li, J. Gao and C.-J. Li, ACS Catal., 2018, 8, 4622 CrossRef CAS; (b) A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856 CrossRef PubMed; (c) H. N. Nguyen, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11818 CrossRef CAS PubMed; (d) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 7369 CrossRef CAS; (e) D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993 CrossRef CAS PubMed; (f) S. D. Ramgren, A. L. Silberstein, Y. Yang and N. K. Garg, Angew. Chem., Int. Ed., 2011, 50, 2171 CrossRef CAS PubMed; (g) P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2013, 52, 7242 CrossRef CAS PubMed.
- (a) J. Masson-Makdissi, J. K. Vandavasi and S. G. Newman, Org. Lett., 2018, 20, 4094 CrossRef CAS PubMed; (b) J. Yang, T. Chen and L.-B. Han, J. Am. Chem. Soc., 2015, 137, 1782 CrossRef CAS PubMed; (c) B.-T. Guan, Y. Wang, B.-J. Li, D.-G. Yu and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 14468 CrossRef CAS PubMed; (d) B.-J. Li, Y.-Z. Li, X.-Y. Lu, J. Liu, B.-T. Guan and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 10124 CrossRef CAS PubMed; (e) T. Shimasaki, M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2010, 49, 2929 CrossRef CAS PubMed; (f) R. Takise, K. Muto, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2014, 53, 6791 CrossRef CAS PubMed; (g) J. Cornella, E. P. Jackson and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 4075 CrossRef CAS PubMed.
- (a) Y. Wang, S.-B. Wu, W.-J. Shi and Z.-J. Shi, Org. Lett., 2016, 18, 2548 CrossRef CAS PubMed; (b) R. Takise, K. Itami and J. Yamaguchi, Org. Lett., 2016, 18, 4428 CrossRef CAS PubMed; (c) A. Antoft-Finch, T. Blackburn and V. Snieckus, J. Am. Chem. Soc., 2009, 131, 17750 CrossRef CAS PubMed; (d) S. Sengupta, M. Leite, D. S. Raslan, C. Quesnelle and V. Snieckus, J. Org. Chem., 1992, 57, 4066 CrossRef CAS; (e) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251 CrossRef CAS PubMed; (f) T. Mesganaw, A. L. Silberstein, S. D. Ramgren, N. F. F. Nathel, X. Hong, P. Liu and N. K. Garg, Chem. Sci., 2011, 2, 1766 RSC.
- (a) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed; (b) T.-H. Wang, R. Ambre, Q. Wang, W.-C. Lee, P.-C. Wang, Y. Liu, L. Zhao and T.-G. Ong, ACS Catal., 2018, 8, 11368 CrossRef CAS; (c) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754 CrossRef CAS PubMed; (d) M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680 CrossRef CAS PubMed; (e) Z.-K. Yang, D.-Y. Wang, H. Minami, H. Ogawa, T. Ozaki, T. Saito, K. Miyamoto, C. Wang and M. Uchiyama, Chem.–Eur. J., 2016, 22, 15693 CrossRef CAS PubMed; (f) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866 CrossRef CAS PubMed; (g) M. Leiendecker, C.-C. Hsiao, L. Guo, N. Alandini and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 12912 CrossRef CAS PubMed; (h) X. Liu, C.-C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093 CrossRef CAS PubMed; (i) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (j) B.-T. Guan, S.-K. Xiang, T. Wu, Z.-P. Sun, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Chem. Commun., 2008, 44, 1437 RSC.
- (a) Z. Rong, M. Luo and X. Zeng, Org. Lett., 2019, 21, 6869 CrossRef CAS PubMed; (b) F. Kakiuchi, M. Usui, S. Ueno, N. Chatani and S. Murai, J. Am. Chem. Soc., 2004, 126, 2706 CrossRef CAS PubMed; (c) Y. Zhao and V. Snieckus, J. Am. Chem. Soc., 2014, 136, 11224 CrossRef CAS PubMed; (d) X. Cong, H. Tang and X. Zeng, J. Am. Chem. Soc., 2015, 137, 14367 CrossRef CAS PubMed; (e) N. Iranpoor and F. Panahi, Adv. Synth. Catal., 2014, 356, 3067 CrossRef CAS.
- (a) Q. Meng, M. Hou, H. Liu, J. Song and B. Han, Nat. Commun., 2017, 8, 14190 CrossRef CAS PubMed; (b) V. Stavila, R. Parthasarathi, R. W. Davis, F. El Gabaly, K. L. Sale, B. A. Simmons, S. Singh and M. D. Allendorf, ACS Catal., 2016, 6, 55 CrossRef CAS; (c) X. Liu, L. Xu, G. Xu, W. Jia, Y. Ma and Y. Zhang, ACS Catal., 2016, 6, 7611 CrossRef CAS; (d) M. Guo, J. Peng, Q. Yang and C. Li, ACS Catal., 2018, 8, 11174 CrossRef CAS; (e) H. Wu, J. Song, C. Xie, C. Wu, C. Chen and B. Han, ACS Sustainable Chem. Eng., 2018, 6, 2872 CrossRef CAS; (f) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed; (g) W.-B. Wu and J.-M. Huang, J. Org. Chem., 2014, 79, 10189 CrossRef CAS PubMed; (h) Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674 CrossRef CAS PubMed; (i) F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474 CrossRef CAS PubMed; (j) S. Yang, X. Lu, H. Yao, J. Xin, J. Xu, Y. Kang, Y. Yang, G. Cai and S. Zhang, Green Chem., 2019, 21, 597 RSC; (k) Y.-L. Ren, M. Tian, X.-Z. Tian, Q. Wang, H. Shang, J. Wang and Z. C. Zhang, Catal. Commun., 2014, 52, 36 CrossRef CAS; (l) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed.
- (a) H. Zeng, D. Cao, Z. Qiu and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 3752 CrossRef CAS PubMed; (b) D. Cao, H. Zeng and C.-J. Li, ACS Catal., 2018, 8, 8873 CrossRef CAS.
- H. Rao and C.-J. Li, Angew. Chem., Int. Ed., 2011, 50, 8936 CrossRef CAS PubMed.
- D. Janssen-Müller, S. Singha, F. Lied, K. Gottschalk and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 6276 CrossRef PubMed.
- (a) W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621 CrossRef CAS PubMed; (b) N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100 CrossRef CAS PubMed; (c) M. De Carolis, S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 1232 CrossRef CAS PubMed; (d) V. Dichiarante, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 6495 CrossRef CAS PubMed; (e) H. Zeng, Q. Dou and C.-J. Li, Org. Lett., 2019, 21, 1301 CrossRef CAS PubMed.
- For selected review, please see: (a) F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27 CrossRef CASFor recent work about intermolecular Minisci acylation of heteroaromatics, please see: (b) P. Cheng, Z. Qing, S. Liu, W. Liu, H. Xie and J. Zeng, Tetrahedron Lett., 2014, 55, 6647 CrossRef CAS; (c) L. Zhang, G. Zhang, Y. Li, S. Wang and A. Lei, Chem. Commun., 2018, 54, 5744 RSC; (d) L. Guillemard, F. Colobert and J. Wencel-Delord, Adv. Synth. Catal., 2018, 360, 4184 CrossRef CAS; (e) M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Org. Lett., 2019, 21, 7119 CrossRef CAS PubMed.
- (a) D. Cao, C. Yan, P. Zhou, H. Zeng and C.-J. Li, Chem. Commun., 2019, 55, 767 RSC; (b) L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01585g |
| This journal is © The Royal Society of Chemistry 2020 |