
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA visible-light Paternò–Büchi dearomatisation process towards the construction of oxeto-indolinic polycycles†
Javier
Mateos‡
 a,
Alberto
Vega-Peñaloza‡
a,
Pietro
Franceschi‡
a,
Francesco
Rigodanza
a,
Philip
Andreetta
a,
Xavier
Companyó
*a,
Giorgio
Pelosi
b,
Marcella
Bonchio
a and
Luca
Dell’Amico
*a
a,
Alberto
Vega-Peñaloza‡
a,
Pietro
Franceschi‡
a,
Francesco
Rigodanza
a,
Philip
Andreetta
a,
Xavier
Companyó
*a,
Giorgio
Pelosi
b,
Marcella
Bonchio
a and
Luca
Dell’Amico
*a
aDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: luca.dellamico@unipd.it; xavier.companyo@unipd.it
bDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17, 43124 Parma, Italy
First published on 16th April 2020
Abstract
A variety of highly functionalised N-containing polycycles (35 examples) are synthesised from simple indoles and aromatic ketones through a mild visible-light Paternò–Büchi process. Tetrahydrooxeto[2,3-b]indole scaffolds, with up to three contiguous all-substituted stereocenters, are generated in high yield (up to >98%) and excellent site- regio- and diastereocontrol (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The use of visible light (405 or 465 nm) ensures enhanced performances by switching off undesired photodimerisation side reactions. The reaction can be easily implemented using a microfluidic photoreactor with improved productivity (up to 0.176 mmol h−1) and generality. Mechanistic investigations revealed that two alternative reaction mechanisms can account for the excellent regio- and diastereocontrol observed.
:1). The use of visible light (405 or 465 nm) ensures enhanced performances by switching off undesired photodimerisation side reactions. The reaction can be easily implemented using a microfluidic photoreactor with improved productivity (up to 0.176 mmol h−1) and generality. Mechanistic investigations revealed that two alternative reaction mechanisms can account for the excellent regio- and diastereocontrol observed.
Introduction
The generation of new chemical architectures starting from abundant, inexpensive materials is one of the main goals of the synthetic chemical community.1 The evolution of 3-D molecular arrangements starting from planar π-systems is of particular interest due to the possibility of assembling complex polycyclic structures from readily available aromatic feedstocks.1a–c In this context, the polycycles originating from indoles are one of the most relevant classes of pharmacophores2 due to their wide occurrence in natural alkaloids and drugs.1,2 The development of effective methods towards the dearomatisation of indoles is an attractive strategy to access complex polycyclic indoline scaffolds.1b–d Light-driven dearomatisation methods are currently emerging as a complementary sustainable approach to accessing strained polycyclic systems that are difficult or impossible to obtain under classical two-electrons polar reactivity (Fig. 1a).3 Further, oxetanes are present in a variety of biologically active molecules, being promising structural modules in drug discovery.4 Regrettably, the construction of oxetane-based polycyclic scaffolds is a challenging synthetic task owing to the high ring strain. A common strategy for the construction of oxetanes relies on UV-light driven processes. The foremost example is the Paternò–Büchi (PB) reaction, where the direct excitation of a carbonyl compound triggers a [2 + 2]-heterocycloaddition with an olefin (Fig. 1b).5 Due to the high relevance of oxetanes, both in natural products and synthetic molecules, this reaction has immense synthetic potential.5,6 Capitalising on this, a number of total syntheses have found the PB reaction to be the key step for the assembly of complex biorelevant compounds.5c,d,6b However, its progress towards industrial and large-scale synthesis has been largely hampered by the need for UV-light sources (Hg or Xe lamps) together with the corresponding specific reaction setups (e.g. quartz vessels). Further limitations of the reported PB methodologies are posed by the lack of generality and selectivity, leading to high substrate-dependency and poor regio- and diastereocontrol.5c,d These drawbacks have precluded the development of general and scalable PB methods for the dearomatisation of heterocycles,7 including indoles.8 The successful development of a PB reaction with indoles would open the way to the construction of biorelevant indolinic scaffolds in one step from readily available aromatic feedstocks (Fig. 1c). However, the realization of such a general method is complicated due to additional synthetic issues, including: (i) the electronic nature of the indole's alkene moiety, which has to match with the amphoteric nature of the excited carbonyl species; and no less important is (ii) the presence of light-driven side reactions, such as the dimerisation of the carbonyl compound.9 From a broader perspective, a general and scalable light-driven indole dearomatisation process should also involve the use of visible-light (>400 nm), thus avoiding product decomposition as well as the need for sophisticated reaction setups.10 While the vast majority of PB reactions require the use of UV-light (Fig. 1b),6 formal intramolecular versions proceed under visible-light irradiation catalyzed by precious Ir-based complexes.11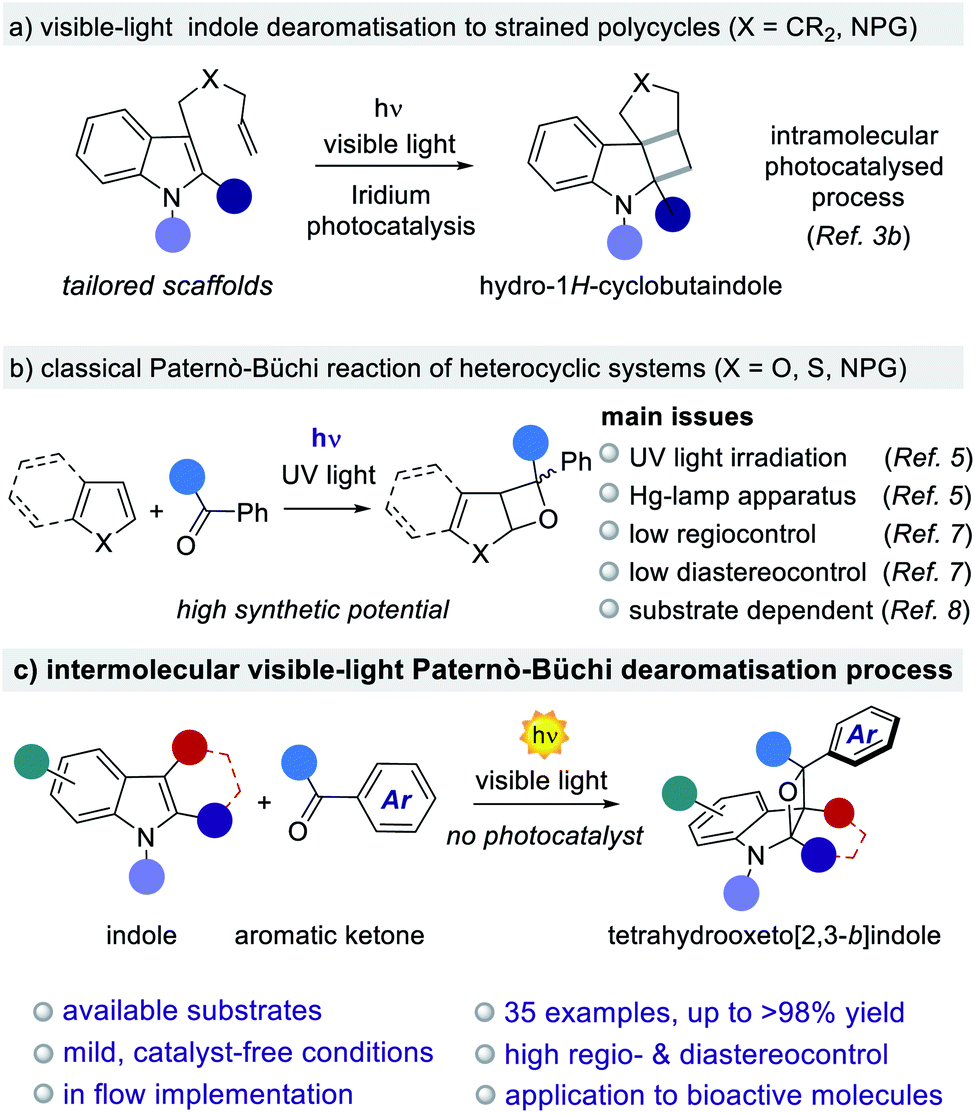 | ||
| Fig. 1 Construction of strained indoline-based polycycles through visible-light indole dearomatisation. | ||
We herein disclose a general, photocatalyst-free visible-light PB process for the dearomatisation of indoles. The method is based on the use of available aromatic ketones, inexpensive illumination sources and simple reaction setups. Structurally strained oxeto-indolinic polycycles are accessed with excellent regio- and diastereocontrol. Importantly, the unprecedented use of visible light (405 or 465 nm) in the PB reaction is key to suppressing the undesired ketone dimerisation side reaction, while guaranteeing a clean, safe and efficient process. The generality of the method is proved for a large variety of indoles and aromatic ketones, including biorelevant pharmacophoric cores and marketed drugs (35 examples, up to >98% yield and >20:1 dr). Implementation using a microfluidic setup allows the gram scale synthesis (up to 1.20 g) of the oxeto-indolinic products with improved synthetic performances. Mechanistic investigations shed light on the origins of the observed regio- and stereoselectivity, revealing the operation of two alternative yet convergent reaction manifolds.
Results and discussion
Reaction optimisation process
We initiated our study by testing different indoles 1a–d in the presence of benzophenone 2a. 3-Benzyl indoles were selected with the aim of generating a valuable all-carbon quaternary stereocenter within product 3 (Table 1). Irradiation at 365 nm of an equimolar mixture of unprotected 3-benzyl indole 1a and benzophenone 2a in toluene resulted in the complete recovery of unreacted starting materials (Table 1, entry 1). A similar fate was observed for the N-methyl indole 1b substrate, although with formation of 4 in trace amounts. We reasoned that reducing the electron density on the indole double bond would facilitate its reactivity with the triplet (T1) biradical excited state of 2a. Indeed, subjecting the N-Boc indole 1c to the reaction conditions furnished the strained fused oxetane 3c, albeit in modest yield (17%). A prolonged reaction time (5, 9 and 12 h) did not result in improved reaction performances. In fact, extensive light irradiation led to increasing amounts of the dimer 4 along with product decomposition. To further modulate the electronic nature of the reactive double bond, we placed a methyl group at the C2-position of the indole (1d). In this case the reactivity was significantly improved and product 3d formed in 55% yield along with 25% of 4 (Table 1, entry 4). At this juncture, an extensive screening of the reaction conditions was performed by testing different solvents, concentrations, and reaction times (see ESI†). Unfortunately, none of these parameters showed beneficial effects on the reaction outcome. When 2a was used in excess (3 equiv.) the NMR yield of 3d was improved up to 90%; however the presence of the homodimer 4 rendered the isolation of the product very complicated. Reasoning on the two competitive photoreactions, we assumed that an inferior amount of T1 ketone excited state could preferentially channel the system towards the intended PB reactivity, since two molecules of ketone 2a are consumed for the formation of a single molecule of 4.8 In order to reduce the amount of T1 excited state, we employed a light source with a maximum emission set at 400 nm (vs. 365 nm), close to the limit of absorption of benzophenone 2a.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya |
|
|
1 | Reaction time | Light source | Yield% (3) | dr | |
| a Reactions in PhMe at rt, 1 0.1 mmol and 2a 1 eq. (see ESI). dr inferred by 1H-NMR analysis on the crude reaction mixture. All yields refer to isolated yields. b Reaction in acetone. | ||||||||
| 1 | H | H | 1a | 3 h | 365 nm | — | 3a | — |
| 2 | Me | H | 1b | 3 h | 365 nm | — | 3b | — |
| 3 | Boc | H | 1c | 3 h | 365 nm | 17 | 3c | >20:1 |
| 4 | Boc | Me | 1d | 5 h | 365 nm | 55 | 3d | >20:1 |
| 5 | Boc | Me | 1d | 9 h | 400 nm | 67 | 3d | >20:1 |
| 6 | Boc | Me | 1d | 12 h | 405 nm | >98 | 3d | >20:1 |
| 7b | Boc | Me | 1d | 7 h | 405 nm | >98 | 3d | >20:1 |
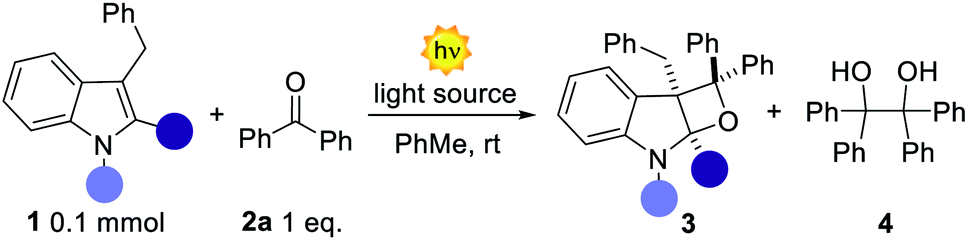


Interestingly, despite the reaction being slower, 4 was only detected in trace amounts along with a promising 67% isolated yield of 3d (Table 1, entry 5). A slightly red-shifted light source resulted in an optimal balance between reactivity and the reaction time, delivering 3d in quantitative yield and complete diastereocontrol after 12 h (Table 1, entry 6). Importantly, neither homodimer 4 nor product decomposition was detected.§ An additional solvent screening revealed acetone to be the best solvent for the present system, with quantitative formation of 3d (>98% yield) in a reduced reaction time (7 h in entry 7 vs. 12 h in entry 6). Under these conditions, product 3d was obtained by simple solvent evaporation. Before evaluating the generality and limits of the present PB process, we investigated the photochemical bases of the observed reactivity. Superimposing the absorption spectrum of benzophenone 2a (under the optimised conditions) with the 405 nm LED emission spectrum, revealed a region (390–400 nm) of possible absorption (Fig. 2a). A more accurate 3-D analysis of the excitation/emission spectra of 2a clearly demonstrates that excitation at 405 nm (Fig. 2b, pink line) results in the generation of the corresponding T1 excited state (green region). As expected, excitation at 365 nm (Fig. 2b, black line) resulted in the generation of a more intense emission signal (red region). These analyses confirm that the direct excitation of 2a can also take place under visible-light illumination. We then evaluated any possible ground- and excited-state association of the reagents.12 The absorption spectra of the isolated species (1d and 2a) and the reaction mixture (1d + 2a) did not present significant spectral variations.
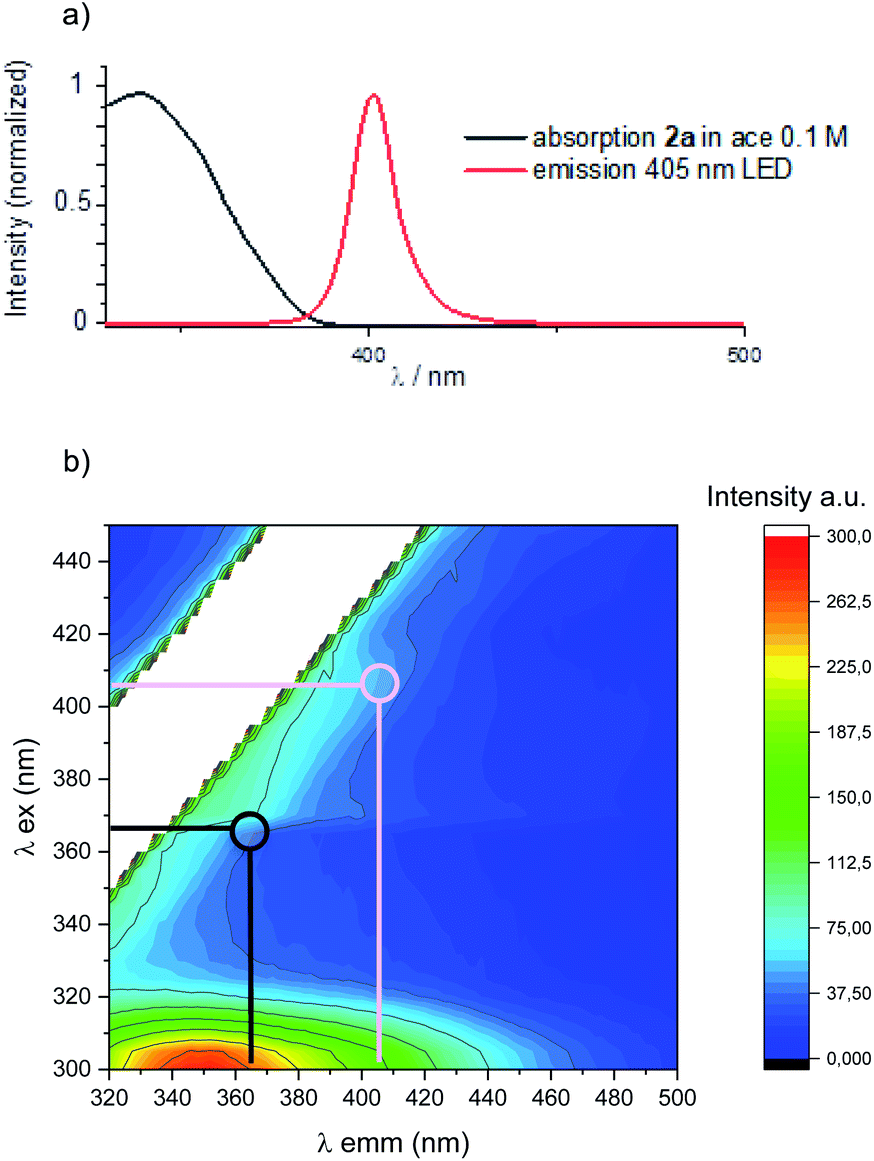 | ||
| Fig. 2 (a) Absorption spectrum of benzophenone and emission spectrum of 405 nm LED used in this study. (b) Emission 3-D-matrix of benzophenone in acetone under the optimised reaction conditions. | ||
Instead, Stern–Volmer analysis of the 2a excited state revealed an efficient quenching of the T1 excited state by 1d (Section F2 in the ESI†), with no detection of excimer emission spectra. Taken together, these observations point to a direct excitation of 2a under visible light towards the formation of its T1 state, followed by indole trapping (vide infra).
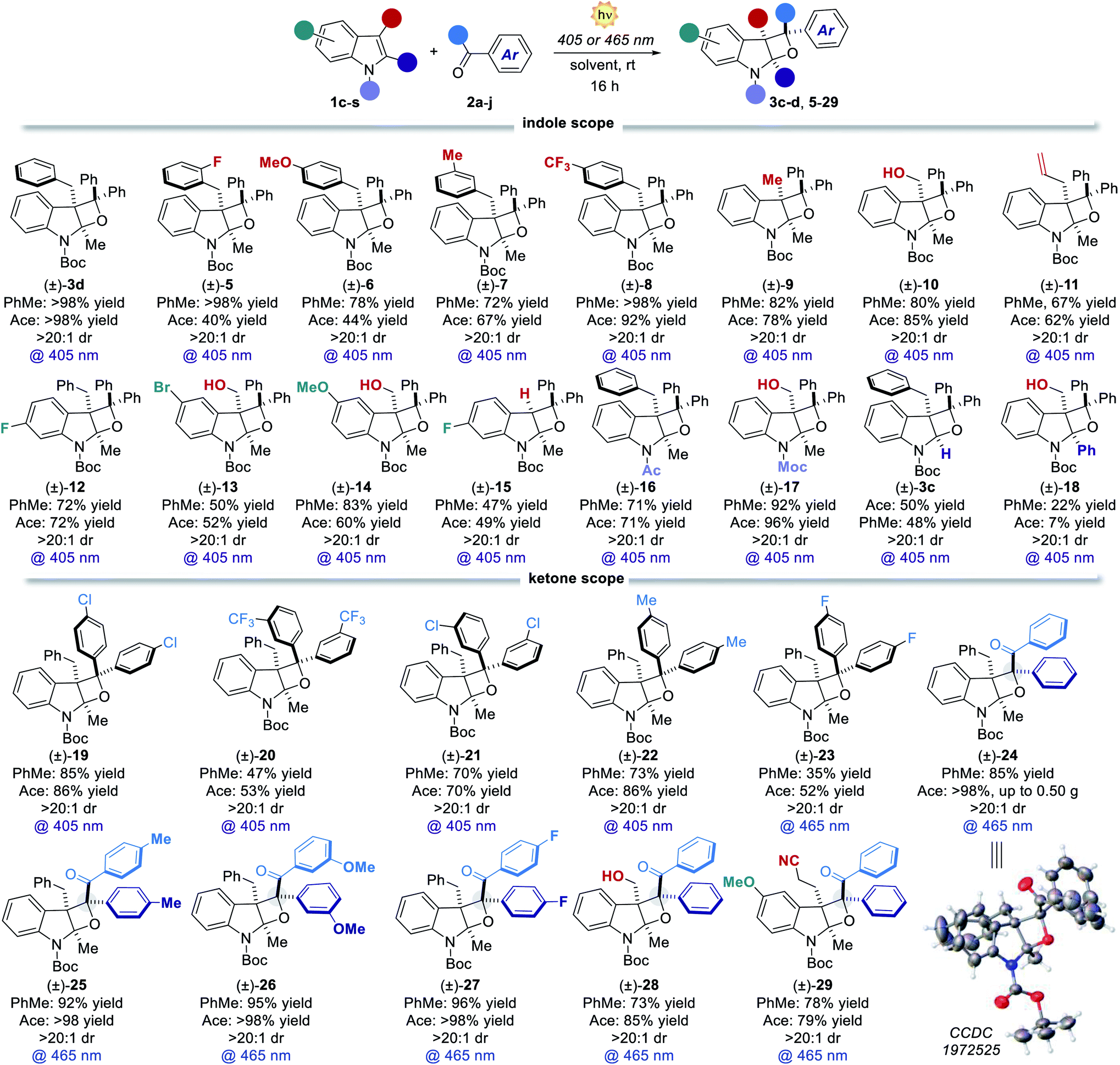
Scope and generality
Having revealed the reason for the observed reactivity, we next explored the generality of the present visible-light PB reaction (Table 2). Toluene (PhMe) and acetone (ace) were selected as the best solvents (Table 1) and all the reactions were performed under both conditions, providing a general evaluation of the synthetic protocol. 3-Benzylindoles with substituents at the orto, meta and para positions furnished the corresponding tricyclic products 5–8 in high yields spanning from 72% to >98% and complete diastereocontrol. 3-Methyl and 3-hydroxymethyl indoles formed products 9 and 10 as single diastereoisomers in excellent yields (up to 85%). Remarkably, the reaction of 3-allylindole produced the product 11 in up to 67% yield with exquisite site-, regio- and diastereoselectivity. Indole derivatives bearing different substituents on the aromatic ring (12–15) and diverse N-protecting groups (16 and 17) as well as various C2-substituents (3c and 18) efficiently participated in the visible-light PB reaction, furnishing diversified strained tetrahydrooxeto[2,3-b]indole scaffolds in yields up to 92% and >20:1 dr. Also, differently substituted benzophenones performed smoothly, forming the corresponding products 19–23 in high yields (up to 86%) and excellent diastereocontrol (>20:1). Remarkably, the prochiral benzil 2g successfully engaged in the [2 + 2] heterocycloaddition process, delivering up to 0.50 g of dearomatised product 24 (>98% yield) with complete stereocontrol over the three generated stereocenters. Its relative stereochemistry was inferred from X-ray analysis of a single crystal. Diverse benzil derivatives were formed quantitatively with complete diastereocontrol (25–27). Also, differently substituted indoles performed well, delivering the products 28 and 29 in up to 85% yield. All the benzil derivatives, with more red-shifted absorption, successfully reacted under 465 nm illumination with no traces of side products. We next sought to apply the developed dearomatisation protocol to accessing relevant bioactive pharmacophoric cores (Fig. 3). A series of oxeto-indolinic derivates 30–33, bearing up to three contiguous stereocenters, were synthesised in good to high yields (48–82%) and complete diastereocontrol. Further, the marketed drug Melatonin proved to be a useful substrate for the present method, affording the corresponding product 34 in 42% yield.
|
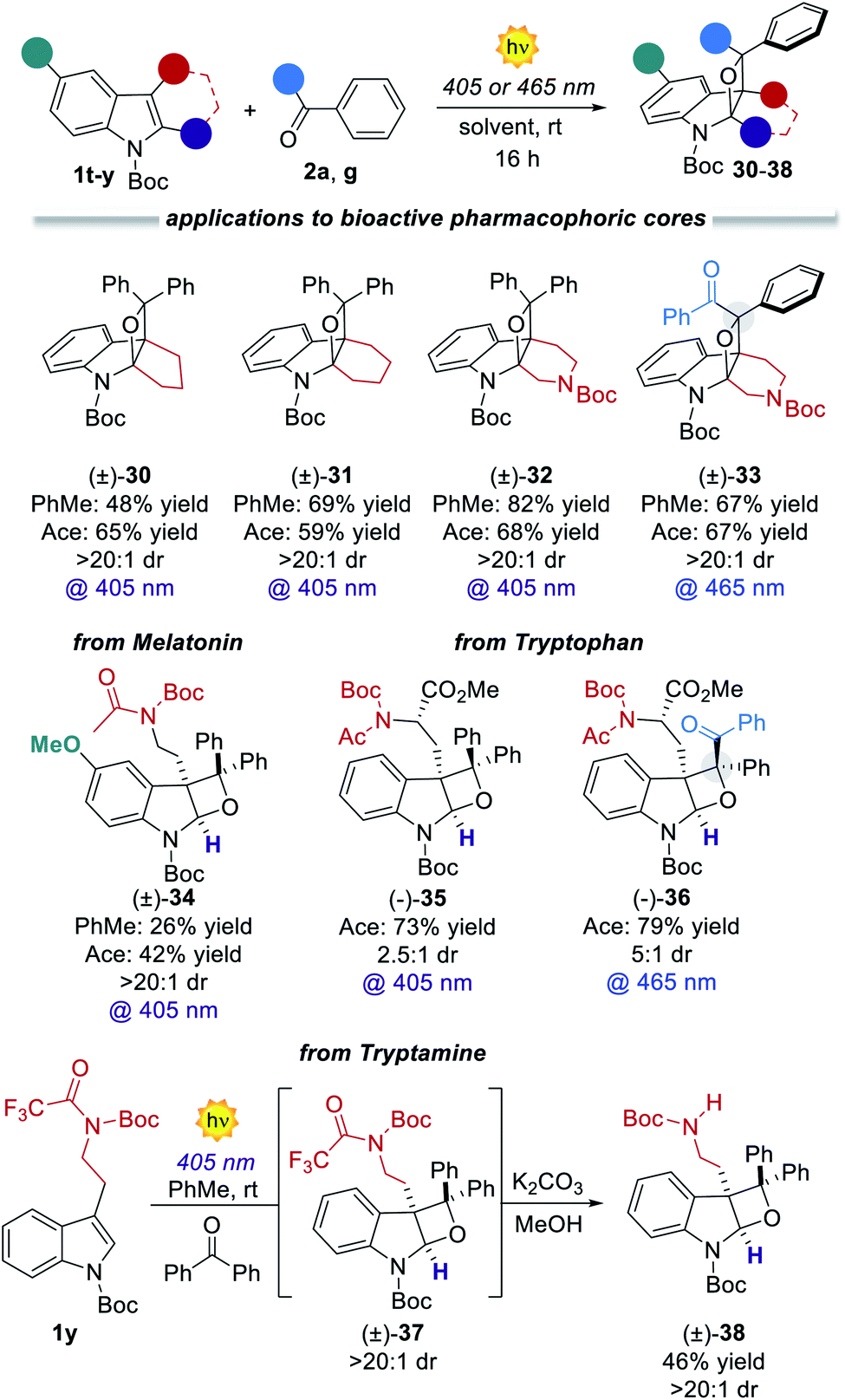 | ||
| Fig. 3 Application of the developed PB process to biorelevant pharmacophoric cores and marketed drugs. | ||
Furthermore, the synthetic potential of the developed method towards structural modifications of bioactive ingredients was demonstrated. Interestingly, enantiopure N-protected tryptophan took part in the developed method both with 2a and benzil 2g, delivering attractive chiral tetrahydrooxeto[2,3-b]indoles 35 and 36 in high yields and 2.5:1 to 5:1 dr, respectively. It is worth mentioning that the chiral compound 36 presents four stereocenters, with two all-substituted centers. Also, tryptamine derivative 1y underwent the dearomatisation process. Product 38 was isolated in 46% yield as a single diastereoisomer. To demonstrate the generality and the operational simplicity of the developed method, the reactions of indole 1d with ketones 2a and 2g were performed under natural sunlight irradiation (Fig. 4a).13 Both reactions proceeded smoothly delivering the corresponding products 3d and 24 in 96% and 86%, within 6 h reaction time. Finally, the synthetic method was implemented using a microfluidic photoreactor (Fig. 4b).14 The oxeto-indolinic products 5 and 24 formed in quantitative yields within 30 min residence time. Encouraged by these results, we performed a large-scale microfluidic synthesis of 10, which was obtained with improved synthetic performance and productivity with respect to the batch setup (96% yield and 0.176 mmol h−1 in flow vs. 80% yield and 0.080 mmol h−1 in batch). The microfluidic setup allowed the production of 1.20 g of 10 in 15 h. Treatment of compound 10 with LiAlH4 delivered the N-methyl protected tetrahydrooxeto[2,3-b]indole 39 in 77% yield (0.68 g) with unexpected formal removal of the hydroxymethyl group. It should be noted that N-methyl indoline-alkaloids possess widespread biological activities,1b–d and the oxeto-scaffold 39 is an unprecedented representative of this class of molecules.
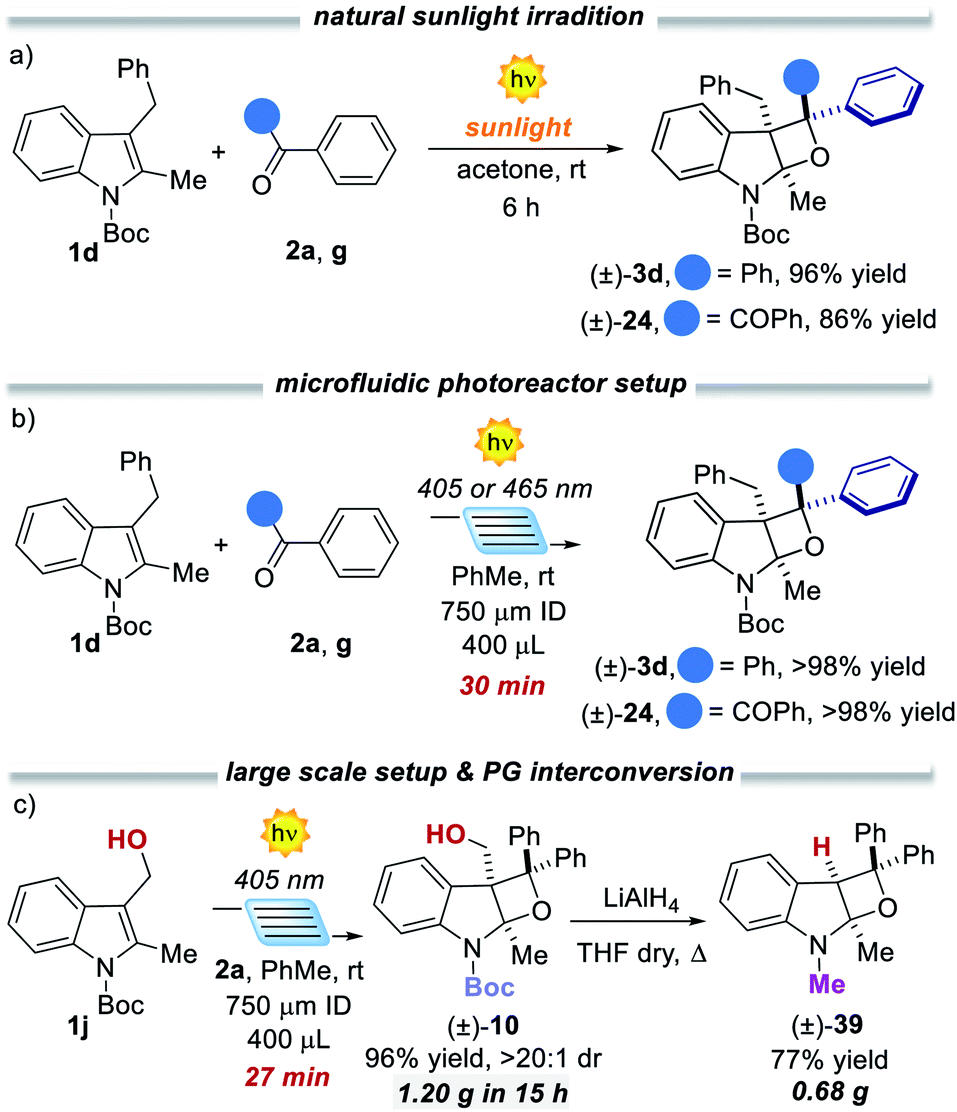 | ||
| Fig. 4 (a) Reactions performed under natural sunlight on 31/01/2020 from 11:00–17:00 (top). (b) Implementation of the reaction using a microfluidic photoreactor and (c) protecting group interconversion. | ||
Mechanistic considerations
As already pointed out, the developed visible-light [2 + 2] heterocycloaddition process proceeds via the direct excitation of the aromatic ketone to its S1 state, which rapidly decays to the T1 excited state. This mechanistic hypothesis was further confirmed by performing control experiments in the presence of an effective tertiary-amine T1 quencher15 such as DABCO15d (1,4-diazabicyclo[2,2,2]octane, Fig. 5a). When the reactions between 1d and ketone 2a or 2g were peformed in the presence of 1 equiv. of DABCO the reactivity dropped dramatically, demonstrating that the T1 excited state of the carbonyl counterpart is the key reactive intermediate under both reaction paths (Fig. 5b). However, diverse reaction mechanisms may originate from the T1 excited state. The conventional PB reaction involves a radical combination between the T1 excited state of the aromatic ketone and the indole. An alternative mechanistic scenario deals with photoinduced electron transfer (PET)16 from the electron-rich indole to the ketone, to generate a radical ion pair which rapidly collapses into the final oxetane (Fig. 5b). To evaluate the feasibility of this process, we calculated the ΔGPET from the Gibbs energy of the PET equation (see the ESI†).5c For the reaction with benzophenone 2a we found a ΔGPET = +1.35 eV, corresponding to 28.83 kcal mol−1. This value indicates that the operation of a PET process is unlikely.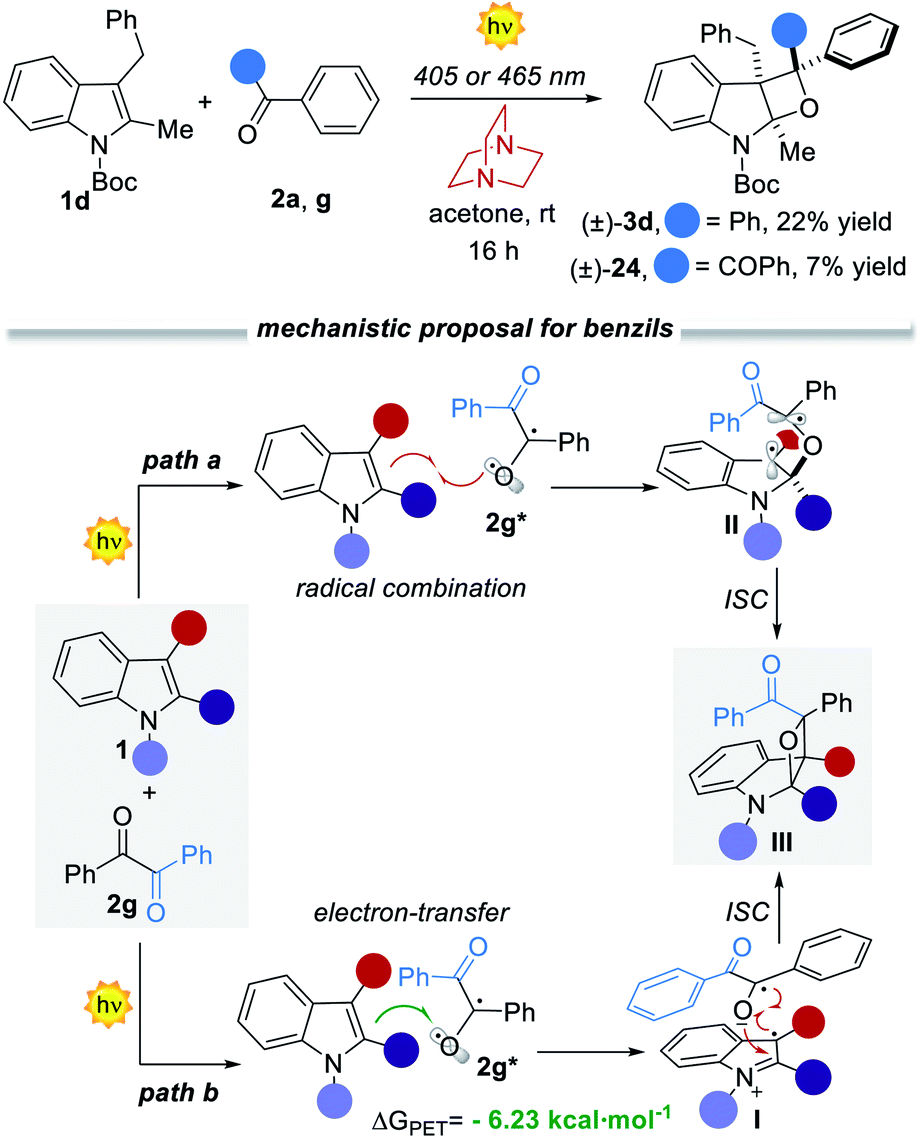 | ||
| Fig. 5 (a) Control experiments and (b) bifurcated mechanistic proposal for belzil 2g. | ||
Having already excluded ground- and excited state associations of the starting materials, the only feasible pathway is a radical trap by the indole 1d of the T1 state of the aromatic ketone, where the regio- and stereocontrol are governed by steric factors (see intermediate II). On the other hand, for benzil 2g, we calculated a ΔGPET = −6.23 kcal mol−1 (−0.27 eV), thus indicating the feasibility of the PET mechanism (path b, Fig. 5).16 On these grounds, we proposed two alternative yet convergent reaction paths that are summarized in Fig. 5b.17 It is worth mentioning that in selective PB reactions either a S1 trapping or a PET mechanism is more likely to be involved.5c,6c These processes are faster with respect to the T1 radical combination manifold, thus precluding structural reorganisations.5c,6 In our case, the operation of a PET process matches well with the observed complete regio- and diastereoselectivity (Fig. 5b, path b). After the initial PET from 1 to 2g*, a reactive radical ion pair I is formed. The regioselectivity of the C–O bond-forming event is controlled by the relative charge distributions and further reinforced by steric factors (Section F4 in the ESI†). The stereoselectivity of the process is secured by the cyclic indole reagent, with the formation of a cis-oxetane. The high stereocontrol over the third stereocenter can be explained with respect to the same intermediate I, which rapidly collapses into the final product III (Fig. 5b and S20 in ESI†). Interestingly, intermediate I is stabilised by a favorable π–π stacking between the two aromatic rings. In fact, when the same reaction was performed with the N-Boc protected pyrrole the final product was obtained as a mixture of diastereoisomers (86% yield and 2.2:1 dr).7d Hence, the presence of the aromatic ring within the indole precursor 1 is essential to ensure the diastereocontrol of the process and corroborates the proposed mechanistic hypothesis.
Conclusions
In summary, we have developed an extremely mild visible-light PB process able to access strained oxeto[2,3-b]indole scaffolds starting from readily available substrates. This has been accomplished by the careful selection of a visible-light source (405 or 465 nm), which enabled the complete shutting down of the ketone dimerisation side reaction. The generality of the PB process has been demonstrated for a large variety of indoles and aromatic ketones with excellent results (up to >98% yield and complete dr). Importantly, the reaction is applicable to the assembly of diverse pharmacophoric cores and for the installation of oxetanes into diverse indole-based marketed drugs. Further, the reaction can be easily performed under natural sunlight as well as in a microfluidic photoreactor with definite advantages in terms of scalability (g scale), generality and productivity (up to 0.176 mmol h−1). Finally, oxetane-indolinic products can be easily converted into biorelevant N-methylated counterparts (up to 0.68 g). Mechanistic insights reveal that the chemistry is driven by the ketone T1 excited states of both benzophenone (2a) and benzil (2g) ketones. In the latter, two distinct yet convergent reaction mechanisms can account for the high regio- and diastereoselectivity observed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Padova, the CariParo Foundation Synergy – Progetti di Eccellenza 2018 (L.D.), the GREEN C-C STARS starting grant (X. C.) and the Seal of Excellence @unipd PLACARD (A. V.). Chiesi Farmaceutici SpA and Dr Ferdinando Vescovi are acknowledged for their support with the D8 Venture X-ray equipment. Stefano Mercanzin is acknowledged for technical assistance. Prof. Andrea Sartorel is acknowledged for insightful discussions.Notes and references
- (a) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC; (b) C. Zheng and S.-L. You, Chem, 2016, 1, 830–857 CrossRef CAS; (c) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (d) J. Mateos, F. Rigodanza, A. Vega-Peñaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyó, M. Bonchio and L. Dell'Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS PubMed; (e) B. Muriel, A. Gagnebin and J. Waser, Chem. Sci., 2019, 10, 10716–10722 RSC.
- (a) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed; (b) W. Gribble, Indole Ring Synthesis: From Natural Products to Drug Discovery, 1st edn, Wiley-VCH, Weinheim, 2016 Search PubMed.
- (a) M. Okumura and D. Sariah, Eur. J. Org. Chem., 2020, 10, 1259–1273 CrossRef; (b) M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed; (c) M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, J. Am. Chem. Soc., 2020, 142, 3094–3103 CAS.
- For a review see: (a) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed; (b) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed.
- (a) E. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341–361 Search PubMed; (b) G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327–4331 CrossRef; (c) M. Fréneau and N. Hoffmann, Org. Biomol. Chem., 2016, 14, 7392–7442 RSC; (d) T. Bach, Synthesis, 1998, 5, 683–703 CrossRef.
- (a) C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046–12055 CrossRef CAS; (b) M. D'Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 Search PubMed.
- For selected examples of PB reactions with heterocycles see: (a) L. Stuart and K. Satake, J. Am. Chem. Soc., 1983, 105, 6123–6124 Search PubMed; (b) M. D'Auria, R. Racioppi, F. Rofrano, S. Stoia and L. Viggiani, Tetrahedron, 2016, 72, 5142–5148 Search PubMed; (c) M. Abe, E. Torii and M. Nojima, J. Org. Chem., 2000, 65, 3426–3431 CrossRef CAS PubMed; (d) C. Rivas and R. A. Bolivar, Heterocycl. Chem., 1973, 10, 967–971 CrossRef CAS; (e) M. D'Auria, L. Emanuele and R. Racioppi, Lett. Org. Chem., 2005, 2, 132–135 CrossRef; (f) M. D'Auria, L. Emanuele, R. Racioppi and A. Valente, Photochem. Photobiol. Sci., 2008, 7, 98–103 RSC.
- For preliminary attempts towards the PB reaction with indoles see: (a) H. Takechi, M. Machida, N. Nishizono and Y. Kanaoka, Chem. Pharm. Bull., 1994, 42, 188–196 CrossRef CAS; (b) M. Machida, H. Takechi and Y. Kanaoka, Tetrahedron Lett., 1982, 23, 4981–4982 CrossRef CAS; (c) D. R. Julian and G. D. Tringham, J. Chem. Soc., Chem. Commun., 1973, 111–112 Search PubMed; (d) T. Haruko, M. Minoru and K. Yuichi, Chem. Pharm. Bull., 1988, 36, 2853–2863 CrossRef; (e) S. G. Cohen and R. J. Baumgarten, J. Am. Chem. Soc., 1967, 89, 3471–3475 CrossRef CAS.
- For the mechanism of light-mediated formation of a ketone homodimer see: (a) Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CAS; (b) G. M. Robertson, Comprehensive Organic Synthesis, in Pinacol Coupling Reactions, 1991, ch. 2.6, vol. 3, pp. 563–611 Search PubMed; (c) Z. Qui, H. D. M. Pham, J. Li, C.-C. Li, D. J. Castillo-Pazos, R. Z. Khaliullin and C.-J. Li, Chem. Sci., 2019, 10, 10937–10943 Search PubMed.
- (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176–1239184 CrossRef PubMed; (b) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- (a) M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095–5103 CrossRef PubMed; (b) E. Kumarasamy, R. Raghunathan, S. K. Kandappa, A. Sreenithya, S. Jockusch, R. B. Sunoj and J. Sivaguru, J. Am. Chem. Soc., 2017, 139, 655–662 CrossRef CAS PubMed.
- Spectroscopic analysis ruled out the hypothesis of indole 1d excitation..
- (a) S. Protti and M. Fagnoni, Photochem. Photobiol. Sci., 2009, 8, 1499–1516 RSC; (b) D. Cambiè, J. Dobbelaar, P. Riente, J. Vanderspikken, C. Shen, P. H. Seeberger, K. Gilmore, M. G. Debije and T. Noël, Angew. Chem., Int. Ed., 2019, 58, 14374–14378 CrossRef PubMed; (c) D. Cambiè and T. Noël, Top. Curr. Chem., 2018, 376, 45 CrossRef PubMed; (d) M. Oelgemöller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed.
- For selected reviews, see: (a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) C. Sambiagio and T. Noël, Trends Chem., 2019, 2, 92–106 CrossRefFor recent selected recent examples, see: (c) J. Mateos, A. Cherubini-Celli, T. Carofiglio, M. Bonchio, N. Marino, X. Companyó and L. Dell'Amico, Chem. Commun., 2018, 54, 6820–6823 RSC; (d) J. Mateos, N. Meneghini, M. Bonchio, N. Marino, T. Carofiglio, X. Companyó and L. Dell'Amico, Beilstein J. Org. Chem., 2018, 14, 2418–2424 CrossRef CAS PubMed; (e) S. Paria, E. Carletti, M. Marcon, A. Cherubini-Celli, A. Mazzanti, M. Rancan, L. Dell'Amico, M. Bonchio and X. Companyó, J. Org. Chem., 2020, 85, 4463–4474 CrossRef CAS PubMed.
- (a) J.-P. Blanchi and A. R. Watkins, J. Chem. Soc., Chem. Commun., 1974, 265–266 RSC; (b) H. Masuhara, N. Mataga and H. Tsubomura, J. Phys. Chem., 1975, 79, 1255–1259 CrossRef; (c) D. Griller, J. A. Howard, P. R. Marriott and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 619–623 CrossRef CAS; (d) K. Peters and J. Lee, J. Phys. Chem., 1993, 97, 3761–3764 CrossRef CAS; (e) L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3318 CrossRef PubMed.
- (a) R. S. J. Mulliken, Phys. Chem., 1952, 56, 801–822 CrossRef CAS; (b) R. Foster, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS; (c) J. Mattay, Angew. Chem., Int. Ed., 1987, 26, 825–845 CrossRef.
- Spectroscopic analysis of benzil and an equimolar mixture of indole 1d and benzil 2b, under the reaction conditions, excluded the formation of any ground- or excited state association between the reagents..
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1972525. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01569e |
| ‡ These authors contributed equally to this work. |
| § We have also evaluated the possibility of reducing the T1 ketone excited state by reducing the intensity of the 365 nm LED. These experiments resulted in extensive product decomposition due to the over irradiation required to reach a decent level of conversion. |
| This journal is © The Royal Society of Chemistry 2020 |