
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransmembrane anion transport mediated by halogen bonding and hydrogen bonding triazole anionophores†
Laura E.
Bickerton
 ,
Alistair J.
Sterling
,
Paul D.
Beer
,
Fernanda
Duarte
* and
Matthew J.
Langton
*
,
Alistair J.
Sterling
,
Paul D.
Beer
,
Fernanda
Duarte
* and
Matthew J.
Langton
*
Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: matthew.langton@chem.ox.ac.uk; fernanda.duartegonzalez@chem.ox.ac.uk
First published on 15th April 2020
Abstract
Transmembrane ion transport by synthetic anionophores is typically achieved using polar hydrogen bonding anion receptors. Here we show that readily accessible halogen and hydrogen bonding 1,2,3-triazole derivatives can efficiently mediate anion transport across lipid bilayer membranes with unusual anti-Hofmeister selectivity. Importantly, the results demonstrate that the iodo-triazole systems exhibit the highest reported activity to date for halogen bonding anionophores, and enhanced transport efficiency relative to the hydrogen bonding analogues. In contrast, the analogous fluoro-triazole systems, which are unable to form intermolecular interactions with anions, are inactive. The halogen bonding anionophores also exhibit a remarkable intrinsic chloride over hydroxide selectivity, which is usually observed only in more complex anionophore designs, in contrast to the readily accessible acyclic systems reported here. This highlights the potential of iodo-triazoles as synthetically accessible and versatile motifs for developing more efficient anion transport systems. Computational studies provide further insight into the nature of the anion-triazole intermolecular interactions, examining the origins of the observed transport activity and selectivity of the systems, and revealing the role of enhanced charge delocalisation in the halogen bonding anion complexes.
Introduction
The development of supramolecular anionophores for transmembrane ion transport is driven by their potential utility as tools for studying ion transport processes and as therapeutics for diseases arising from mis-regulation of protein ion channels.1–4 Significant effort has been devoted to designing mobile carrier systems with high anion transport activity in vesicles (particularly for chloride), and more recently, in cells.5–10 As with naturally occurring ion transporters, anion selectivity is crucial, and depends on the delicate balance between transporter anion binding selectivity and anion desolvation. Selectivity for chloride over proton transport (or the functionally equivalent hydroxide) is particularly necessary for applications in which dissipation of transmembrane pH gradients must be avoided. The naturally occurring anionophore, prodigiosin,11 and its synthetic analogues,12 are known to uncouple H+-ATPases or neutralise organelles by facilitated H+/Cl− symport, and for this reason are promising candidates for anti-cancer agents. Conversely, anionophores designed for treating diseases arising from misregulated chloride channels, including cystic fibrosis and Bartter syndrome, must have high Cl− > H+/OH− selectivity to avoid toxicity arising from disrupting transmembrane pH gradients. However, designing transporters with such selectivity remains a significant challenge. Polar NH hydrogen bond (HB) donors (such as ureas or squaramides) which are typically used in supramolecular anionophores exhibit no intrinsic Cl− > H+/OH− selectivity.10 Improved chloride selectivity has been achieved by designing anionophores which encapsulate the anion, such as tripodal or cholapod-based receptors, but in general, more acidic NH HB-donors that are required for anion binding and efficient transport lead to decreased Cl− > H+/OH− selectivity.10C–H hydrogen bonding or C–I halogen bonding (XB)13–15 interactions have recently emerged as potential alternatives to the classical N–H or O–H HB donors for anion transport. Compared to classical HB, these interactions are less hydrophilic, encounter diminished dehydration penalties, and are less inclined to promote detrimental transporter aggregation.16 XB in particular can exhibit superior anion binding affinity in competitive polar organic17 or aqueous media18–23 to hydrogen bonding,18–23 pointing to its potential utility in transmembrane anion transport systems. However, to date only a handful of XB-mediated anion transport systems have been reported, exploiting iodo-perfluoro alkane and arene derivatives for anion recognition and transport,24–28 and examples of transporters mediating anion transport solely through C–H HB interactions are also rare.16,29–31 The potential power of XB for anion transport was exemplified by Matile and co-workers who reported that gaseous iodotrifluoromethane facilitates anion transport when bubbled through the vesicle solution.25
Herein we show that simple acyclic XB or HB triazoles (Fig. 1) can efficiently mediate anion transport across lipid bilayer membranes with an unusual anti-Hofmeister selectivity. Importantly, we demonstrate unprecedented activity for XB anionophores with up to two orders of magnitude improvement over the previously reported iodo-perfluorobenzene systems, and reveal their remarkable intrinsic Cl− > OH− selectivity. Iodo- and proto-triazoles are versatile motifs readily accessible via Cu-catalysed azide–alkyne click chemistry,32 and these results demonstrate their potential for applications in synthetic anion transport systems.
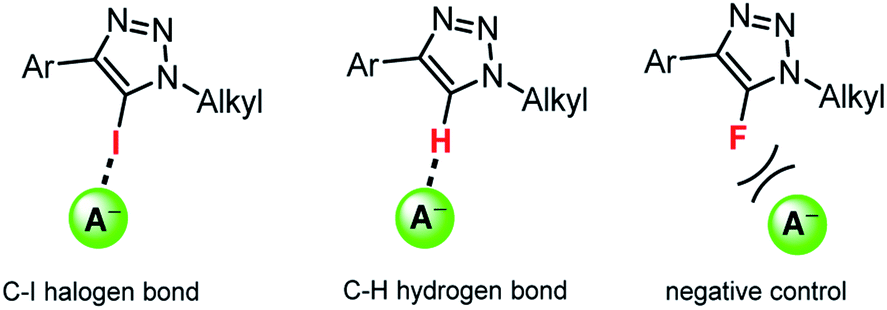 | ||
| Fig. 1 Representative acyclic halogen bonding (XB) and hydrogen bonding (HB) triazole anion transporters, and the analogous fluorotriazole negative controls. | ||
Results and discussion
Synthesis and anion recognition properties
Acyclic 5-iodo/proto/fluoro-1,4-disubstituted-1,2,3-triazoles, of the general form shown in Fig. 1, were prepared as minimalistic scaffolds with which to explore the intrinsic activity and selectivity of the hydrophobic C–I XB- and C–H HB-mediated anion transport processes. The family of compounds spans a range of iodo-triazole derivatives, and their proto-triazole analogues, with varied electron-withdrawing/donating substituents on the aryl group and varying alkyl chain lengths. The library of 5-iodo-1,4-disubstituted-1,2,3-triazoles (compounds 1b–9b) were prepared by Cu-catalysed azide–alkyne cycloaddition of the respective alkyl azide and iodo-aryl-alkyne, in the presence of tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (Scheme 1). The analogous proto-triazoles (compounds 1a–9a) were prepared from the corresponding alkyne. As control compounds, the fluoro-triazole analogues of 1 and 9, namely 1c and 9c, which are unable to form HB or XB interactions, were also prepared from the corresponding iodo-triazole by heating with potassium fluoride. Full synthetic procedures and characterisation are available in the ESI.†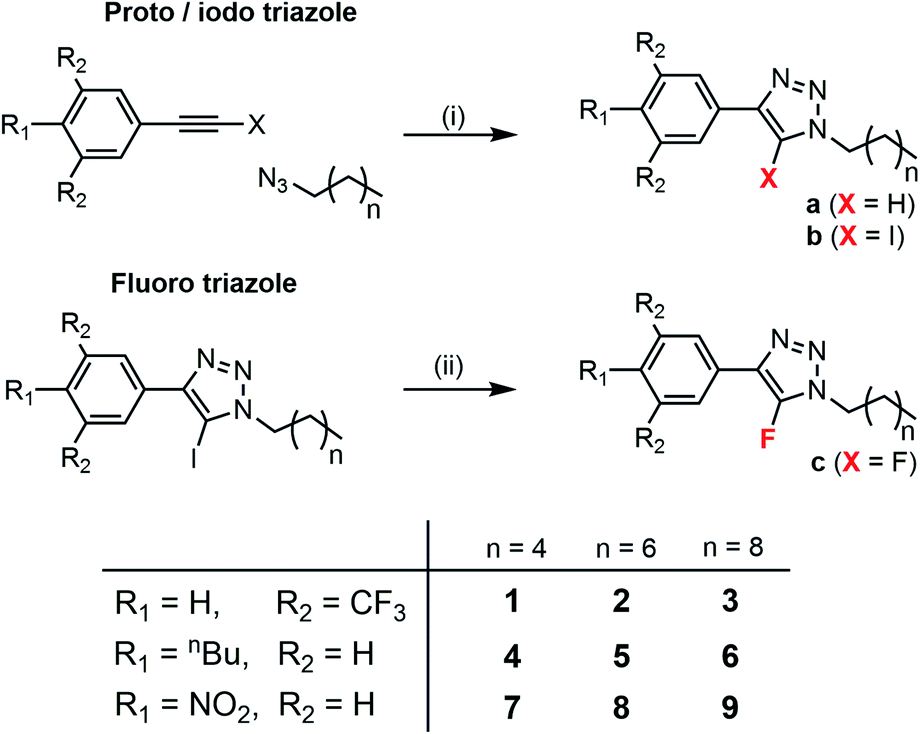 | ||
| Scheme 1 Synthesis of triazole derivatives and compound numbering scheme. Conditions (i) Cu(CH3CN)4PF6, TBTA, THF, N2, rt (ii) KF, CH3CN, μ-wave 180 °C. | ||
The anion recognition capability of representative proto-triazole 1a and iodo-triazole 1b were first investigated by 1H NMR binding titrations with Bu4N+X− in d6-acetone, monitoring the binding induced chemical shift perturbations of the proto-triazole H and adjacent aryl protons. For each anion, the data could be fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm (see ESI†) and the 1:1 stoichiometric association constants determined using the Bindfit program (Table 1).33,34 The data revealed an overall selectivity trend of Cl− > Br− > I− for both 1a and 1b, and approximately one order of magnitude enhancement of halide binding affinity for the XB iodo-triazole receptor 1b compared to HB proto-triazole 1a. For both compounds, no measurable binding of nitrate was observed under these conditions.
:1 binding isotherm (see ESI†) and the 1:1 stoichiometric association constants determined using the Bindfit program (Table 1).33,34 The data revealed an overall selectivity trend of Cl− > Br− > I− for both 1a and 1b, and approximately one order of magnitude enhancement of halide binding affinity for the XB iodo-triazole receptor 1b compared to HB proto-triazole 1a. For both compounds, no measurable binding of nitrate was observed under these conditions.
Transmembrane anion transport activity
The ability of anionophores 1–9a/b to mediate OH−/Cl− transmembrane anion transport was first determined in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine large unilamellar vesicles (POPC LUVs), loaded with 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) and buffered to pH 7.0 in NaCl solution. HPTS is a pH-sensitive fluorophore, which allows for ratiometric determination of the internal pH of the LUVs. A pH gradient was applied across the membrane by addition of a base pulse, followed by addition of the carrier as a DMSO solution (<0.5% v/v). The ability of the anionophore to dissipate the pH gradient by transmembrane OH−/Cl− exchange was determined by recording the change in the HPTS emission, Irel (λem = 510 nm), with time following excitation at λex = 405/465 nm (Fig. 2a). At the end of each experiment, excess detergent (Triton X-100) was added to lyse the vesicles for calibration of the emission intensity. The HPTS assay was used to determine the concentration dependence of the activity of each anion carrier. Fig. 2a shows representative data for the activity of compound 1b at a range of concentrations. The fractional activities (y, the relative intensity at 288 s, immediately prior to lysis) were plotted as a function of concentration (Fig. 2b) and fitted to the Hill equation (eqn (1)). Original data for all other compounds is available in the ESI.†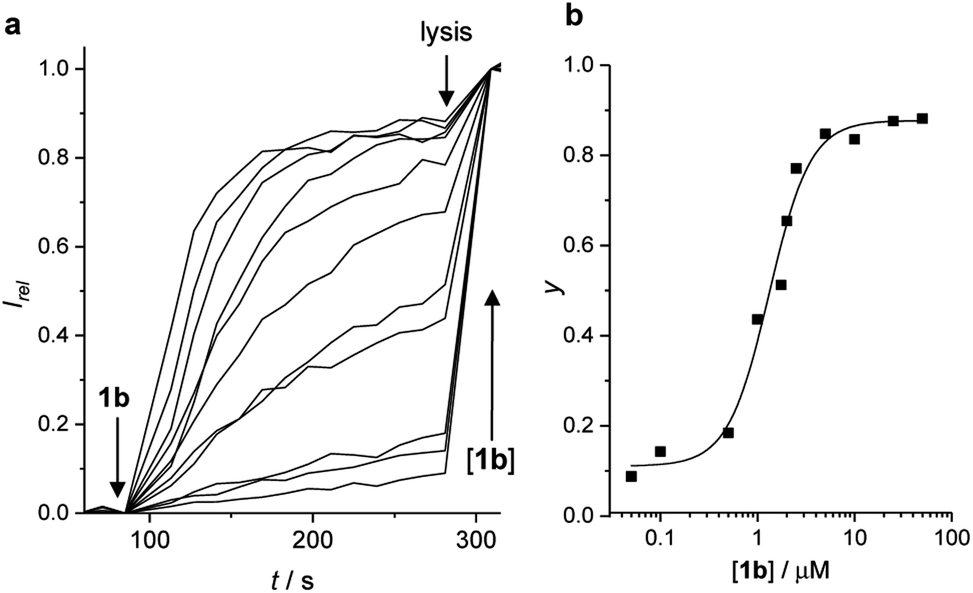 | ||
| Fig. 2 Anion transport by XB transporter 1b in the HPTS assay. (a) Change in ratiometric emission (λem = 510 nm; λex1 = 405 nm, λex2 = 460 nm) upon addition of 1b in DMSO (0.05–50 μM) to POPC vesicles containing 1 mM HPTS, 100 mM internal and external NaCl, buffered with 10 mM HEPES at pH 7.0. A pH gradient is generated by addition of a NaOH base pulse (0.5 M), followed by 1b at t = 0. Vesicle lysis by Triton X-100 calibrates the assay. (b) Dependence on fractional activities (y, the relative intensity at t = 288 s just prior to lysis) on concentration of 1b (black squares), and fit to the Hill equation (black line). | ||
The Hill equation here is used to describe the dependence of the fractional activities y on the nth power of the anionophore concentration x, and facilitates comparison of the relative activities of transporters through an effective concentration value (EC50) required to reach 50% activity in the given assay (eqn (1)). Compatibility with the Hill equation reveals endergonic self-assembly of the active supramolecule, and Hill coefficients n > 1 are indicative of the stoichiometry of the unstable supramolecular assembly that exists in the presence of an excess of uncomplexed anionophores.35
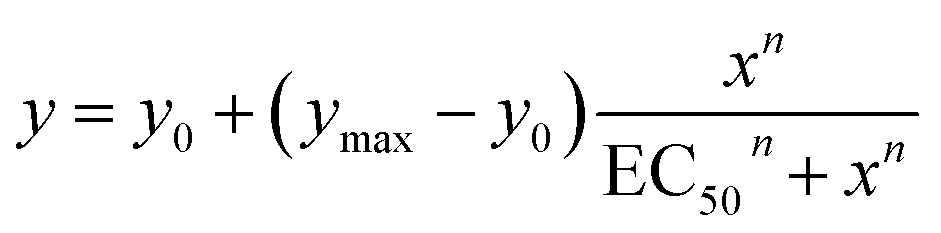 | (1) |
P) values for the proto- and iodo-triazoles are shown in Table 2. clogP values were calculated using the VCCLab software.36
| n = 4 | n = 6 | n = 8 | |||||
|---|---|---|---|---|---|---|---|
| X = H | X = I | X = H | X = I | X = H | X = I | ||
|
a Effective concentration to reach 50% of maximal activity in the HPTS assay, determined from Hill analysis of the relative fluorescence intensity at t = 288 s, immediately prior to vesicle lysis. Experiments conducted in LUVs of POPC (mean diameter 200 nm) loaded with 1 mM HPTS, NaCl (100 mM) and buffered at pH 7.0 with 10 mM HEPES. For compounds of low activity, estimated lower bounds for the EC50 value are given.
b Hill coefficient.
c Calculated logP.
d Insufficient activity for Hill analysis.
|
|||||||
|
1a | 1b | 2a | 2b | 3a | 3b | |
| EC50a/μM | 2.0 (0.1) | 1.3 (0.1) | 0.9 (0.1) | 1.1 (0.1) | >100 | >100 | |
| n | 2.4 (0.3) | 2.1 (0.4) | 3.7 (0.8) | 2.6 (0.4) | —d | —d | |
| clogPc |
4.4 | 5.4 | 5.1 | 6.1 | 5.8 | 6.8 | |
|
|||||||
|
4a | 4b | 5a | 5b | 6a | 6b | |
| EC50a/μM | 6.2 (0.2) | 3.3 (0.2) | >200 | >200 | >200 | >200 | |
| n | 5.6 (0.5) | 2.0 (0.2) | —d | —d | —d | —d | |
| clogPc |
5.7 | 5.7 | 6.5 | 6.3 | 7.2 | 6.3 | |
|
|||||||
|
7a | 7b | 8a | 8b | 9a | 9b | |
| EC50a/μM | 14.8 (0.4) | 1.8 (0.2) | 2.9 (0.2) | 3.6 (0.3) | 2.7 (0.05) | 1.2 (0.1) | |
| n | 5.7 (0.8) | 3.1 (0.7) | 3.6 (0.7) | 2.7 (0.6) | 7.7 (1.2) | 2.0 (0.2) | |
| clogPc |
3.9 | 4.4 | 4.8 | 5.2 | 5.6 | 6.0 | |
In general, the electron withdrawing 3,5-bis(trifluoromethyl)benzene and 4-nitrobenzene substituents led to excellent activity despite the simplicity of the anionophores (compounds 1–3, and 7–9, respectively). The electron deficient halogen bonding transporters 1b and 9b are approximately 3× more active than the most active XB anionophore reported to date, iodoperfluorohexane (EC50 = 3.1 μM);25 and notably over two orders of magnitude more active than the archetypal XB donor iodoperfluorobenzene under comparable assay conditions (EC50 = 260 μM).25,28 Electron donating 4-n-butylbenzene substituents (compounds 4–6) conversely decreased activity, consistent with the expected decreased XB and HB donor strength. The correlation between activity and logP is complex, because transport efficiency is also strongly dependent on other factors including anion binding affinity and molecular size, which are not easily decoupled.37 Nevertheless, the observed trend of maximum activity of compounds with logP of 4–6, and reduced activity observed for compounds with higher and lower lipophilicities, is broadly consistent with previous reports on the role of lipophilicity on HB-mediated anion carriers.38 For compounds in which both XB and HB derivatives fall within these optimum parameters (compounds 1, 4, 7 and 8), a general enhancement of activity of around 2×, and up to 8×, is observed for XB systems over their HB analogues. The most lipophilic transporters (3, 5, 6) are inactive, likely arising from reduced mobility and reorganization of longer chain derivatives within the lipid bilayer.39
Calculation of the electrostatic potential (ESP) surface for methyl-truncated analogues‡ of 1a/b, 4a/b and 7a/b (denoted 1a′/b′, 4a′/b′ and 7a′/b′ respectively) reveals the characteristic iodine-centred sigma-hole40,41 associated with halogen bonding donors, and the accumulation of δ+ surrounding the triazole protons active in hydrogen bonding (Fig. 3). Comparison of the Hammett σ values for each substituent demonstrate that the more electron-withdrawing substituents result in a more positive value for the ESP maximum,42 leading to a stronger XB-/HB-anion intermolecular interaction. This correlates with the observed greater activity of 1a/b and 7a/b over 4a/b (Table 2).
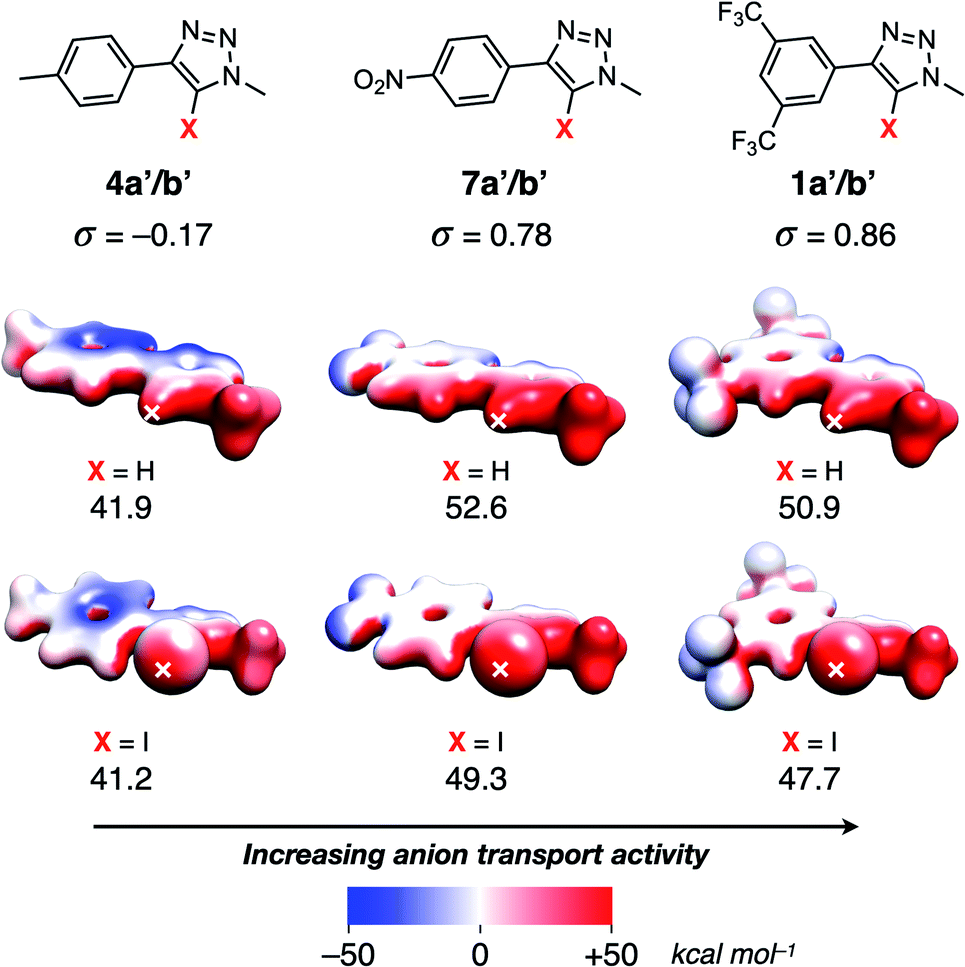 | ||
| Fig. 3 Calculated electrostatic potential surfaces (isovalue = 0.04 a.u.) for truncated transporter ligands 4a′/b′, 7a′/b′ and 1a′/b′ calculated at the SMD(CHCl3)-ωB97X-D/def2-TZVP level of theory. Maximum ESP values are located at the position of each cross, and reported beneath each transporter in kcal mol−1. | ||
Evidence for XB/HB-anion interactions and mobile carrier mechanism
To confirm the role of the C–I XB and C–H HB interactions in the anion transport process, we investigated the anion transport capability of fluoro-triazole derivative 1c, which is unable to form XB or HB interactions through the triazole motif. Inactivity of 1c in the HPTS assay confirmed the role of the XB and HB intermolecular interactions in the transport processes (Fig. 4a). Similar behaviour was observed for nitro-phenyl fluoro-triazole derivative 9c.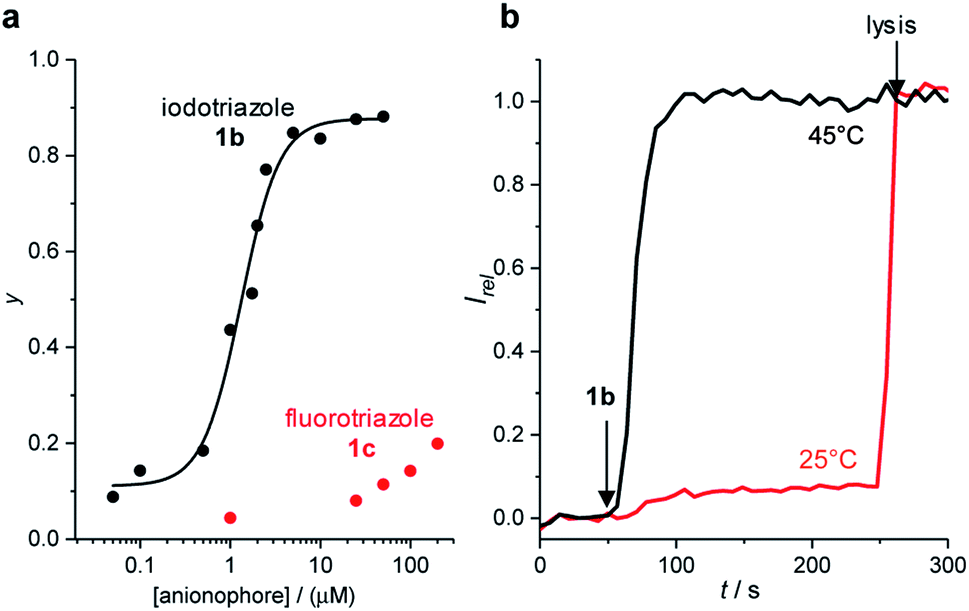 | ||
| Fig. 4 (a) Activity of fluoro-triazole 1c (red) in comparison with iodo-triazole 1b (black) in the HPTS assay. Plot shows dependence on Irel at t = 288 s immediately prior to lysis on anionophore concentration (points) and fit to the Hill equation (line). (b) Temperature dependence of anion transport by 1b (2 μM) in DPPC lipid vesicles in the HPTS assay at 25 °C (black line) and 45 °C (red line). | ||
Inactivity of the transport systems 1–9 reported here in the carboxyfluorescein dye leakage assay rules out non-specific leakage by these systems (ESI†). Replacement of the zwitterionic POPC lipids in the HPTS assay with anionic egg yolk phosphatidylglycerol (EYPG) lipids led to a significant decrease in observed activity, consistent with the requirement for formation of an anionic complex in the rate limiting transport process. Transport activity at 25 °C with XB transporter 1b and dipalmitoylphosphatidylcholine (DPPC) lipids, under otherwise identical conditions, was negligible (Fig. 4b). The lipid gel phase inhibits translation of mobile carriers through the membrane which are otherwise mobile in a fluid lipid phase. The gel to fluid phase transition for DPPC lipids is 41 °C, and repeating the assay above this temperature (45 °C) restored anion transport activity. The observed temperature dependence is indicative of a mobile carrier mechanism, and rules out formation of a membrane-spanning supramolecular channel structure whose activity would be independent of the lipid phase.
Anion selectivity
Dissipation of the transmembrane pH gradient measured in the HPTS assay by the carrier species can in principle occur through either cation (H+/M+) or anion (OH−/A−) antiport (exchange), or H+/A− symport (co-transport) mechanisms. The activities of HB transporter 1a and XB transporter 1b were not affected by iso-osmolar replacement of the external Na+ cation with Li+, K+, Rb+ or Cs+ (see ESI†), indicative of selective anion transport (OH−/A− antiport or H+/A− symport) rather than H+/M+ cation antiport. Further evidence to support the cation-independent transport mechanism was provided by conducting analogous experiments in the presence of sodium gluconate, a large hydrophilic anion (ESI†).10 The absence of detectable transport indicates that, as expected, neither OH−/gluconate antiport or H+/gluconate symport mechanisms are active because of the insurmountable dehydration penalty of gluconate, and also that the alternative H+/Na+ cation antiport process is negligible.OH−/A− antiport and H+/A− symport mechanisms are functionally equivalent and cannot be distinguished through these transport assays. However, the low basicity of triazole derivatives (pKaH ∼ 0–1)43 suggests that H+/A− symport (achieved via triazole protonation and XB-/HB-mediated anion recognition) is unlikely to contribute to any significant extent to the ion transport process at neutral pH. This is consistent with the activity of previously reported XB iodofluoroalkene/arene transporters in the same assay, which do not possess any basic atoms and are therefore most likely operate through an anion antiport mechanism.25 For simplicity, we refer to the transport process from here on in as OH−/A− antiport.
To examine the relative rates of Cl−vs. OH− transport, we repeated the HPTS assay with the addition of carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), a weak acid protonophore at a low concentration (0.25 μM) insufficient to cause activity alone. FCCP transports protons via transmembrane shuttling of both protonated and deprotonated forms of the molecule. Enhancement of activity by FCCP is indicative of a rate limiting electrogenic OH− transport process, because FCCP decouples the anionophore-mediated OH−/Cl− antiport process (Fig. 5a) by facilitating rapid electrogenic H+ transport in an overall coupled Cl−/H+ symport process (Fig. 5b). In this scenario, the observed rate of pH gradient dissipation reports on the rate-limiting electrogenic Cl− transport process. As such, if the activity of a given carrier is invariant to FCCP addition, Cl− transport must therefore be rate limiting in the antiport mechanism (i.e. slower than electrogenic OH− transport). The EC50 values for compounds 1a and 1b with FCCP are shown in Table 3. The activity of HB anionophore is invariant to FCCP addition, demonstrating rapid OH− transport and rate limiting Cl− transport by 1a. In contrast, a four-fold increase in activity for XB transporter 1b is observed in the presence of FCCP, revealing rate limiting OH− transport in its absence. The ratio of EC50 values in the absence and presence of FCCP (Table 3, column 4) therefore reports on the relative Cl− > OH− selectivity of the system (for the given conditions and ion concentration gradient), and reveals the unusual Cl− > OH− selectivity of the XB transporter 1b. Notably, transporter 1b in the presence of FCCP achieved nanomolar activity (EC50 = 300 nM) in the HPTS assay, which to the best of our knowledge is the highest activity reported to date for an XB anion transporter under comparable experimental conditions. This highlights the potential of iodo-triazoles as synthetically accessible and versatile motifs for developing more efficient anion transport systems.
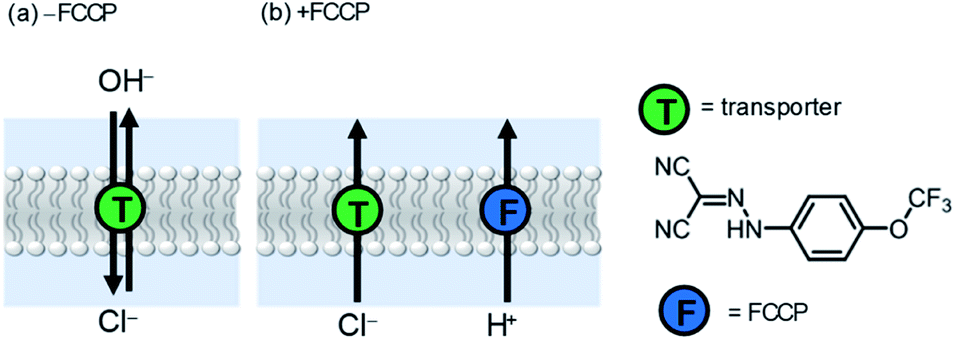 | ||
| Fig. 5 Ion transport mechanisms for anionophore-mediated dissipation of a transmembrane pH gradient (a) in the absence and (b) presence of cyanide-p-trifluoromethoxyphenylhydrazone (FCCP). | ||
In the presence of FCCP, the observed transport kinetics report on the rate limiting A− transport for a given anionophore. This allows the relative selectivity of both XB and HB transporters for each anion to be investigated, enabling direct comparison of the relative rates of electrogenic A− transport (Fig. 6). HB transporter 1a exhibits an overall selectivity profile of Cl− > Br− > I− > NO3− (Fig. 6, black data). This selectivity is remarkable because in general hydrogen bonding anion transporters exhibit Hofmeister selectivity, whereby more weakly hydrated anions are selectively transported across the membrane due to ease of desolvation.44–47 The analogous XB anionophore 1b exhibits a selectivity profile of NO3− > Cl− > Br− > I−. Comparison of the relative activities for both transporters in the absence of FCCP allows placement of OH− into the overall selectivity trends, revealing OH− > Cl− > Br− > I− > NO3− behaviour for 1a and NO3− > Cl− > Br− > OH− > I− behaviour for 1b. Similar selectivity trends for analogous compounds 9a and 9b were also observed in these assays (see ESI†).
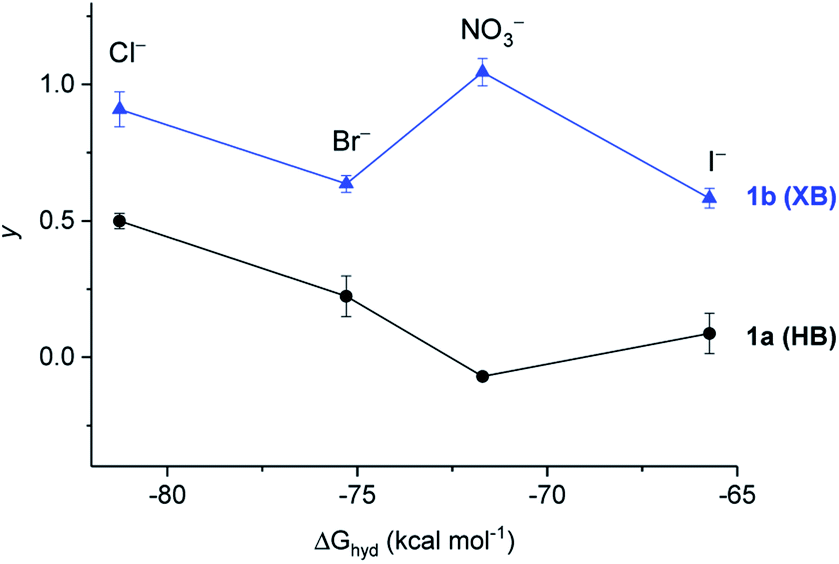 | ||
| Fig. 6 Dependence of the activity of 1a (black circles) and 1b (blue triangle) on the hydration energy ΔGhyd of extravesicular anions (100 mM NaX, inside 100 mM NaCl) in the presence of 0.25 μM protonophore FCCP, reporting on relative rate of electrogenic X− transport. Error bars represent standard deviations. | ||
Overall these transport experiments reveal (i) enhanced activity of halogen bonding anionophores vs. hydrogen bonding analogues across all anions, (ii) unusual Cl− > OH− selectivity for the halogen bonding anionophores and (iii) anti-Hofmeister bias for halide transport, demonstrating the dominant role of ion binding to the anionophore and correlating with NMR determined anion binding affinities (see Table 1).§
Computational studies into anionophore–anion interactions
Computational studies were used to probe the binding energies and structures of the anionophore–anion complexes. Calculation48–53 of the relative binding enthalpies for both 1:1 and 2:1 model anionophore–anion complexes in CHCl3 implicit solvent as a membrane-mimetic environment reveals that ΔHbind(2:1) > 2 × ΔHbind(1:1) (Fig. 7), and that 2:1 binding is enhanced by π-stacking between the arenes (see ESI†).¶ Bidentate binding was found to be preferred in the model 1:1 HB system 1a′ involving hydrogen bonds from both the alkyl C(sp3)–H1 and triazole C(sp2)–H2 (Fig. 7a). This finding is in line with the result of the ESP calculations in Fig. 3. Monodentate XB interactions are observed with 1b′ (Fig. 7b). Interestingly, our results indicate that in the 2:1 complex, four HB C–H⋯anion interactions per anion exists for 1a′ (Fig. 7a), while only one XB C–I⋯anion interaction is present for 1b′ (Fig. 7b).
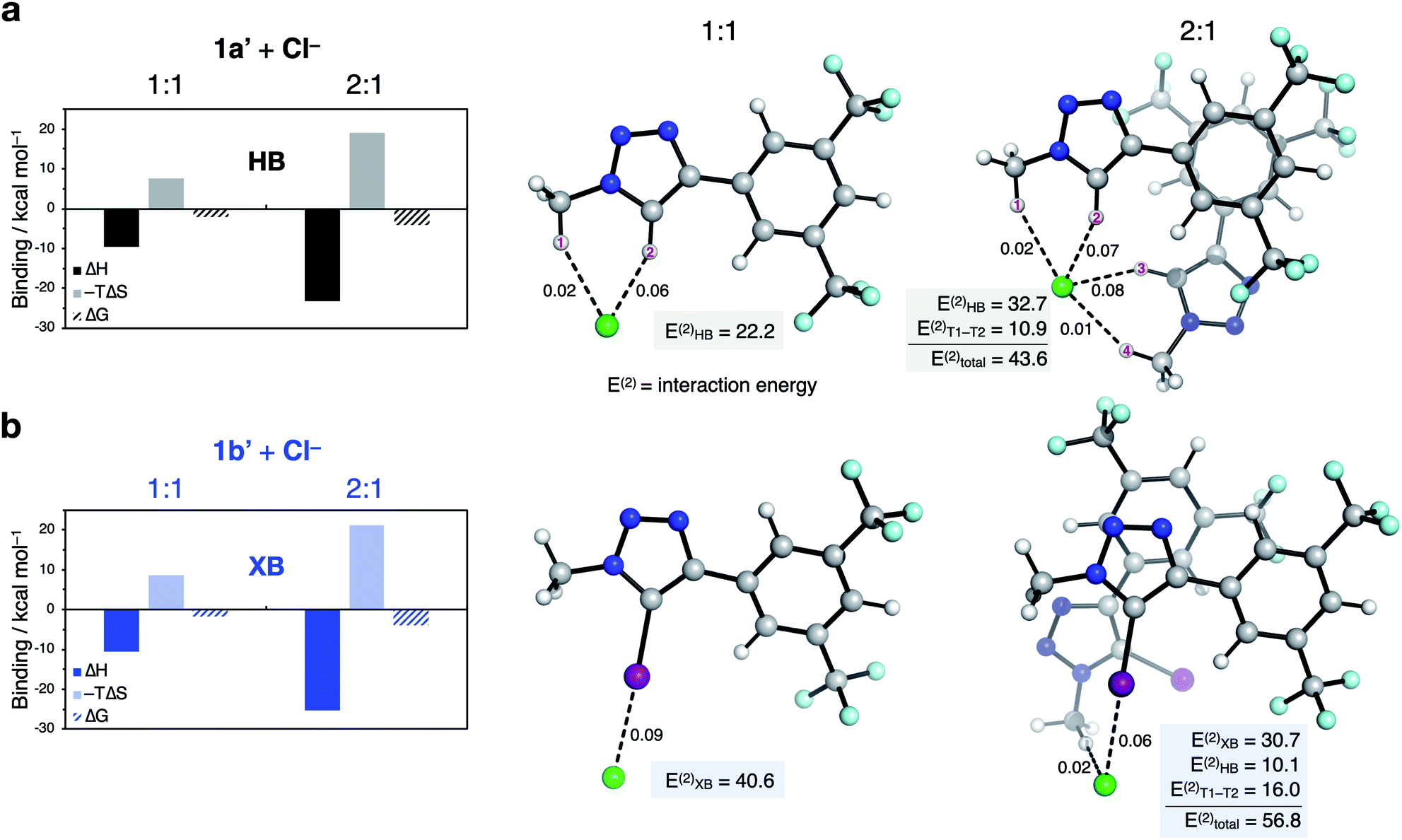 | ||
| Fig. 7 Characterisation of 1:1 and 2:1 binding of (a) 1a′ and (b) 1b′ to chloride. Enthalpies, entropies and free energies of binding (kcal mol−1) were calculated at the [SMD(CHCl3)-DLPNO-CCSD(T)/def2-TZVP (ma-def2-TZVP on Cl, I)//ωB97X-D3/def2-SVP (ma-def2-SVP on Cl, I)] level of theory. Mayer bond orders (dashed lines), their associated HB/XB bond energies (E(2)HB/XB), and interaction energy between ligands (E(2)T1–T2) in kcal mol−1 were calculated at the [NBO/SMD(CHCl3)-ωB97X-D3/def2-SVP (ma-def2-TZVP on Cl, I)//ωB97X-D3/def2-SVP (ma-def2-SVP on Cl, I)] level of theory. Free energies were calculated using a quasi-RRHO approximation53 and corrected for a 1 M standard state at 298.15 K. | ||
Additionally, bond order and second-order perturbation theory analysis, which allows the quantification of bond energies (E(2))54 indicates that addition of the second transporter ligand weakens the total XB interactions compared with the 1:1 complex (E(2)XB = 40.6 vs. 30.7 kcal mol−1). However, this is partially compensated by dispersion interactions between the two transporter ligands (ET1–T2 = 16.0 kcal mol−1) and a C–H interaction (10.1 kcal mol−1), which leads to an overall larger stabilisation energy (E(2)total = 56.8 kcal mol−1).
Analysis of the partial charges on each anion in the 1:1 and 2:1 binding modes suggest a correlation between the rate of transport and the ability of the transporter to stabilise the anion charge (Fig. 8). For example, for Cl−, Br− and I−, the negative charge is more effectively delocalised over the XB anionophore than the HB analogue. This is consistent with previous results from Donor K-edge X-ray Absorption Spectroscopy experiments which revealed significant covalency in the XB interaction between related iodo-triazole XB donors and chloride anions (comparable to that in transition metal chloride complexes), in contrast to proto-triazole analogues with negligible charge transfer contribution to the HB interaction.55 Improved charge delocalisation over the anion-triazole complex is expected to decrease the barrier to transport across the hydrophobic membrane (Fig. 8), and this is consistent with the experimentally observed selectivity trend of the XB anionophores enhancing rates of anion transport over the analogous HB system. A different behaviour is observed for nitrate, where the observed efficient nitrate transport does not correlate with the increased calculated partial charge. We suggest that this behaviour is due to the greater number of thermally-accessible conformations for the binding of the trigonal nitrate anion, each of them having a different charge distribution. In this case, the most stable conformer in solution is likely not the most active for membrane transport (see ESI†).
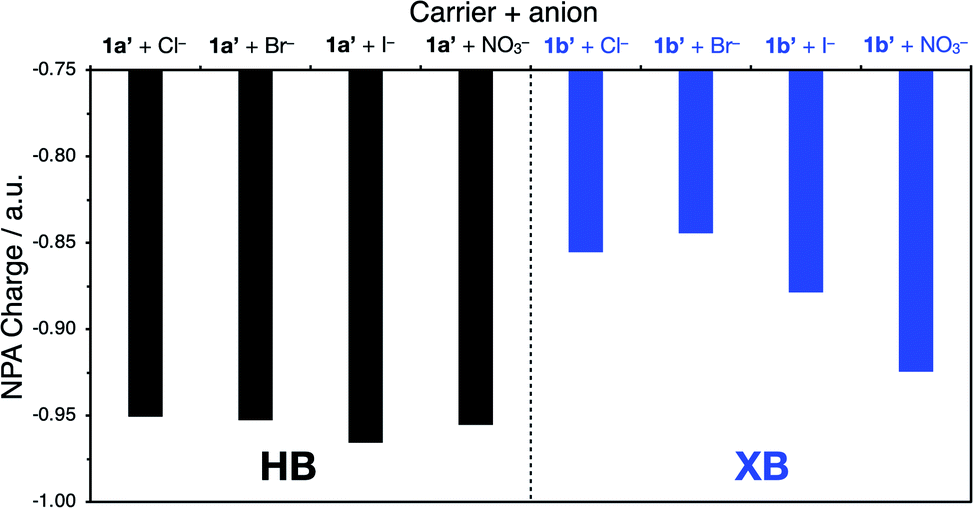 | ||
| Fig. 8 Partial charges on each anion for 1:1 anionophore–anion complexes of 1a′ and 1b′, calculated using natural population analysis (NPA) at the [NBO/SMD(CHCl3)-ωB97X-D3/def2-SVP (ma-def2-SVP on anion)] level of theory. | ||
Conclusions
We have shown that acyclic halogen bonding iodo-triazoles can efficiently transport anions across lipid bilayer membranes, with enhanced activity in comparison to hydrogen bonding proto-triazole analogues. Despite the relative simplicity of the design, the iodo-triazole systems are the most active halogen bonding anionophores reported to date. Anion transport experiments reveal unusual anti-Hofmeister selectivities, and a remarkable intrinsic Cl− > OH− selectivity for the halogen bonding systems. Experimental and computational studies provide further insight into the binding modes of the anion–anionophore complexes. Calculations demonstrate that the strength of the non-covalent interactions are sufficient to overcome the entropic cost of 2:1 complex formation, and reveal the remarkable ability of the halogen bonding anionophores to delocalise the anion charge over the complex, which correlates with the enhanced anion transport capability observed by experiment. This work demonstrates the utility of the synthetically accessible and highly versatile XB and HB triazole motif for anionophore design. The observed Cl− > OH− selectivity of the iodotriazole derivatives also suggest that such motifs may provide the starting point for designing novel ionophores with enhanced selectivity for potential future application in channelopathy therapeutics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. S. thanks the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1), generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. A. J. S. also thanks the Oxford-Radcliffe Scholarship for a studentship. A. J. S. and F. D. thank the EPSRC Centre for Doctoral Training for Theory and Modelling in Chemical Sciences (EP/L015722/1) for providing access to the Dirac cluster at Oxford. This work used the Cirrus UK National Tier-2 HPC Service at EPCC (http://www.cirrus.ac.uk) funded by the University of Edinburgh and EPSRC (EP/P020267/1). M. J. L. acknowledges funding from the Royal Society. F. D. and M. J. L. thank the John Fell Oxford University Press Research Fund for financial support. M. J. L. is a Royal Society University Research Fellow.Notes and references
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- S. Matile, A. V. Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- B. Díaz de Greñu, P. I. Hernández, M. Espona, D. Quiñonero, M. E. Light, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem.–Eur. J., 2011, 17, 14074–14083 CrossRef PubMed.
- B. Shen, X. Li, F. Wang, X. Yao and D. Yang, PLoS One, 2012, 7, e34694 CrossRef CAS PubMed.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed.
- V. Soto-Cerrato, P. Manuel-Manresa, E. Hernando, S. Calabuig-Fariñas, A. Martínez-Romero, V. Fernández-Dueñas, K. Sahlholm, T. Knöpfel, M. García-Valverde, A. M. Rodilla, E. Jantus-Lewintre, R. Farràs, F. Ciruela, R. Pérez-Tomás and R. Quesada, J. Am. Chem. Soc., 2015, 137, 15892–15898 CrossRef CAS PubMed.
- H. Li, H. Valkenier, L. W. Judd, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurček, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24–32 CrossRef CAS PubMed.
- X. Wu, L. W. Judd, E. N. W. Howe, A. M. Withecombe, V. Soto-Cerrato, H. Li, N. Busschaert, H. Valkenier, R. Pérez-Tomás, D. N. Sheppard, Y.-B. Jiang, A. P. Davis and P. A. Gale, Chem, 2016, 1, 127–146 CAS.
- S. Ohkuma, T. Sato, M. Okamoto, H. Matsuya, K. Arai, T. Kataoka, K. Nagai and H. H. Wasserman, Biochem. J., 1998, 334, 731–741 CrossRef CAS PubMed.
- P. I. Hernández, D. Moreno, A. A. Javier, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem. Commun., 2012, 48, 1556–1558 RSC.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed.
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem.–Eur. J., 2016, 22, 185–192 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- C. J. Massena, D. A. Decato and O. B. Berryman, Angew. Chem., Int. Ed., 2018, 57, 16109–16113 CrossRef CAS PubMed.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119–4129 CrossRef CAS PubMed.
- A. Vargas Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef CAS PubMed.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed.
- A. Vargas Jentzsch and S. Matile, J. Am. Chem. Soc., 2013, 135, 5302–5303 CrossRef CAS PubMed.
- C. Ren, X. Ding, A. Roy, J. Shen, S. Zhou, F. Chen, S. F. Y. Li, H. Ren, Y. Yan Yang and H. Zeng, Chem. Sci., 2018, 9, 4044–4051 RSC.
- L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS PubMed.
- J. Shang, W. Si, W. Zhao, Y. Che, J.-L. Hou and H. Jiang, Org. Lett., 2014, 16, 4008–4011 CrossRef CAS PubMed.
- S. Chen, S. Zhang, C. Bao, C. Wang, Q. Lin and L. Zhu, Chem. Commun., 2016, 52, 13132–13135 RSC.
- H. Valkenier, O. Akrawi, P. Jurček, K. Sleziaková, T. Lízal, K. Bartik and V. Šindelář, Chem, 2019, 5, 429–444 CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- BindFit v0.5 | Supramolecular, http://app.supramolecular.org/bindfit/.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- S. Bhosale and S. Matile, Chirality, 2006, 18, 849–856 CrossRef CAS PubMed.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed.
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Félix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC.
- V. Saggiomo, S. Otto, I. Marques, V. Félix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC.
- S. J. Edwards, I. Marques, C. M. Dias, R. A. Tromans, N. R. Lees, V. Félix, H. Valkenier and A. P. Davis, Chem.–Eur. J., 2016, 22, 2004–2011 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J.-L. M. Abboud, C. Foces-Foces, R. Notario, R. E. Trifonov, A. P. Volovodenko, V. A. Ostrovskii, I. Alkorta and J. Elguero, Eur. J. Inorg. Chem., 2001, 2001, 3013–3024 CrossRef.
- S. K. Berezin and J. T. Davis, J. Am. Chem. Soc., 2009, 131, 2458–2459 CrossRef CAS PubMed.
- S. K. Berezin, Supramol. Chem., 2013, 25, 323–334 CrossRef CAS.
- Y. Yang, X. Wu, N. Busschaert, H. Furuta and P. A. Gale, Chem. Commun., 2017, 53, 9230–9233 RSC.
- X. Wu, J. R. Small, A. Cataldo, A. M. Withecombe, P. Turner and P. A. Gale, Angew. Chem., Int. Ed., 2019, 58, 15142–15147 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis, and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01467b |
| ‡ Truncated alkyl chains to methyl were used for computational studies. This does not affect the calculated ESP (see ESI† for details). |
| § Detailed comparison between the measured association constants in solution and transport kinetics in membranes should be avoided, given that the observed rate of transport is also dependent on many other factors including desolvation, anionophore–ion complex mobility in the membrane, and competing binding interactions of the transporter with the phosphate head groups.37 |
| ¶ Discussed here are the lowest energy conformers obtained from the sampling procedure described in the ESI.† |
| This journal is © The Royal Society of Chemistry 2020 |