
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-activated Rh–Zr mixed oxide as a nonhazardous cocatalyst for photocatalytic hydrogen evolution†
Toshio
Nishino
a,
Masaki
Saruyama
 *b,
Zhanzhao
Li
c,
Yoshie
Nagatsuma
d,
Mamiko
Nakabayashi
e,
Naoya
Shibata
e,
Taro
Yamada
d,
Ryo
Takahata
b,
Seiji
Yamazoe
f,
Takashi
Hisatomi
g,
Kazunari
Domen
gh and
Toshiharu
Teranishi
*b
*b,
Zhanzhao
Li
c,
Yoshie
Nagatsuma
d,
Mamiko
Nakabayashi
e,
Naoya
Shibata
e,
Taro
Yamada
d,
Ryo
Takahata
b,
Seiji
Yamazoe
f,
Takashi
Hisatomi
g,
Kazunari
Domen
gh and
Toshiharu
Teranishi
*b
aGraduate School of Science and Technology, Nara Institute of Science and Technology, Ikoma, Nara 630-0192, Japan
bInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: saruyama@scl.kyoto-u.ac.jp; teranisi@scl.kyoto-u.ac.jp
cDepartment of Chemistry, Graduate School of Science, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
dDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
eInstitute for Engineering Innovation, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
fDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
gResearch Initiative for Supra-Materials, Shinshu University, 4-17-1, Wakasato, Nagano, 380-8553, Japan
hOffice of University Professor, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 19th June 2020
Abstract
Efficient, robust and environmentally friendly cocatalysts for photocatalysts are important for large-scale solar hydrogen production. Herein, we demonstrate that a Rh–Zr mixed oxide is an efficient cocatalyst for hydrogen evolution. Impregnation of Zr and Rh precursors (Zr/Rh = 5 wt/wt%) formed RhZrOx cocatalyst particles on Al-doped SrTiO3, which exhibited 31× higher photocatalytic water-splitting activity than a RhOx cocatalyst. X-ray photoelectron spectroscopy proved that the dissociation of Cl− ions from preformed Rh–Cl–Zr–O solid led to formation of the active phase of RhZrOx, in which the Zr/Rh ratio was critical to high catalytic activity. Additional CoOx loading as an oxygen evolution cocatalyst further improved the activity by 120%, resulting in an apparent quantum yield of 33 (±4)% at 365 nm and a long durability of 60 h. Our discovery could help scale up photocatalytic hydrogen production.
Introduction
Photocatalytic water-splitting is a promising technology for sustainable hydrogen production.1,2 Various photocatalysts, such as TiO2, SrTiO3, TaON, GaN:ZnO, Ta3N5 and Y2Ti2O5S2, have been developed for effective utilization of solar energy.3–8 Developing cocatalysts for photocatalysis is equally indispensable for improving photocatalytic activity;9 this is because the cocatalysts promote extraction of excited charges from the photocatalysts and reduce the overpotential of the water-splitting reaction. Noble metals, particularly platinum group metals, are widely used for the hydrogen evolution reaction (HER); they reduce H2O because of the metals' high activity and stability.10 However, noble metals also catalyze backward reactions, such as O2 photoreduction and H2O formation, from evolved H2 and O2.11 Suppressing the backward reactions is central for developing an efficient HER cocatalyst. Based on this concept, Pt@SiO2 core@shell,12 Rh@Cr2O3 core@shell11,13 and Rh–Cr mixed oxide (RhCrOx)14 cocatalysts have been developed. In these systems, H2O can access the noble metal surface, whereas oxygen access is prevented to dramatically suppress the backward reactions. Although both Rh@CrOx and RhCrOx are commonly used because of their excellent activity, long-duration photo-irradiation causes dissolution of Cr6+ ions,15 resulting in not only decreased catalytic activity but also environmental damage and health hazards. To meet increasing demands on solar-to-energy conversion, many scientists are trying to replace hazardous elements in solar active materials, as observed in organic–inorganic lead halide perovskite solar cells (e.g., replace Pb with Sn or Bi).16 Similarly, alternatives to Cr are necessary for large-scale, environmentally friendly photocatalytic water-splitting systems.17In this paper, we demonstrate that a novel Rh–Zr mixed oxide (RhZrOx) is an efficient HER cocatalyst for an Al-doped SrTiO3 (SrTiO3:Al) photocatalyst. SrTiO3:Al is the best water-splitting photocatalyst under UV irradiation to date,18 and can work in sunlight because of the high apparent quantum yield (AQY) in the UV region. Therefore, we chose SrTiO3:Al synthesized by the flux-mediated method15 as a model photocatalyst to explore new efficient cocatalysts. Surface characterizations of the cocatalysts using HRTEM, STEM-EDS and XPS indicate that the active RhZrOx phase forms by activation of the Rh–Cl–Zr–O mixed solid. A systematic study of loading conditions and electrocatalytic activity assessments of the cocatalyst uncovered the effect of Zr in the cocatalyst. Our novel, robust and nonhazardous Zr-based HER cocatalysts will be useful for large-scale solar hydrogen production.
Results and discussion
To find a new partner metal for Rh, we conducted element screening experiments (Fig. S1†). We chose impregnation to deposit cocatalysts on SrTiO3:Al because impregnation is the easiest way to test a number of combinations without varying other experimental conditions. We fixed the weight ratio of metal to Rh (0.1 wt% vs. SrTiO3:Al) at 5. Of 18 elements except for Cr, only five (Zr, Fe, Sm, La and Ce) showed higher activity than Rh alone. Among them, Zr exhibited outstanding H2 evolution activity, 3.6× higher than the next most active element, Fe. The H2/O2 evolution molar ratio was approximately 2, indicating efficient overall water splitting. Zr is a nonhazardous element and its oxide is widely used in functional ceramics for bio-related applications. Therefore, Zr is a promising environmentally friendly alternative to Cr.Then we optimized various parameters of RhZrOx cocatalysts on SrTiO3:Al. We first varied the Zr/Rh weight ratio from 0 to 7 at a constant weight percent Rh of 0.1 wt% vs. SrTiO3:Al (Fig. 1a). The catalytic activity increased in accordance with increasing Zr/Rh weight ratio from 0 to 5, and showed the highest H2 evolution rate of 249 μmol h−1 at Zr/Rh = 5 wt/wt%, which is 31× higher than RhOx/SrTiO3:Al (Zr/Rh = 0). At Zr/Rh < 3 wt/wt%, the H2 evolution rate drastically decreased after 1.5 h, indicating a Zr/Rh weight ratio >3 improves the catalyst's stability. Although the sample synthesized at Zr/Rh = 7 wt/wt% was stable, the overall activity was less than half (110 μmol h−1) that at Zr/Rh = 5 wt/wt%. We also optimized the total quantity of cocatalyst at a fixed Zr/Rh weight ratio of 5 wt/wt% (Fig. 1b). When the loading quantity of cocatalyst was less than standard (Rh = 0.1 and Zr = 0.5 wt%), the H2 evolution activity decreased considerably (22 μmol h−1) because of the insufficient number of water reduction sites. However, loading a larger quantity of cocatalyst (Rh = 0.2 and Zr = 1.0 wt%) only slightly lessened the activity (221 μmol h−1). Based on these results, the optimum quantity of cocatalyst for SrTiO3:Al is Rh = 0.1 wt% and Zr = 0.5 wt%.
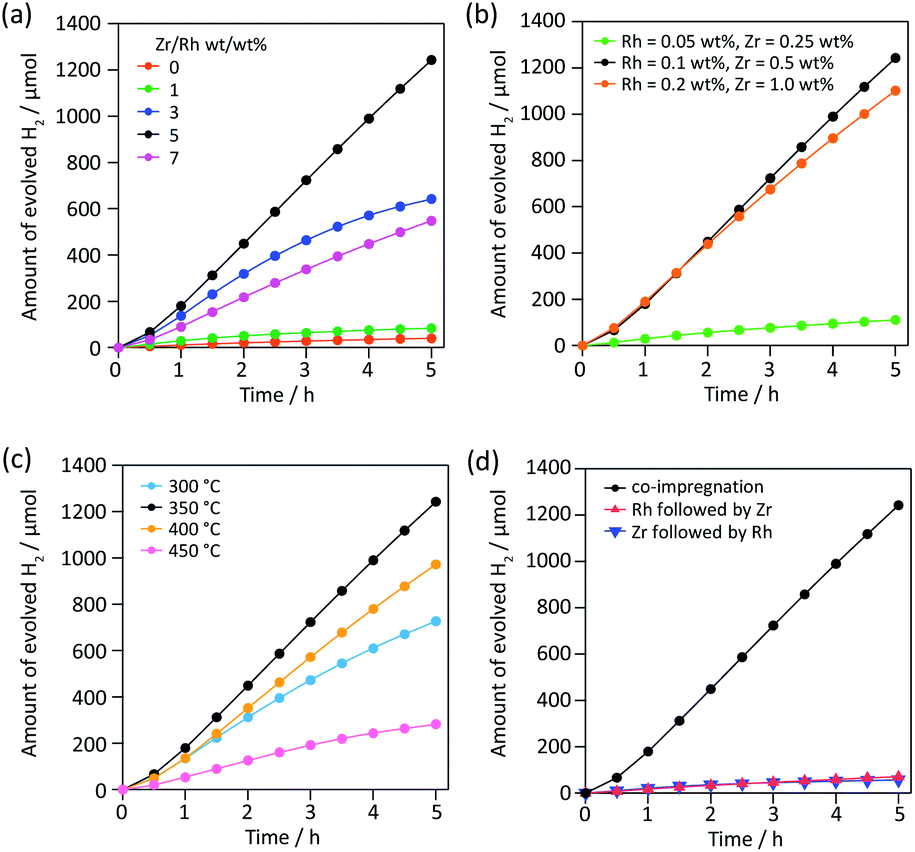 | ||
| Fig. 1 Photocatalytic activities of RhZrOx/SrTiO3:Al synthesized in various experimental conditions. The quantity of evolved H2 of RhZrOx/SrTiO3:Al synthesized (a) at various Zr/Rh weight ratios (Rh = 0.1 wt%), (b) at various quantities of Rh (Zr/Rh = 5 wt/wt%), (c) at various annealing temperatures (Rh = 0.1, Zr = 0.5 wt%) and (d) by co-impregnation and sequential impregnation (Rh = 0.1, Zr = 0.5 wt%). Reaction conditions: catalyst, 10 mg; solution, 20 mL of H2O; light source, 300 W Xe lamp (λ = 300–450 nm). | ||
Then we investigated the effect of calcination temperature in the range of 300–450 °C. RhZrOx/SrTiO3:Al calcined at 350 °C showed the highest H2 evolution (Fig. 1c). A calcination temperature of 300 °C is too low to transform ZrOCl2·8H2O into ZrO2 (Fig. S2 and Table S1†). The Cl/Zr molar ratio decreases with increasing the calcination temperature (Table S1†), indicating that higher calcination temperature assists the formation of ZrO2. SEM images revealed that the cocatalyst size increased from several nanometres to hundreds of nanometres when we calcined the sample at a temperature higher than 400 °C, which decreased the number of active sites on the photocatalyst (Fig. S3†). The order of impregnation of two metal species is important, because the nanostructure of the cocatalyst affects the catalytic activity.11 We compared a co-impregnated (Rh and Zr precursors mixed and loaded simultaneously) with sequentially impregnated (Rh and Zr precursors loaded sequentially) photocatalyst. The co-impregnated photocatalyst showed much higher activity than the sequentially impregnated photocatalyst (Fig. 1d), implying that the Rh–Zr mixed oxide was the active species for overall water splitting rather than the mixture of RhOx and ZrOx. Fig. S4† summarizes all aforementioned catalytic activities including O2 evolution activity.
Next, we characterized the optimized RhZrOx cocatalysts on SrTiO3:Al. We conducted HRTEM to observe the hetero-interface between RhZrOx and SrTiO3:Al phases. We observed no appreciable spots in a FFT image from the cocatalyst region (light-blue square), whereas the SrTiO3:Al region (orange square) exhibited spots that are attributable to crystalline SrTiO3:Al (Fig. 2a), which indicates that the RhZrOx cocatalyst is amorphous. STEM-EDS elemental mapping analysis revealed that the elements Rh, Zr and O coexist in the cocatalysts and uniformly disperse on the photocatalysts (Fig. 2b).
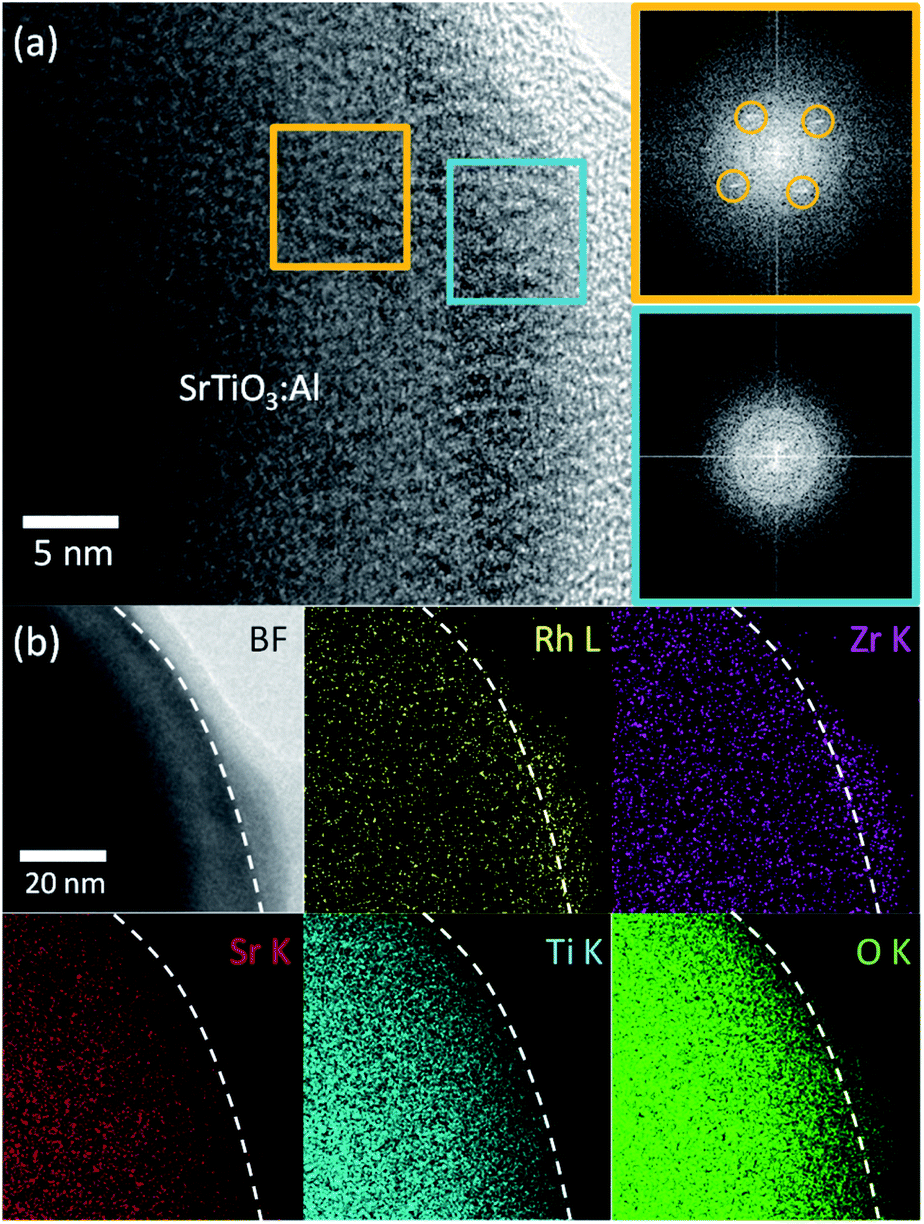 | ||
| Fig. 2 Electron microscopy analyses of Rh–Zr oxide cocatalyst on SrTiO3:Al. (a) HRTEM image and FFT images of RhZrOx/SrTiO3:Al at the interface. Outer bright layer: carbon-related contamination formed during observation. Orange and light-blue squares: SrTiO3:Al and cocatalyst, respectively. (b) STEM-EDS elemental mapping images of RhZrOx/SrTiO3:Al at the interface. White broken lines: surface of SrTiO3:Al. | ||
For loading cocatalysts, we used RhCl3·3H2O and ZrOCl2·8H2O as Rh and Zr precursors, respectively. Considering the exceptional stability of anhydrous RhCl3,19 Cl may contaminate the Rh–Zr mixed oxide cocatalysts. At the impregnation conditions (350 °C for 1 h), ZrOCl2·8H2O was mostly converted into ZrO2 (Fig. S2†), whereas RhCl3·3H2O released only H2O to give anhydrous RhCl3 (Fig. S5†), which is stable until 600 °C in air.19 Thus Cl should exist in the RhZrOx cocatalyst; however, the XRD measurement for RhZrOx solid (Zr/Rh = 5 wt/wt%) did not indicate any metal chloride peaks (Fig. S6†). EDS analysis cannot indicate the coexistence of Rh and Cl, because the characteristic X-ray energies from Rh-L and Cl-K largely overlap (2.6–2.9 keV). Therefore, we used XPS to confirm the presence of Cl in the cocatalyst, because the Rh 3d and Cl 2p peaks separately appear at approximately 310 and 198 eV, respectively. The XPS spectra of RhZrOx/SrTiO3:Al showed Cl 2p peaks along with Rh 3d and Zr 3d peaks (Fig. 3), indicating the presence of Cl in the cocatalyst. Rh 3d peaks have an intermediate feature between Rh2O3 and RhCl3·3H2O, suggesting that the Rh is partially oxidized to a Rh–Cl–O species. Zr 3d and Cl 2p peaks can be assigned to Zr4+ of ZrO2 and Cl− of RhCl3·3H2O, respectively. These results indicate the formation of Rh–Cl and Zr–O bonds. In addition, water-washed RhZrOx/SrTiO3:Al still has clear Cl 2p peaks along with Rh 3d and Zr 3d peaks, which is consistent with the intrinsic insolubility of anhydrous RhCl3 in water. We also applied Ar bombardment to remove a several-nanometre surface extract of the samples to obtain the XPS spectra in the inner regions of the cocatalyst.20 We detected three elements after Ar bombardment, indicating they were uniformly distributed in the cocatalyst. Thus, as-prepared cocatalyst can be described as a Rh–Cl–Zr–O solid. Because the Ar bombardment sometimes induces an XPS peak shift, we do not discuss the valence state of the elements in these experiments.
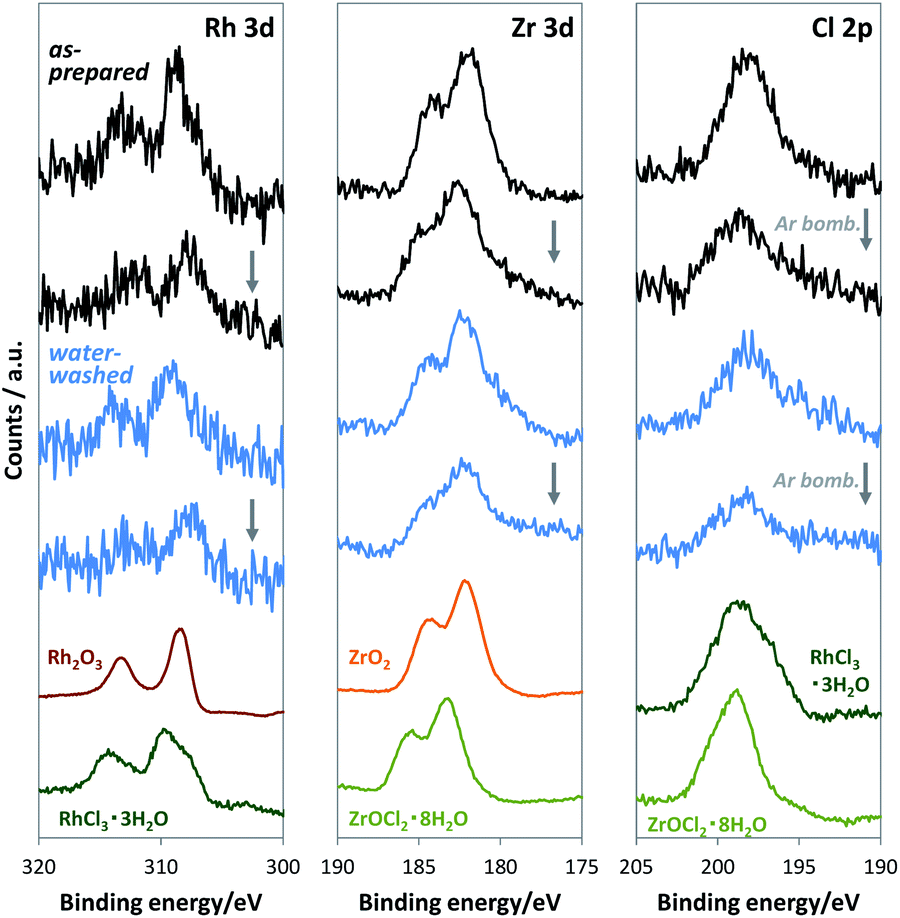 | ||
| Fig. 3 XPS spectra of RhZrOx/SrTiO3:Al and reference materials at the energy ranges of (left) Rh 3d, (middle) Zr 3d and (right) Cl 2p. Black and blue spectra: as-prepared and water-washed samples, respectively, before and after Ar bombardment. | ||
To understand why Rh–Cl–Zr–O solid-loaded SrTiO3:Al exhibits high catalytic activity and durability, we investigated the structural change of Rh–Cl–Zr–O during the photocatalytic reaction. First, we examined the valence state of Rh during the photocatalytic reaction by in situ XANES measurements (Fig. S7†). We obtained XANES spectra every 11 min over the course of a 200 min photocatalytic reaction. The Rh K-edge spectra indicates that the valence of Rh retained nearly a trivalent state, which in turn indicates that the photo-excited electrons in SrTiO3:Al do not reduce Rh3+ into Rh0, as observed in previously reported RhCrOx cocatalysts.21
We conducted XPS measurements of RhZrOx/SrTiO3:Al after a 5 h reaction to further clarify the valence and composition changes of the elements in the cocatalyst (Fig. 4a). The Rh 3d5/2 peak at 309 eV showed no considerable shift after the reaction, which is consistent with our in situ XANES results. The Zr 3d5/2 peak at 182 eV was also unchanged after the reaction. However, the Cl 2p peak completely disappeared after the reaction, indicating the dissolution of Cl− ions into the water during the reaction. Notably, the Rh 3d peak intensity considerably decreased after Ar bombardment, whereas the Zr 3d peak intensity remained unchanged. We did not observe the Cl 2p peak even inside the cocatalyst. ICP measurements revealed that the Zr/Rh weight ratio in the entire cocatalyst was retained after the reaction (Table S2†). These results strongly suggest that the Rh3+ ions are concentrated at the outer surface of the cocatalysts during the reaction without eluting into the water. This noteworthy concentration phenomenon does not result from the photoreduction of once-eluted Rh3+ on SrTiO3:Al; this is because in situ XANES and XPS measurements revealed that Rh3+ was not reduced during the photocatalysis.
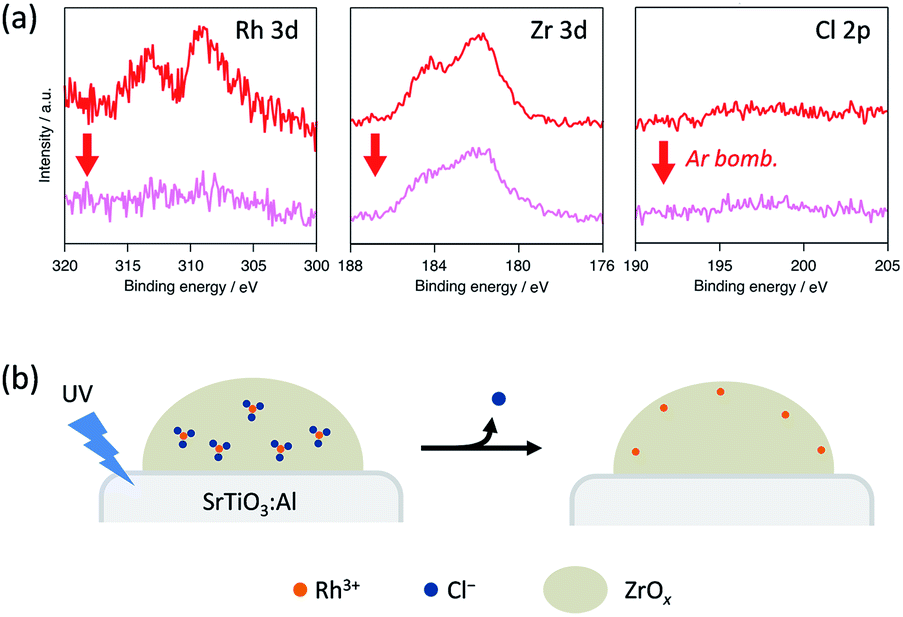 | ||
| Fig. 4 (a) XPS spectra of Rh 3d, Zr 3d and Cl 2p peaks in a RhZrOx cocatalyst on SrTiO3:Al after the reaction. (b) Schematic of the atomic rearrangement in Rh–Cl–Zr–O solid during photocatalysis. | ||
Considering the aforementioned results, we summarize the structural change of RhZrOx cocatalysts during photocatalysis as follows (Fig. 4b). Both Rh and Zr are uniformly dispersed in as-synthesized Rh–Cl–Zr–O solid in the forms of RhCl3 and ZrO2, respectively. As the reaction proceeds, the Cl− ions move to the surface and elute to water. The Rh3+ ions also migrate together with Cl− ions to the surface because of the strong Rh–Cl bond. As a result, the Rh3+ ions are condensed near the water/cocatalyst interface. However, the driving force for Cl− dissolution remains to be clarified. In the resulting structure, the insulating ZrOx layer may hinder the electron transfer from SrTiO3:Al to Rh3+ reaction centres. Recently, it has been reported that the photoelectrodes coated with the insulating layers (ZrO2, SiOx, Al2O3, HfO2, Ta2O5, etc.) exhibited enhanced photocatalytic activity, in which the photoexcited electrons can penetrate a few nanometre insulating overlayers.22–26 Because our RhZrOx cocatalysts are several nanometres in size, it is reasonable that the photoexcited electrons can migrate through ZrOx layers to reach Rh3+.
The atom migration (induced by Cl−) in the cocatalysts seems to affect the catalysts' catalytic properties. To confirm this migration effect, we synthesized RhZrOx/SrTiO3:Al catalysts from Cl-free precursors, such as Rh(NO3)3·xH2O and ZrO(NO3)2·2H2O, which were converted into mixed oxide cocatalysts through co-impregnation (Fig. S8†). The photocatalysts synthesized from Rh(NO3)3·xH2O have considerably lower activity than those from RhCl3·3H2O (Fig. S9†). Because the diameters of the cocatalysts were similar regardless of the precursors, a morphological effect on the activity can be excluded (Fig. S10†). These results strongly suggest that Cl− elution from the Rh–Cl–Zr–O solid formed the RhZrOx active species with the optimal spatial distribution of Rh, Zr and O for the overall water-splitting reaction.
The ZrOx-loaded SrTiO3:Al showed almost no H2 evolution activity (1 μmol h−1), therefore, the ZrOx does not function as a HER-active centre (Fig. S11†). To understand how Zr contributes to the activity enhancement, we conducted electrochemical catalysis measurements of RhZrOx. We loaded the RhZrOx cocatalysts on carbon powder (XC-72) by the same impregnation method adopted for SrTiO3:Al. We evaluated the catalytic activity of the backward reaction by measuring the current density of the oxygen reduction reaction (ORR) in O2-saturated 1 M aqueous Na2SO4. Linear sweep voltammograms showed that the onset potential for the ORR shifted in the negative direction, and the current density at 0.4 V vs. reversible hydrogen electrode decreased from 2.5 to 1.3 mA cm−2 as the Zr/Rh weight ratio increased from 0 to 7 (Fig. S13a and b†). This indicates that increasing the Zr/Rh weight ratio suppresses the ORR. We also evaluated the HER activity from an overpotential for the water reduction reaction in Ar-saturated 1 M aqueous Na2SO4. Overpotential of HER at −8 mA cm−2 increased from 445 to 524 mV in accordance with increasing Zr/Rh weight ratio (Fig. S13c and d†), which suggests that the larger quantity of Zr also suppresses the water reduction activity. This tendency accounts for the reason why Zr/Rh = 5 wt/wt% showed the highest water-splitting activity; that is, at Zr/Rh = 0–5 wt/wt%, suppression of the backward reaction is the main contributor to the improvement of the catalytic activity. In the range of Zr/Rh > 5 wt/wt%, deactivation of HER exceeds the suppression of the backward reaction.
From the structural point of view, we have to mention that it is hard to clarify whether the structure of RhZrOx on XC-72 is exactly the same as that of activated RhZrOx on SrTiO3:Al, because the amount of RhZrOx used in the electrochemical test was not enough for XPS measurement (Rh + Zr = 0.12 μg). Although the Cl− effect is unclear in electrochemical catalyses without structural characterization, the RhZrOx/XC-72 prepared from Rh(NO3)3·xH2O showed different ORR and HER activities from that prepared from RhCl3·3H2O (Fig. S13b and d†). One clear trend is that both ORR and HER activities decrease with increasing the Zr/Rh weight ratio for each catalyst (Fig. S13b and d†), which suggests that the addition of ZrOx hinders both ORR and HER of Rh3+ regardless of the Cl−-mediated activation.
The ORR blocking ability of ZrOx was confirmed in the photocatalysis of RhZrOx/SrTiO3:Al (Zr/Rh = 5 wt/wt%) by monitoring the concentrations of accumulated H2 and O2 in a reaction vessel after turning off the light irradiation (Fig. S14†). H2 and O2 concentrations in the reactor remained almost unchanged (>95%) over the subsequent 9 h in the dark, indicating that the backward reaction was strongly suppressed on RhZrOx/SrTiO3:Al with Zr/Rh = 5 wt/wt%. This result also suggests that the ratio of Zr/Rh = 5 wt/wt% is sufficient to suppress the backward reaction, which is qualitatively linked to the less ORR activity of RhZrOx cocatalyst with Zr/Rh = 5 wt/wt% in electrochemical results (Fig. S13a†). Additionally, this result also supports the conclusion that the less activity of RhZrOx/SrTiO3:Al with Zr/Rh = 7 wt/wt% results from the decreased HER activity, as shown in Fig. S13c.†
Co-loading of both HER and oxygen evolution reaction (OER) cocatalysts synergistically enhances the photocatalytic overall water splitting.27 For example, co-loading of cobalt oxide (CoOx) with RhCrOx on SrTiO3:Al enhances both the catalytic activity and durability.15 Encouraged by these results, we co-loaded CoOx and RhZrOx on SrTiO3:Al (CoOx + RhZrOx/SrTiO3:Al) by sequential impregnation. Initially, we loaded CoOx (Co = 0.1 wt% vs. SrTiO3:Al) on SrTiO3:Al, followed by loading RhZrOx (Rh = 0.1 and Zr = 0.5 wt% vs. SrTiO3:Al). The HER activity of CoOx + RhZrOx/SrTiO3:Al was 296 μmol h−1, which is 1.2× higher than that of RhZrOx/SrTiO3:Al (Fig. 5a). We confirmed that 0.1 wt% vs. SrTiO3:Al is an optimum amount of Co in the range of 0.05–0.2 wt% (Fig. S15†). Other well-known OER cocatalysts like NiOx and IrOx were also tested as additives on RhZrOx/SrTiO3:Al but their activities did not exceed the CoOx + RhZrOx/SrTiO3:Al (Fig. S16†). We calculated the AQY of CoOx + RhZrOx/SrTiO3:Al to be 33 (±4)% at 365 nm. The solar-to-hydrogen efficiency (STH) was estimated to be 0.21% (Fig. S17†). The stability was also improved by co-loading CoOx with RhZrOx (Fig. 5b). Because CoOx/SrTiO3:Al showed much lower activity for HER (Fig. 5a and b), we conclude that coexistence of CoOx and RhZrOx has a synergetic effect on the catalytic activity. In the case of the CoOx + RhCrOx combination, CoOx promotes hole extraction from SrTiO3:Al, which disrupts hole migration to RhCrOx to suppress the degradation,21 and also catalyzes water oxidation. We consider that this positive effect would also be effective for RhZrOx/SrTiO3:Al. A 60 h long-term stability test in 10 h intervals revealed that the gas evolution rate was relatively constant to the end of the reaction, maintaining a H2/O2 molar ratio of approximately 2 (Fig. 5c and d), indicating the robustness of CoOx + RhZrOx/SrTiO3:Al.
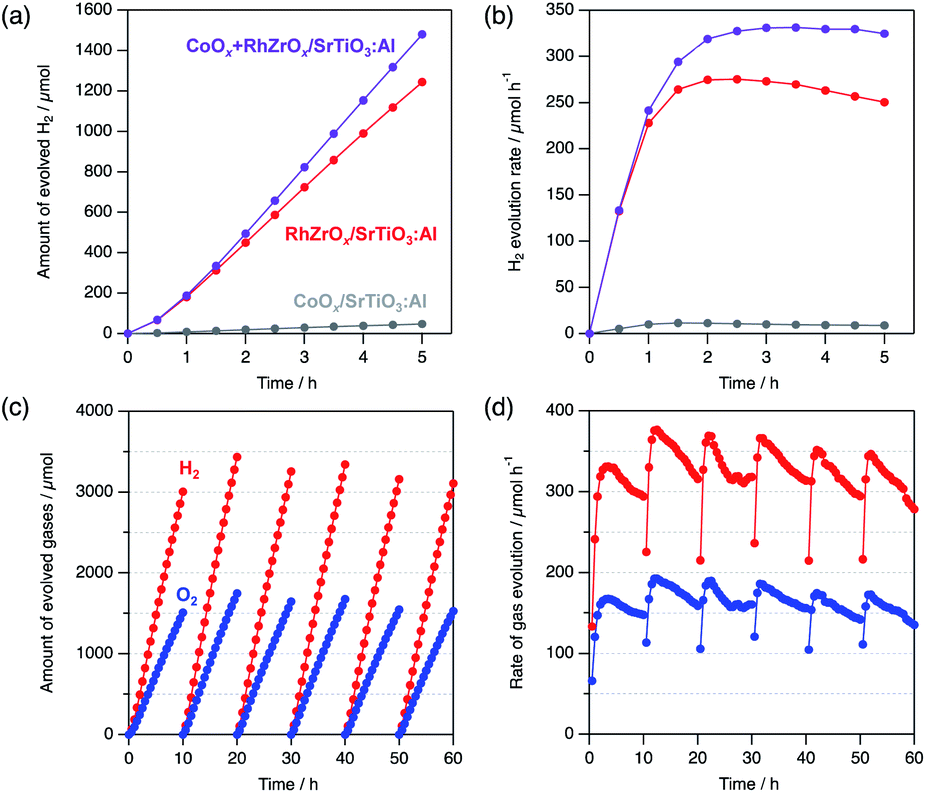 | ||
| Fig. 5 (a) Quantity of evolved H2 and (b) H2 evolution rate of CoOx + RhZrOx/SrTiO3:Al, RhZrOx/SrTiO3:Al and CoOx/SrTiO3:Al. (c) Quantity of evolved gases and (d) evolution rate of gases of CoOx + RhZrOx/SrTiO3:Al in a long-term test for 60 h. Red and blue circles represent H2 and O2, respectively. Reaction conditions: catalyst, 10 mg; solution, 20 mL of H2O; light source, 300 W Xe lamp (λ = 300–450 nm). | ||
Although we have shown the superior catalytic performance of RhZrOx/SrTiO3:Al, the activity is still less than that of RhCrOx/SrTiO3:Al, which has a >50% AQY at 365 nm (Table S3†).15 In the Rh@Cr2O3 cocatalyst, the Cr2O3 layer suppresses only the ORR and does not hinder the HER of Rh.11 This seems to be one of the reasons why RhZrOx/SrTiO3:Al has less activity than RhCrOx/SrTiO3:Al. To enhance the activity of RhZrOx/SrTiO3:Al, doping foreign metals into ZrO2 may increase proton conductivity.28 This strategy may help proton penetration towards the Rh reaction centres to evolve a larger quantity of H2. Robot-operating combinatorial experiments will help researchers discover such novel ternary or quaternary oxides cocatalyst materials from enormous numbers of candidates.29
Conclusions
We demonstrated that a Rh–Zr mixed oxide efficiently functions as a HER cocatalyst on SrTiO3:Al. The active phase for HER, RhZrOx, formed by spontaneous Cl-dissolution-assisted activation of the Rh–Cl–Zr–O solid solution. Unexpected Cl− contamination was found to sacrificially assist the formation of suitable spatial distributions of Rh3+ and Zr4+ in RhZrOx cocatalyst during photocatalysis. Electrochemical measurements showed that addition of Zr suppresses not only the ORR, but also the HER of Rh. We used this fact to determine the proper weight ratio of Zr/Rh (=5) for overall water splitting. Additional deposition of an OER cocatalyst, CoOx, further improved the photocatalytic activity and stability of RhZrOx/SrTiO3:Al. Our nonhazardous Zr-based cocatalyst will be useful for large-scale applications to photocatalytic water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank H. Tokudome and S. Okunaka of TOTO Ltd. for preparing SrTiO3:Al. This work was partly supported by the Artificial Photosynthesis Project of the New Energy and Industrial Technology Development Organization (NEDO) of Japan and JSPS KAKENHI for Scientific Research S (Grant No. JP19H0563) (T. T.), and Young Scientists B (Grant No. 17K14081) (M. S.).References
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83 CrossRef CAS.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827 CrossRef CAS PubMed.
- K. Takanabe, ACS Catal., 2017, 7, 8006 CrossRef CAS.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151 CrossRef CAS.
- J. A. Bau and K. Takanabe, ACS Catal., 2017, 7(11), 7931 CrossRef CAS.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753 CrossRef CAS PubMed.
- H. Lyu, T. Hisatomi, Y. Goto, M. Yoshida, T. Higashi, M. Katayama, T. Takata, T. Minegishi, H. Nishiyama, T. Yamada, Y. Sakata, K. Asakura and K. Domen, Chem. Sci., 2019, 10, 3196 RSC.
- F. Giustino and H. J. Snaith, ACS Energy Lett., 2016, 1, 1233 CrossRef CAS.
- A. T. Garcia-Esparza, T. Shinagawa, S. Ould-Chikh, M. Qureshi, X. Peng, N. Wei, D. H. Anjum, A. Clo, T. C. Weng, D. Nordlund, D. Sokaras, J. Kubota, K. Domen and K. Takanabe, Angew. Chem., Int. Ed., 2017, 56, 5780 CrossRef CAS PubMed.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386 CrossRef CAS.
- W. Balcerowiak, J. Therm. Anal. Calorim., 2003, 71, 559 CrossRef CAS.
- S. P. Phivilay, C. A. Roberts, A. D. Gamalski, E. A. StachShiran, Z. Luan, N. Y. Tang, A. Xiong, A. A. Puretzky, F. F. Tao, K. Domen and I. E. Wachs, ACS Catal., 2018, 8, 6650 CrossRef CAS.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107 CrossRef CAS PubMed.
- J. Liu, T. Hisatomi, D. H. K. Murthy, M. Zhong, M. Nakabayashi, T. Higashi, Y. Suzuki, H. Matsuzaki, K. Seki, A. Furube, N. Shibata, M. Katayama, T. Minegishi and K. Domen, J. Phys. Chem. Lett., 2017, 8, 375 CrossRef CAS PubMed.
- S. Y. Lim, D. Han, Y. R. Kim and T. D. Chung, ACS Appl. Mater. Interfaces, 2017, 9, 23698 CrossRef CAS PubMed.
- R. Fan, J. Mao, Z. Yin, J. Jie, W. Dong, L. Fang, F. Zheng and M. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 6123 CrossRef CAS PubMed.
- J. Quinn, J. Hemmerling and S. Linic, ACS Energy Lett., 2019, 4, 2632 CrossRef CAS.
- T. Wang, S. Liu, H. Li, C. Li, Z. Liu and J. Gong, Ind. Eng. Chem. Res., 2019, 58, 5510 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed.
- J. A. Dawson and I. Tanaka, Langmuir, 2014, 30, 10456 CrossRef CAS PubMed.
- R. Saito, Y. Miseki, W. Nini and K. Sayama, ACS Comb. Sci., 2015, 17, 592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization and results. See DOI: 10.1039/d0sc01363c |
| This journal is © The Royal Society of Chemistry 2020 |