
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning naphthopyran mechanophores with tunable mechanochromic behavior†
Brooke A.
Versaw
 ,
Molly E.
McFadden
,
Corey C.
Husic
and
Maxwell J.
Robb
*
,
Molly E.
McFadden
,
Corey C.
Husic
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 17th April 2020
Abstract
Mechanochromic molecular force probes conveniently report on stress and strain in polymeric materials through straightforward visual cues. We capitalize on the versatility of the naphthopyran framework to design a series of mechanochromic mechanophores that exhibit highly tunable color and fading kinetics after mechanochemical activation. Structurally diverse naphthopyran crosslinkers are synthesized and covalently incorporated into silicone elastomers, where the mechanochemical ring–opening reactions are achieved under tension to generate the merocyanine dyes. Strategic structural modifications to the naphthopyran mechanophore scaffold produce dramatic differences in the color and thermal electrocyclization behavior of the corresponding merocyanine dyes. The color of the merocyanines varies from orange-yellow to purple upon the introduction of an electron donating pyrrolidine substituent, while the rate of thermal electrocyclization is controlled through electronic and steric factors, enabling access to derivatives that display both fast-fading and persistent coloration after mechanical activation and subsequent stress relaxation. In addition to identifying key structure–property relationships for tuning the behavior of the naphthopyran mechanophore, the modularity of the naphthopyran platform is demonstrated by leveraging blends of structurally distinct mechanophores to create materials with desirable multicolor mechanochromic and complex stimuli-responsive behavior, expanding the scope and accessibility of force-responsive materials for applications such as multimodal sensing.
Introduction
The rapidly growing field of polymer mechanochemistry explores the use of mechanical force to activate chemical transformations in stress-responsive molecules called mechanophores.1 Mechanical stress is transmitted through polymer chains to covalently attached mechanophores where the transduction of mechanical energy elicits a chemoselective reaction.2,3 In particular, mechanophores that produce a visible color change in response to mechanical force are of particular interest for stress sensing applications, enabling the straightforward detection of critical stress and/or strain in polymeric materials.4 Mechanochromic mechanophores have been developed that generate color through a variety of mechanisms. Spiropyran is one of the most widely investigated mechanophores, which undergoes a 6π electrocyclic ring–opening reaction under force to generate a highly colored merocyanine species.5,6 The mechanochromic behavior of other chromene derivatives including naphthopyran7,8 and spirothiopyran,9 the ring–opening reaction of a rhodamine-based mechanophore,10,11 and mechanochemically-induced unzipping of polyladderene12,13 have also been recently demonstrated. Mechanophores that dissociate homolytically under force to generate a pair of colored stable free radicals in polymers have also been reported.14–18In addition to their recently discovered mechanochemical activity,7 naphthopyrans have been widely developed for their excellent photochromic properties, which can be extensively modified through chemical substitution to control the color and thermal reversion behavior of the merocyanine.19 For the 3,3-diaryl-3H-naphtho[2,1-b]pyran scaffold illustrated in Scheme 1, conversion of the colorless naphthopyran to the colored merocyanine form proceeds via a 6π electrocyclic ring–opening reaction that is mediated by UV light (or force), while the ring–closing reaction is driven by visible light or heat. Substitution of the aryl rings attached at the 3-position of the naphthopyran has a significant impact on the absorption and electrocyclization behavior of the photochemically generated merocyanine. Electron-donating substituents in the para position of the phenyl rings cause a bathochromic shift of the merocyanine absorption, which ranges from approximately 30 nm for an alkoxy substituent to greater than 100 nm for a secondary amine.20,21 Substituents at the ortho position dramatically reduce the rate of thermal electrocyclization through putative steric interactions, leading to significantly longer lifetimes of the merocyanine state in solution.20,22 In addition, substituents on the naphthopyran skeleton have been shown to affect the photochromic properties. Methoxy substitution at the 5-position, for example, results in a hypsochromic shift of approximately 40 nm in the ring-closed form, but does not significantly affect the color of the merocyanine state.23
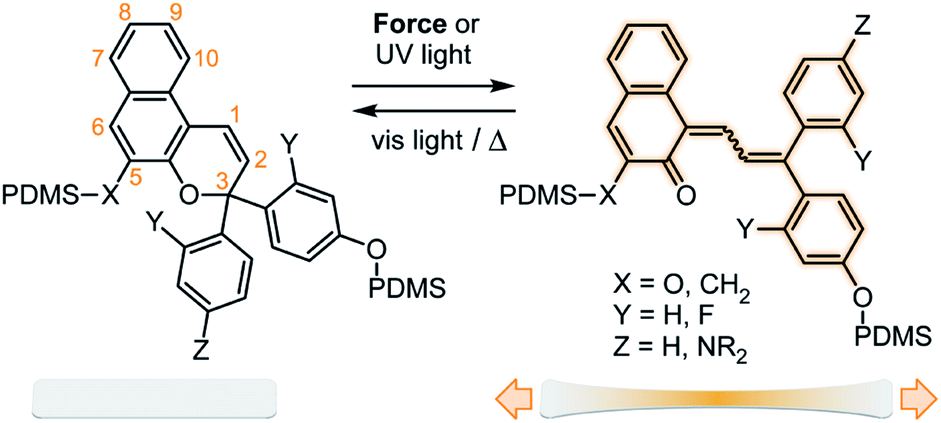 | ||
| Scheme 1 Reaction of naphthopyran in PDMS materials generates colored merocyanine dyes with substituent-dependent mechanochromic properties. | ||
Here we investigate the influence of substitution on the naphthopyran mechanophore framework, establishing a modular platform for accessing polymeric materials with highly tunable mechanochromic properties. Structure–activity relationships for the photochromic and mechanochromic properties of a series of naphthopyran mechanophores are investigated in solution and in crosslinked polydimethylsiloxane (PDMS) materials. Strategic structural modifications to the naphthopyran mechanophore result in significant changes to the visible absorption properties and rates of electrocyclization (i.e., color fading) in solution that translate to the mechanochromic behavior of polymeric materials activated in tension. We leverage the diverse mechanochromic and photochromic properties of the naphthopyran mechanophores to create stimuli-responsive polymers capable of complex reporting functionality including multicolor mechanochromism and visually orthogonal reactivity under photochemical and mechanical stimulation.
Results and discussion
We designed a series of naphthopyran mechanophores with different substituents tailored to control both the color and fading kinetics of the merocyanine generated under force (Chart 1). The regiochemistry of naphthopyran is critical for mechanochemical activity, which is achieved only when mechanical force is effectively transferred across the labile C–O pyran bond.7 Naphthopyran 1a with polymer attachment at the 5-position generates an orange-yellow merocyanine dye under mechanical stress in polymeric materials, while regioisomers attached at the 8- and 9-position were previously demonstrated to be mechanochemically inert due to geometrical incongruity with the direction of applied force (see Scheme 1). Therefore, each naphthopyran in the current study was designed with the same polymer attachment geometry employed for known mechanophore 1a. Naphthopyrans were synthesized via the acid-catalyzed reaction between appropriately substituted 2-naphthols and propargyl alcohols and appended with vinyl-functional tethers to allow for their incorporation as crosslinkers into elastomeric PDMS networks (see ESI† for details). A strongly electron-donating pyrrolidine substituent was introduced at the para position of one of the phenyl rings on the naphthopyran to bathochromically shift the absorption of the merocyanine relative to 1a. From these two basic scaffolds with variably colored merocyanines (1a and 2a), we sought to further modulate the thermal reversion kinetics. An ortho-fluoro group was incorporated on one of the aromatic rings to reduce the rate of thermal electrocyclization and extend the lifetime of the merocyanine state in mechanochemically activated materials (1b and 2b). Conversely, faster thermal reversion could be useful for achieving materials with rapid switching capabilities. While the influence of alkoxy substituents at the 5-position of naphthopyran on the absorption properties has been characterized,19 relatively little is known about the impact of substitution at this position on the lifetime of the merocyanine state. As described below, replacing the electron-donating alkoxy group at the 5-position of the naphthopyran with an alkyl tether results in faster electrocyclization (1c and 2c). Encouragingly, density functional theory (DFT) calculations performed using the constrained geometries simulate external force (CoGEF) method24 predict the desired electrocyclic ring–opening reaction for each naphthopyran derivative upon mechanical elongation (see ESI† for details).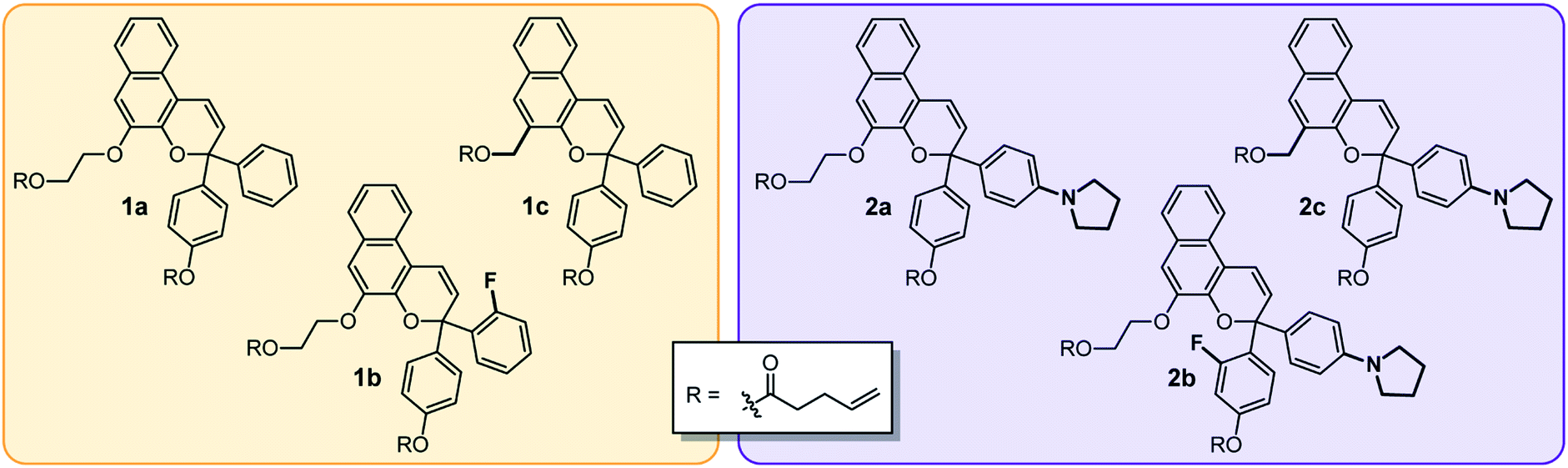 | ||
| Chart 1 Naphthopyran mechanophore crosslinkers used in the preparation of PDMS materials with tunable mechanochromic behavior. | ||
The absorption properties of the small molecule naphthopyran crosslinkers were first characterized in solution to evaluate the impact of substitution (Fig. 1). UV-vis absorption spectra were acquired for each compound in THF before and after irradiation with UV light (311 nm for 30 s). The UV-vis absorption properties are summarized in Table 1. Each naphthopyran exhibits an absorption peak around 320 nm, while naphthopyrans containing an alkyl tether at the 5-position (1c and 2c) have a second absorption feature at approximately 350 nm, resulting in an overall bathochromic shift relative to the 5-alkoxy derivatives. As expected, substituents on the aryl rings attached at the 3-position of the naphthopyran scaffold do not significantly affect the absorption properties of the ring-closed naphthopyran at wavelengths longer than ∼320 nm due to connectivity through the sp3-hybridized carbon. Irradiation of the naphthopyran solutions with UV light results in new absorption peaks in the visible region corresponding to the ring-opened merocyanine state. For compounds 1a–1c, the associated merocyanine species exhibit absorption peaks between 422 and 435 nm and the solutions appear orange-yellow in color. In contrast to the ring-closed naphthopyrans, the pyrrolidine substituents on compounds 2a–2c result in a substantial bathochromic shift in the absorption of the merocyanine dyes with peaks at 530–553 nm, producing solutions that appear purple in color. For naphthopyrans 1b and 2b, the ortho-fluoro group results in greater absorbance in the visible region of the spectra after UV irradiation, consistent with a significantly longer-lived merocyanine state.
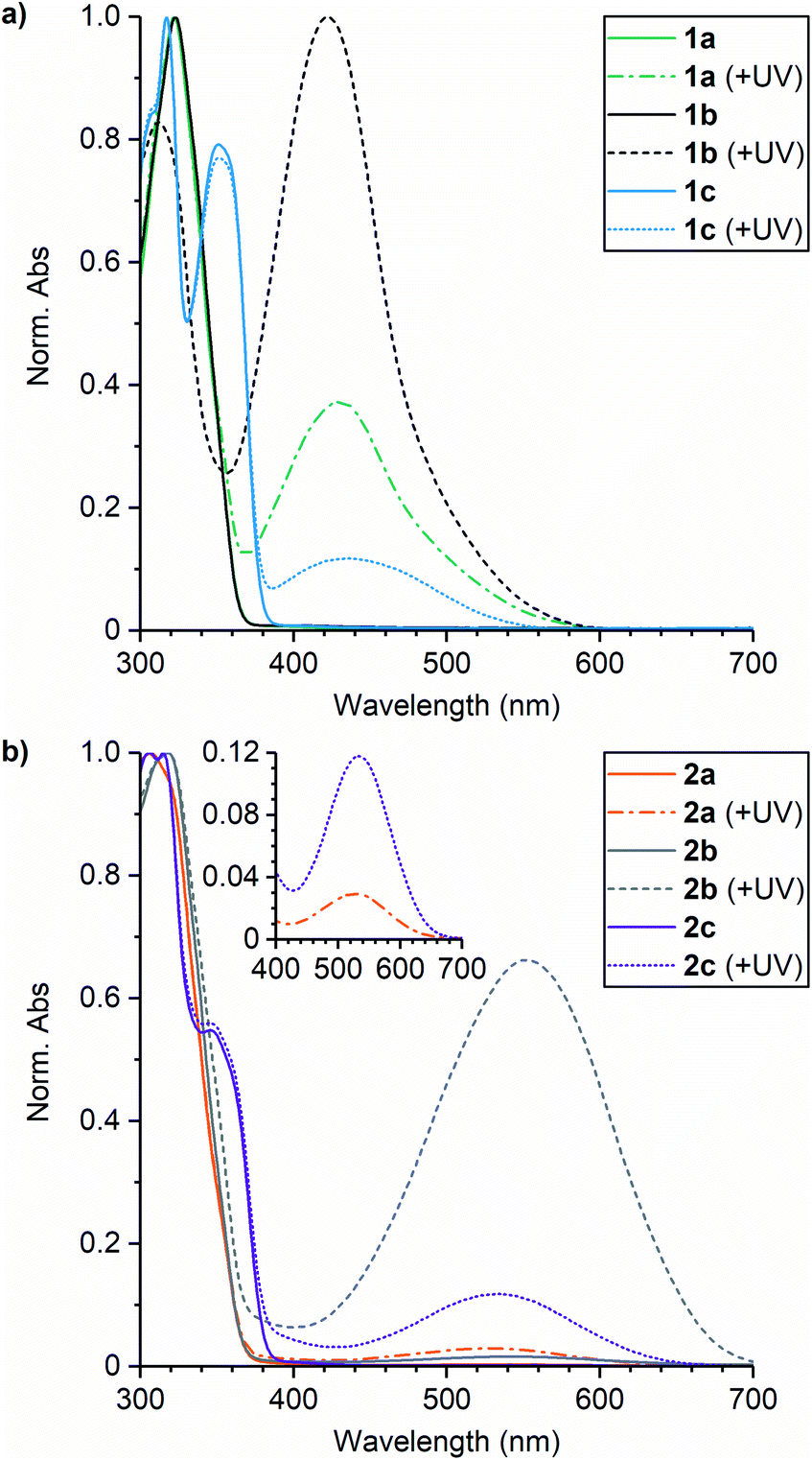 | ||
| Fig. 1 UV-vis absorption spectra acquired for solutions of (a) naphthopyrans 1a–1c, and (b) pyrrolidine-substituted naphthopyrans 2a–2c (0.1 mM in THF) before and after irradiation with UV light (λ = 311 nm, 30 s). The introduction of a pyrrolidine substituent results in a significant bathochromic shift of the merocyanine absorption. | ||
| Absorption propertiesa | Thermal reversion kinetics | |||||||
|---|---|---|---|---|---|---|---|---|
| Before hν, λmax (nm) | After hν, λmax (nm) | k r,soln (s−1) | t 1/2,soln (s) | k r1,solid (s−1) | k r2,solid (s−1) | A 1,solid | A 2,solid | |
| a Absorption maxima measured in THF (0.1 mM) before and after irradiation with UV light (λ = 311 nm, 30 s). b Average rate constant and half-life (t1/2) from 1st order thermal reversion kinetics in THF at room temperature after UV photoactivation. c Rate constants and pre-exponential factors from biexponential fitting of thermal reversion kinetics measured by digital color analysis of PDMS films containing 1.5 wt% mechanophore after UV photoactivation. Kinetic data for 1a were fitted to monoexponential decay. d Not determined. See ESI for additional details. | ||||||||
| 1a | 322 | 430 | 0.033 | 21 | 0.02 | — | — | — |
| 1b | 323 | 422 | 0.0023 | 300 | 0.002 | 0.02 | 0.2 | 0.04 |
| 1c | 317, 351 | 435 | 0.075 | 9 | 0.02 | 0.09 | 0.07 | 0.03 |
| 2a | 318 | 530 | 0.19 | 4 | 0.01 | 0.1 | 0.04 | 0.03 |
| 2b | 318 | 553 | 0.010 | 69 | 0.01 | 0.002 | 0.2 | 0.1 |
| 2c | 315, 346 | 533 | ndd | ndd | 0.02 | 0.1 | 0.04 | 0.02 |
The kinetics of thermal electrocyclization for each merocyanine dye were quantified to further characterize the impact of substitution on the color-fading behavior. Solutions of each naphthopyran in THF were initially irradiated with 311 nm UV light, and the absorbance at wavelengths corresponding to the λmax of each merocyanine was subsequently monitored at room temperature in the dark. Plots of the time-dependent merocyanine absorbance were fit to first-order exponential decay to determine the rate constant (kr,soln) and corresponding half-life (t1/2,soln) for thermal reversion (Fig. S1†). The results are presented in Table 1 as averages from three separate trials. Comparing the rates of thermal ring-closure for merocyanines derived from 1a and 2a, for example, illustrates the impact of the electron-donating para-pyrrolidine substituent. The pyrrolidine substituent increases the rate of electrocyclization approximately six-fold, corresponding to a reduction in t1/2 from 21 s to 4 s. Conversely, the introduction of an ortho-fluoro group decreases the value of kr,soln by approximately one order of magnitude with measured half-lives of t1/2 = 300 s for 1b and t1/2 = 69 s for 2b. This effect has previously been attributed to a steric interaction that hinders formation of the requisite geometry for ring-closure.20 Replacing the alkoxy group with an alkyl tether at the 5-position of the naphthopyran approximately doubles the rate of thermal reversion from the merocyanine state, as illustrated for 1a and 1c, respectively. Electrocyclization of the merocyanine derived from 2c was too rapid for the initial fading rate to be effectively characterized, although a persistent merocyanine isomer remains after an extended period of time post-irradiation that is responsible for the visible absorption peak in Fig. 1b. The persistent color observed after UV photoactivation of some naphthopyrans has been attributed to the relative thermal stability of the merocyanine isomer with trans configuration of the exocyclic double bond, which isomerizes slowly in the dark to the cis isomer prior to ring-closure.25–27 This isomerization is efficiently promoted with visible light, however, and subsequent irradiation of the above solution with white light results in complete attenuation of the visible absorption peak, indicating full conversion of the merocyanine back to the original ring-closed naphthopyran (see ESI† for details).
Following evaluation of their solution-phase properties, the mechanochromic behavior of the series of naphthopyran derivatives was investigated in elastomeric PDMS materials. Naphthopyrans were incorporated as crosslinkers (∼1.5 wt% loading) into PDMS films via platinum-catalyzed hydrosilylation according to the method reported by Craig and coworkers,28 which provides an ideal materials testing platform (Fig. 2a). With the exception of the material containing naphthopyran 2b that exhibits some background color, the films are optically clear and nearly colorless. Stretching each naphthopyran-containing PDMS film causes the gauge region of the material to change color, characteristic of the mechanochemical ring–opening reaction to generate the merocyanine dye (Fig. 2b). After mechanical activation, films containing naphthopyrans 1a–c are orange-yellow in color, while films containing pyrrolidine-substituted naphthopyrans 2a–c appear purple, consistent with the photochromic behavior of the molecules in both solution and in the solid-state after irradiation with UV light. Qualitative differences in thermal reversion kinetics are also evident in the photographs of the films acquired 2 min after initial mechanical activation and subsequent stress relaxation. The color of the merocyanine dye is still apparent in films containing mechanophores 1b and 2b, which produce the slowest fading merocyanines due to the ortho-fluorophenyl substituents. In contrast, PDMS films incorporating mechanophores 1c and 2c that contain the alkyl tether at the 5-position and produce the fastest-fading merocyanines exhibit nearly complete thermal reversion in the same period of time following mechanical activation.
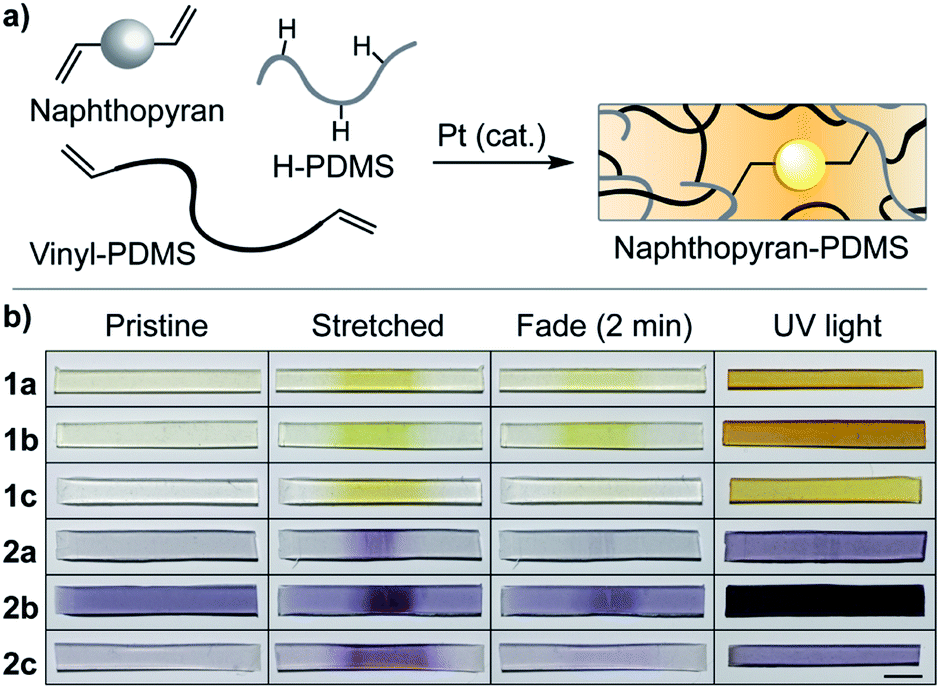 | ||
| Fig. 2 (a) PDMS materials covalently crosslinked with naphthopyran mechanophores are prepared via platinum-catalyzed hydrosilylation. (b) Photographs of PDMS films incorporating naphthopyran mechanophores (1.5 wt%) immediately after mechanical activation (tension) and subsequent stress relaxation (2 min), and after irradiation with UV light (λ = 311 nm, 90 s). Scale bar = 5 mm. | ||
To further characterize the kinetics of thermal ring-closure in the solid-state, PDMS films were uniformly irradiated with UV light and photographs were subsequently acquired at regular time intervals to monitor the disappearance of color associated with the conversion of the merocyanine to the naphthopyran (see ESI† for details). Time-dependent changes in the ratio of the green and red color channel intensities were extracted from the digital images and fit to models of exponential decay (Fig. S2†).6 The thermal reversion kinetics for mechanophore 1a are well described by monoexponential decay; however, a biexponential decay equation was required to accurately model the thermal fading behavior of the other merocyanine dyes in the solid state. Previous studies of naphthopyran–merocyanine systems suggest that the biexponential kinetics originate from multiple merocyanine stereoisomers, each of which exhibits a unique rate of thermal reversion.25–27 The rate constants determined for thermal reversion of each merocyanine in solid PDMS materials (kr1,solid and kr2,solid) are summarized in Table 1, along with the corresponding pre-exponential factors (A1,solid and A2,solid) that express the relative weight of each term (see ESI† for details). On average, the observed rates of merocyanine reversion are slower in solid PDMS materials than in solution, which we attribute to differences in polarity and conformational constraints imposed by the polymer network.29–31 The overall trends in fading kinetics, however, reflect the structure–property relationships observed for the small molecules in solution, despite the more complex fading behavior in the solid state that potentially arises from differences in merocyanine isomerization that are more pronounced in solid PDMS materials.
Beyond the typical binary response of most mechanochromic mechanophores, materials that are capable of reporting on the state of stress or strain through discrete visual signals are desirable targets. Rhodamine11 and spiropyran28,32 mechanophores display differences in absorption under active tension and after stress relaxation due to presumed torsional effects and changes in cis–trans isomerization, respectively. We recently developed a bis-naphthopyran mechanophore that exhibits gradient multicolor mechanochromism resulting from force-dependent changes in the distribution of uniquely colored merocyanine products.8 In addition, the differential activation of distinct mechanophores localized in hard and soft domains of phase-separated polymer and composite materials has been demonstrated using different types of mechanical stimuli (e.g., stretching and grinding).33,34 Here, the substantial differences in merocyanine absorption and reversion kinetics provided by the series of naphthopyran mechanophores affords opportunities to create materials capable of multicolor mechanochromism and other complex reporting functions. To demonstrate the potential of this approach, a PDMS film was prepared using a blend of mechanophore crosslinkers 1b and 2c, which generate a slow-fading orange-yellow and a fast-fading purple-colored merocyanine, respectively (Fig. 3a). Under active tension the material appears red-orange in color due to the combined contribution of both merocyanines. Upon stress relaxation, the purple component quickly fades to reveal a more persistent orange-yellow color, providing a clear visual response that distinguishes between the different stress states with temporal resolution. A 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (by wt) of 1b/2c was chosen to optimize the visual contrast between the different stress states.
:1 ratio (by wt) of 1b/2c was chosen to optimize the visual contrast between the different stress states.
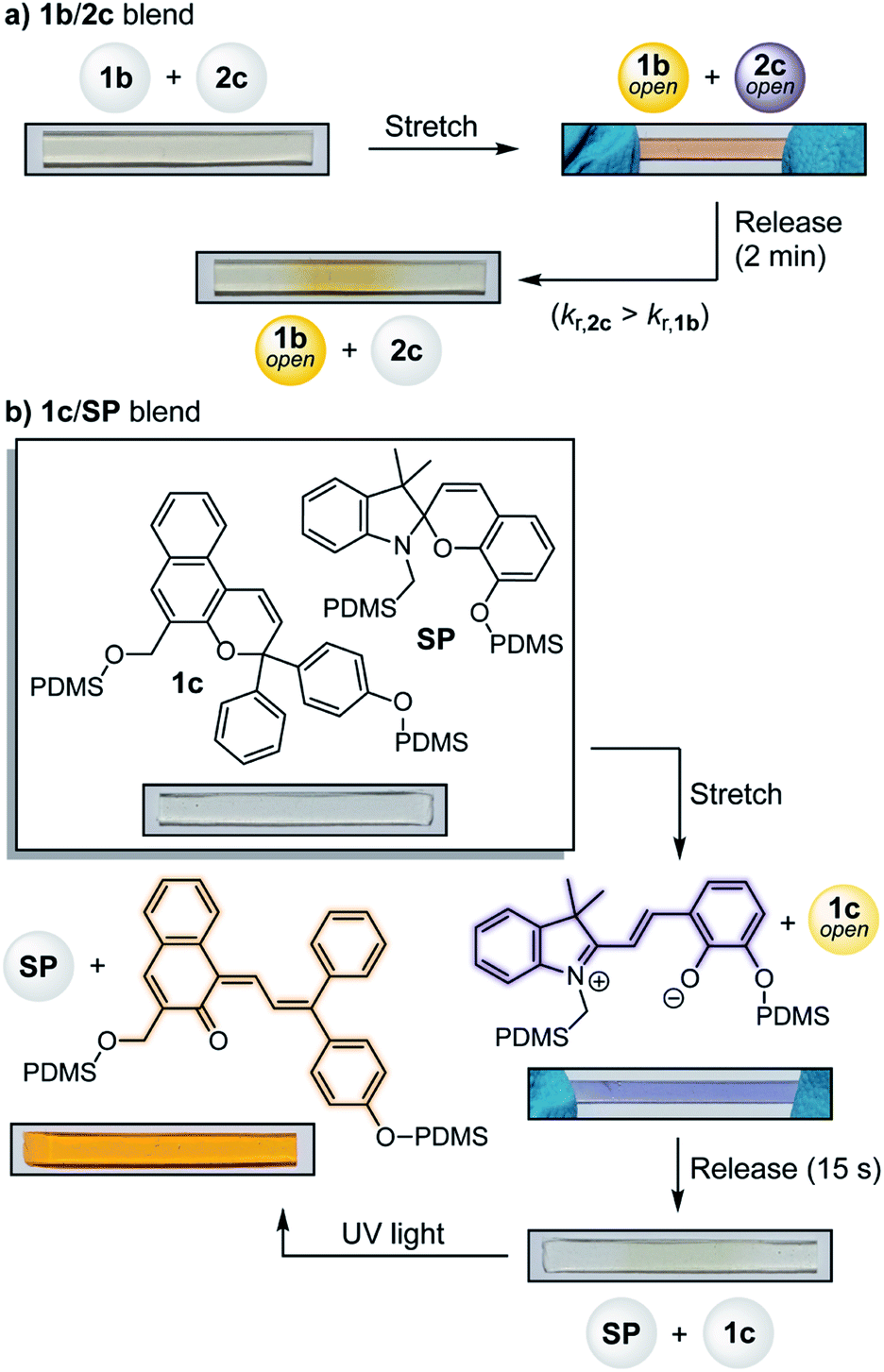 | ||
| Fig. 3 Demonstration of multicolor mechanochromism and photochromism from PDMS materials incorporating blends of different mechanophores. (a) PDMS containing a 4:1 mixture (by wt) of naphthopyrans 1b and 2c, respectively, exhibits variation in color under tension and after stress relaxation due to pronounced differences in merocyanine absorption and reversion rates. (b) PDMS film containing a 1:1 blend (by wt) of naphthopyran 1c and non-photochromic spiropyran mechanophore SP exhibits distinct responsive behavior when activated mechanochemically in tension or with UV light (λ = 311 nm, 90 s). Samples measure 25 mm × 3 mm and contain a total mechanophore loading of 1.5 wt%. | ||
We further sought to access materials that respond to multiple types of stimuli by taking advantage of the photochromic properties of naphthopyran combined with a non-photochromic spiropyran mechanophore35 (Fig. 3b, see ESI† for details). A PDMS material containing a 1:1 blend (by wt) of naphthopyran 1c and spiropyran mechanophore SP was prepared and activated mechanically in tension, causing the film to turn blue-purple in color due to the dominant appearance of the spiropyran-derived merocyanine. Upon stress relaxation, the color quickly disappears due to the rapid thermal reversion of both merocyanine dyes. The same film was then irradiated with UV light, which selectively activates the ring–opening reaction of naphthopyran 1c to give a vibrant orange-yellow readout. These examples illustrate the types of stimuli-responsive materials that are accessible for multimodal sensing applications by capitalizing on the highly tunable and modular properties of the naphthopyran platform.
Conclusions
In summary, we have demonstrated the versatility of the naphthopyran scaffold as a modular platform for designing polymeric materials with tunable mechanochromic and stimuli–responsive properties. Diverse naphthopyran mechanophores are accessed through straightforward synthetic methods with strategic structural modifications that provide control over both the color and fading kinetics of the merocyanine dyes generated under photochemical and mechanochemical activation. The rates of thermal reversion are tunable through both steric and electronic factors while the color of the merocyanine dyes is varied from orange-yellow to purple upon the installation of an electron donating pyrrolidine substituent. The absorption and thermal reversion properties of the small molecules in solution translate to the mechanochromic behavior of the naphthopyran mechanophores in crosslinked PDMS elastomers activated in tension. The variation in electrocyclization kinetics and visible absorption behavior provided by judicious substitution of the naphthopyran scaffold enables the development of polymeric materials that exhibit desirable multicolor mechanochromic and complex stimuli–responsive properties from a simple and modular molecular platform. The structure–activity relationships presented for the naphthopyran mechanophore expand the scope and accessibility of light and force-responsive materials for a number of applications, including multimodal sensing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Caltech and the Dow Next Generation Educator Fund is gratefully acknowledged. B. A. V. and M. E. M. were supported by NSF Graduate Research Fellowships (DGE-1745301). We thank the Center for Catalysis and Chemical Synthesis of the Beckman Institute at Caltech for access to equipment and Dr Mona Shahgholi and the CCE Multiuser Mass Spectrometry Laboratory for assistance with mass spectrometry.References
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- R. W. Barber, M. E. McFadden, X. Hu and M. J. Robb, Synlett, 2019, 30, 1725–1732 CrossRef CAS.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS PubMed.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- F. Verstraeten, R. Göstl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem.–Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed.
- H. Sakai, T. Sumi, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1359–1363 CrossRef CAS.
- B. Van Gemert, in Organic Photochromic and Thermochromic Compounds: Volume 1: Main Photochromic Families, ed. J. C. Crano and R. J. Guglielmetti, Springer US, Boston, MA, 2002, pp. 111–140 Search PubMed.
- C. D. Gabbutt, B. M. Heron and A. C. Instone, Heterocycles, 2003, 60, 843–855 CrossRef CAS.
- C. D. Gabbutt, B. M. Heron, A. C. Instone, P. N. Horton and M. B. Hursthouse, Tetrahedron, 2005, 61, 463–471 CrossRef CAS.
- B. Van Gemert and M. P. Bergomi, US Pat., US5066818A, 1991.
- B. Van Gemert, M. Bergomi and D. Knowles, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 246, 67–73 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- G. Ottavi, G. Favaro and V. Malatesta, J. Photochem. Photobiol., A, 1998, 115, 123–128 CrossRef CAS.
- S. Delbaere, B. Luccioni-Houze, C. Bochu, Y. Teral, M. Campredon and G. Vermeersch, J. Chem. Soc., Perkin Trans. 2, 1998, 1153–1158 RSC.
- S. Delbaere, J.-C. Micheau, Y. Teral, C. Bochu, M. Campredon and G. Vermeersch, Photochem. Photobiol., 2001, 74, 694–699 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS.
- W. Sriprom, M. Néel, C. D. Gabbutt, B. M. Heron and S. Perrier, J. Mater. Chem., 2007, 17, 1885–1893 RSC.
- N. Malic, J. A. Campbell and R. A. Evans, Macromolecules, 2008, 41, 1206–1214 CrossRef CAS.
- R. A. Evans, T. L. Hanley, M. A. Skidmore, T. P. Davis, G. K. Such, L. H. Yee, G. E. Ball and D. A. Lewis, Nat. Mater., 2005, 4, 249–253 CrossRef CAS PubMed.
- T. A. Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2018, 51, 9177–9183 CrossRef CAS.
- K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 556–560 CrossRef CAS.
- T. Kosuge, X. Zhu, V. M. Lau, D. Aoki, T. J. Martinez, J. S. Moore and H. Otsuka, J. Am. Chem. Soc., 2019, 141, 1898–1902 CrossRef CAS PubMed.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data, and density functional theory calculations. See DOI: 10.1039/d0sc01359e |
| This journal is © The Royal Society of Chemistry 2020 |