From toxic amyloid oligomers to ion channel formation, in recent years peptide self-assembly provided novel biomaterials with potential for diverse applications ranging from tissue engineering through antiviral and antibacterial therapeutics to supramolecular polymer tubes with various properties.1–9 Considering the general sensitivity of natural peptides to enzymatic degradation, proteolytically stable compounds built from exotic synthetic amino acids receive an increased interest. Consequently, in the last two decades peptidic foldamers, composed of non-natural amino acids, were intensively studied reaching structural diversity and widespread applicability similar to those of the natural biomolecules.10–19 Their most commonly studied members are β-peptides, composed of β-amino acids, where an additional methylene group is introduced between the peptide bonds. These compounds can adopt various secondary structures (helical, sheet-like conformations or hairpins)15,17,20–27 and even higher ordered aqueous assemblies such as helical bundles,10,28–30 protein-like assemblies31–35 and nanofibers.36–38 Their applicability has been tested in nanotechnology,39–41 biomedical fields,15,42–48 biopolymer surface recognition,49–51 catalysis28,52–55 and biotechnology.56–58 It has also been demonstrated using theoretical calculations that in principle β-peptides could form energetically stable assemblies similar to those of natural β-barrel proteins.59,60 However, despite the significant progress in these areas, controlling the process of self-assembly of non-natural peptides, especially in relation to lipid bilayers is still challenging. The few applications employed only cyclic compounds so far, which drastically limits their conformational flexibility and adaptability to environmental changes.61–64 In contrast, for antimicrobial, host-defense or anticancer peptides a growing amount of evidence suggests, that temporary assembly may play a crucial role in exerting biological function. Note, that complexity of these systems is clearly shown by the fact that recent studies still provide new insights and examples for self-assembled constructs even for otherwise well-studied peptides, such as melittin.65
Accordingly, for progress towards the controlled formation of oligomers with membrane activity, here we focused on short β-peptide sequences. The obtained β3-peptides readily form oligomers which are soluble in water and in hydrophobic organic solvents such as octanol and can insert into a lipid bilayer.
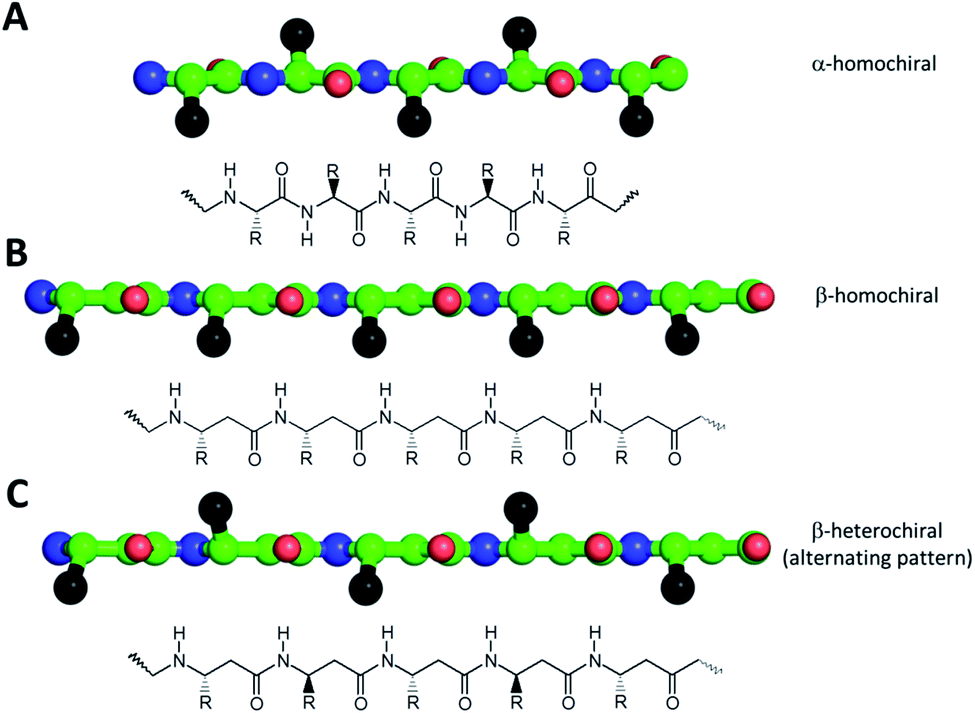
One of the simplest, yet efficient scaffold for natural proteins which can separate hydrophobic and hydrophilic residues is the β-barrel, where both membrane soluble and water-soluble constructs can be found. In these proteins the elongated sheet conformation results in a finite number of β-strands, where the extended conformation together with homochiral α-amino acids provides an alternating positioning of the amino acid side chains (Scheme 1). This allows efficient environment-dependent adjustment of the entire protein in the lipid bilayer, as well as diverse functionality. The separation of inner and outer surfaces in such a straightforward tertiary structure could potentially be reproduced by self-assembling peptide units. We have earlier confirmed by quantum chemical calculations that similar, energetically stable barrels could be formed by β-peptides as well.59,60 However, since β-peptides have an additional methylene group in their amino acid backbone, the homochiral sequences in an elongated conformation render side chains to be on the same side (Scheme 1).18,66,67 This additional methylene group is also the reason to some extent, why intermolecular hydrogen bonds can be formed easier, which results in dominance of helical conformations.68 Thus, to obtain oligomers which mimic the above natural constructs where the hydrophobic and hydrophilic sides are separated, but simultaneously avoid helical monomers, we employed short β3-peptides with alternating side chain chirality (Scheme 1). Furthermore, to prevent infinite fibril formation69 the alternating chirality pattern was combined with a homochiral unit at the N-terminus.
Accordingly, five β3-hexapeptides (1–5, named here as “RS” set) were designed and synthesized (Fig. 1). Initially, peptide 1 was synthesized containing aromatic residues and leucines on the apolar side, whereas glutamines were chosen for the polar side. To enhance membrane activity and result discrete small oligomers, instead of infinite nanofibers,69 the N-terminal was kept more hydrophobic with the first two residues being homochiral for all sequences. The phenyl and 2-naphthyl-homoalanine residues were selected to test anchoring properties upon potential membrane interactions. Sequence 2 was designed to be less hydrophobic by introducing charged residues on the hydrophilic side but maintaining a neutral total charge with only [R]-β3-homoleucines on the apolar side. To increase water solubility, three additional sequences were synthesized (3–5). The peptide synthesis were performed using solid phase peptide synthesis (SPPS) and Fmoc strategy, in a continuous-flow apparatus, whereas peptide purity was determined and analyzed using HPLC-MS as described in the Materials and methods section of the ESI (Fig. S1–S5).†
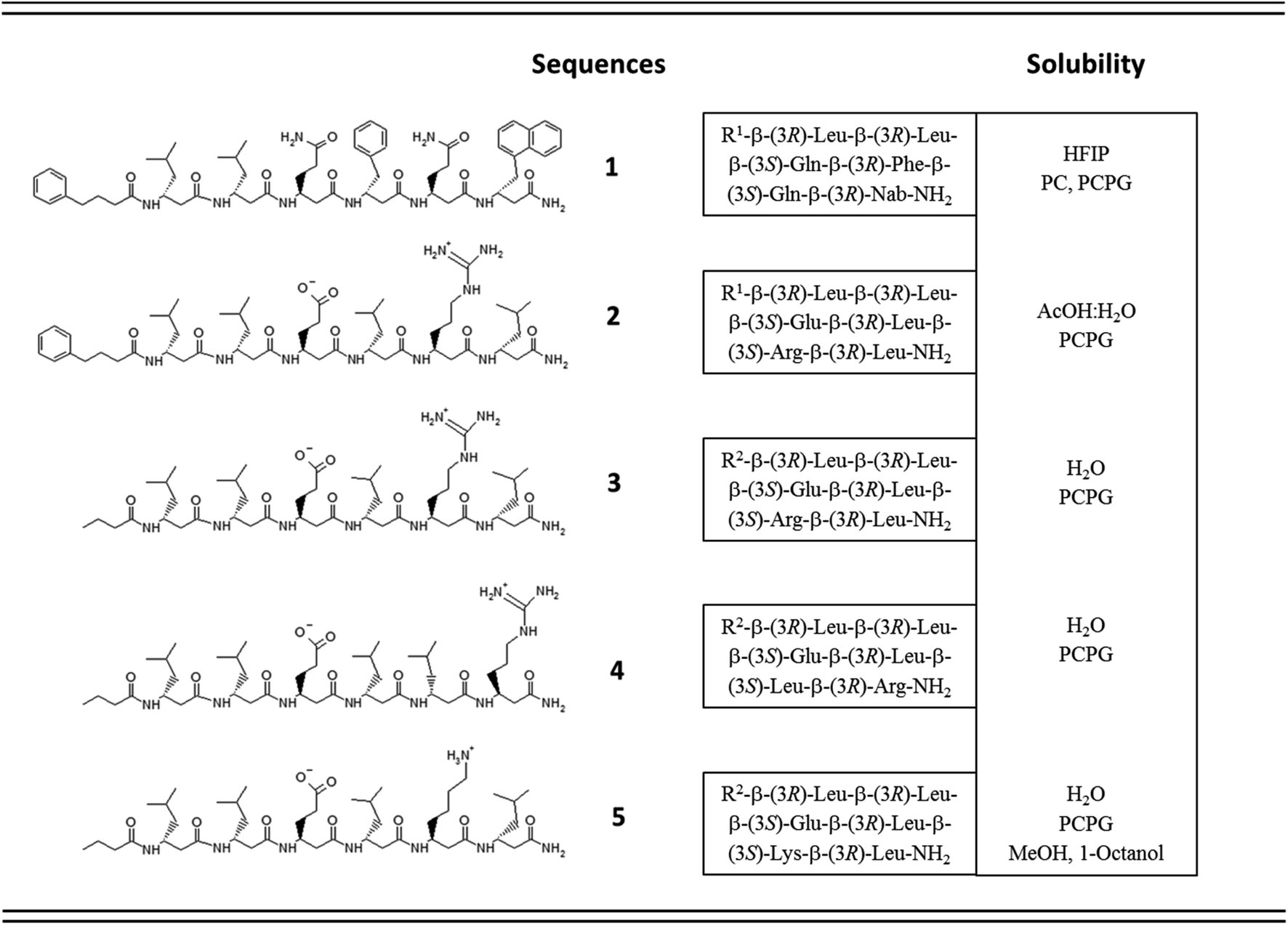
Solubility test of the designed sequences has shown that 1, the most hydrophobic sequence, was soluble only in hexafluoroisopropanol (HFIP), whereas 2 could be dissolved in 10% acetic acid (Fig. 1). The more hydrophilic sequences 3–5 were highly water-soluble.
Structural properties of the foldamers were initially characterized in aqueous solution using a combination of FTIR, NMR and CD spectroscopy.
FTIR spectroscopy can be used for characterization of β-peptide sequences.70,71 Similarly, to IR spectra of natural proteins and peptides, the amide I and II absorption bands, originating mainly from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and N–H bending vibrations (∼1651 and ∼1540 cm−1) of the peptide backbone were considered.72–74 The amide I vibration depends on the backbone structure and is commonly used for secondary structure analysis by fitting with components bands or by decomposing into basis spectra.75
O stretching and N–H bending vibrations (∼1651 and ∼1540 cm−1) of the peptide backbone were considered.72–74 The amide I vibration depends on the backbone structure and is commonly used for secondary structure analysis by fitting with components bands or by decomposing into basis spectra.75
The most intense contribution of deconvoluted amide I band of 1 in HFIP is at 1648 cm−1 which could be assigned to a random coil conformation (Table S1†). For peptide 2 in 10% acetic acid solution the unordered structure was dominant too, and only a very small amount of sheet-like conformation was observed (Table S1†). The band maximum at 1646 cm−1 in the spectra of 3 in water (Fig. 2A) referred to a random coil structure. The band component at ∼1630 cm−1 could be assigned to the vibration of the guanidinium group (νs(CN3H6+)) of the arginine side chain.75 Similar structure was observed for foldamer 4 in water with small shoulders at 1628 and 1688 cm−1 which was attributed to sheet-like conformation (Fig. 2A). In the aqueous solution of 5 the random coil structure was also dominant, while an additional band at 1690 cm−1 could be assigned to turn-like conformations.76,77 Among these, the small bands ∼1567 and 1534 cm−1 originating from deprotonated glutamic acid carboxylate and protonated lysine side chains70,75,78,79 together gave the possibility of salt bridge formation. In the deconvoluted spectra of all three water-soluble foldamers (3–5) (Fig. S6A–C†) a band at 1658 cm−1 with medium intensity was observed, which was previously identified as originating from H-bond formation between the β-peptides.20 These results suggest that random coil structure predominates in “RS” peptides while there is a small amount of sheet-like structures with the presence of inter- and intramolecular H-bonds.

To further study secondary structure, CD spectroscopy was also applied for all water-soluble foldamers (3–5). However, in contrast to α-peptides, for β-peptides, helical and sheet secondary structures often produce similar spectra.80 Consequently, absolute structure determination of these new heterochiral molecules is challenging. Thus, CD spectra were used to compare foldamer conformations to each other (Fig. 2B), rather than to identify individual secondary structures. In each case, positive signals were observed below 206 nm, whereas slightly weaker negative peaks were detected at longer wavelengths. Nevertheless, some differences, between these spectral signatures should be noted, such as the intensity, shape and spectral position of the bands. The CD curve of peptide 4 exhibits similar curve pattern to that of alternating heterochiral trans-2-aminocyclopentanecarboxylic acid (trans-ACPC) hexamer which forms an elongated strand-like structure.25 Peptide 3 shows similar properties, however with lower signal intensities, suggesting a less ordered structure. The CD curve of 5 is similar to those of elongated strand-like β-peptides composed of cis-ACPC with monotonic chirality.69
To obtain further structural information, NMR measurements were also carried out for peptides 3–5. In line with expectations, the dynamic nature of these assemblies resulted in low signal resolution of the 1H NMR spectra (Fig. S8†), which is also a typical property for non-helically ordered structures.25,69 The complete signal assignation of the backbone protons using 2D NMR (Fig. S9 and S10†) measurements cannot be made unequivocally due to the similar chemical shift of the second and fourth amino acids. Thus, we performed DOSY experiments for these peptides (Fig. S11–S13†) which proved more informative. The results revealed the formation of small assemblies in aqueous solution. The average number of molecules in the assemblies were determined as detailed previously20 using 1,4-dioxane as an internal standard.81,82 Based on molar mass calculation of peptides 3–5 it results in oligomers with low number of peptide molecules, with estimated tetrameric to decameric composition (Fig. S11–S13†).
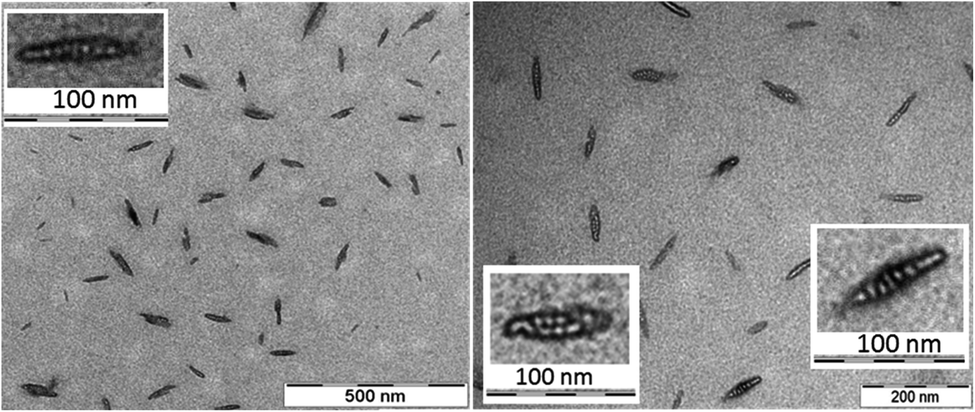
The morphology and the size of the assemblies were investigated by using transmission electron microscopy (TEM) and dynamic light scattering (DLS) techniques. The freshly dissolved peptide 5 showed the formation of rod-or needle-like assemblies in the range of 60–90 nm size (Fig. 3). Detailed investigation of these particles shows that this morphology is formed from smaller individual bundles of ∼8–10 nm in size. Assemblies similar to those of peptide 5 were reported earlier for longer polyglutamine sequences with β-hairpin and for peptides of amyloid β.83–85 This not only suggests capacity of the method to identify these oligomeric species, but also that combination of residues with hydrophobic and hydrogen bonding character can result similar discrete assemblies for otherwise distinct peptidic compounds. DLS measurements also support the size range of formations observed on the TEM images (Fig. S14†).
To support the experimental observations and provide molecular level insight, molecular dynamics (MD) simulations were performed on self-assembly of peptides 3–5. For this we employed our recently developed parameters for β-peptide backbone dihedrals, which enables compounds to explore non-standard conformations as well.86 Besides simulations on the monomer state, spontaneous assembly of eight individual peptides was also tested in explicit water where the computations started with eight individual molecules placed in random orientation at the corners of a cube of 4.5 nm edge length (for more details see the Materials and methods section of the ESI†). Interestingly, after very short simulation time (∼100 ns), the peptide molecules quickly assembled, forming a sandwich-like octameric bundle (Fig. 4). Similar assemblies for peptides 3 and 4 were also obtained (Fig. S15 and S16†). These oligomers consist of two nearly parallel layers (I and II), each of them built from peptides aligned parallel, where the layers are stabilized separately with intermolecular hydrogen bonds. The alignment of the peptide backbones in the two layers is nearly orthogonal (Fig. 4A). The two layers are formed by keeping the Leu side chains in the inner hydrophobic core, while the external ones are hydrophilic with Lys and Glu side chains. The two layers face each other with the hydrophobic side inwards, “protecting” the Leu side chains from the polar solvent (Fig. 4B and C).

The secondary structure of the peptides in the bundle can be assessed by conformational clustering. For analysis, the part of the trajectory corresponding to the aggregated state has been split into eight parts after 400 ns, corresponding to the individual peptides. These sub-trajectories were concatenated, giving a set of frames sampling the secondary structures of all peptides. Cluster analysis was performed87 which identified the most common fold as an elongated conformation termed as zig-zag conformation,59,88,89 accounting for nearly 53% population of the conformations explored throughout the simulation (Fig. S17 and S18†).
The charged Lys and Glu side chains can form salt bridges (Fig. S19 and Table S2†) which are also supported by FTIR results. There are no salt bridges found between chains on opposite (I and II) layers of the aggregate, nonetheless, besides intramolecular salt bridges, almost all neighboring chains in the same layer form intermolecular Lys–Glu side chain hydrogen bonds as well. This suggests that the separate layers are rather stable, with ∼10 intermolecular peptide H-bonds and several intermolecular salt bridges per layer formed between the Lys–Glu side chains.
To determine the membrane activity of synthesized foldamers uni- and multilamellar liposomes were employed. Initially, the effect of these compounds on the lipid phase transition was tested using differential scanning calorimetry (DSC) by focusing on the more hydrophilic peptide compounds (3–5). DSC measurements are typically used to better understand the peptide–lipid interactions by indicating whether guest molecules perturb the phase transitions of the original bilayer.90,91 To get insight into the mechanism of interaction, the 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipid was used, which form multilamellar vesicles (MLVs) in aqueous solution. The fully hydrated DPPC/water system undergoes two phase transitions: the pre-transition from the gel to the rippled gel phases (∼34 °C) and the main transition between the rippled gel and liquid crystalline phases (∼41 °C). The pre-transition exhibits a weak enthalpy change (ΔH ∼ 5.0 kJ mol−1), mainly due to the rotation of the polar headgroups, whereas the main transition shows a well-defined transition temperature (Tm = 41.1 °C) and a larger enthalpy change (ΔH ∼ 35.0 kJ mol−1).92 In the presence of peptides 3–5 (Fig. 5) we observed the disappearance of the pre-transition signal for all three water soluble foldamers. Loss of this signal is typically seen for the perturbed head groups affected by the inserted molecules from the aqueous outer side.93,94 Further on, strongly perturbed signal shape and change in the main transition Tm was detected (Fig. 5 and Table S4†). The shoulder in the right side of the transition signal is caused most likely by domains enriched in foldamers.

To better understand β-peptide–lipid interactions, we investigated the two major regions of the lipid bilayer using IR spectroscopy. The band position of the symmetric (νsCH2) at 2853 cm−1 and antisymmetric (νasCH2) at 2922 cm−1 methylene stretching vibrations are a sensitive parameter to lipid conformational order and acyl chain packing.75,95 No significant changes were observed in the CH2 region of DOPC and DOPC/DOPG liposomes when peptides 1 and 2 were added (data not shown), suggesting that these peptides do not perturb the hydrophobic lipid chains in the membrane. The phosphate group vibrations with antisymmetric (νasPO2−) and symmetric (νsPO2−) stretching modes are at around 1240–1230 cm−1 and 1087 cm−1, respectively. The R–O–P–O–R′ stretching vibration generally appears as a shoulder near the νsPO2− band at ∼1066 cm−1. For 1 and 2, νasPO2−, the most sensitive band for H-bonding interactions, showed a shift towards lower wavenumbers compared to the pure lipid systems (Fig. S7A†). This indicates that peptides 1 and 2 are situated on the surface of the lipid bilayer, perturbing the primary hydration shell of the lipid molecules, and inducing a conformational change of the lipid headgroup moiety. The amide I band, tracked regularly for natural peptides,96–98 was also analyzed (Fig. S7B†). In the presence of DOPC liposomes, the main band around 1648 cm−1 assigned to the random coil structure remained dominant for 1 and 2 and a new band component appeared around 1688 cm−1. More pronounced changes were observed with the negatively charged DOPC/DOPG liposomes. The spectrum showed a more ordered structure with a pronounced band at 1688 cm−1 and a small one at 1629 cm−1, which could be assigned to a sheet-like conformation. The most intense contribution of amide I band of 2 in the presence of DOPC/DOPG liposomes is shifted to 1653 cm−1 and two shoulders could be observed at 1671 and 1638 cm−1, which may again suggest sheet-like conformations.
Based on IR spectra of peptides 1 and 2, the more water-soluble RS compounds (3–5) were investigated only in the presence of DOPC/DOPG liposomes. Interestingly, for peptide 5 a shift was observed for the acyl chain vibrations in both the antisymmetric (νasCH2) (from 2922.1 to 2924.1 cm−1) and the symmetric (νsCH2) (from 2852.7 to 2853.7 cm−1) methylene stretching vibrations compared to that of pure lipids (Fig. 6A). The extent of these shifts agrees well in magnitude with those observed for other membrane inserted peptides.99 These results suggest that peptide 5 interacts with the lipid acyl chain region by inserting into the lipid bilayer, which cannot be observed for peptides 3 and 4. For the latter two, the antisymmetric and symmetric PO2− stretching vibrations showed a shift towards lower wavenumbers (from 1243 to 1233 cm−1 and from 1091 to 1087 cm−1), which refers to the “on surface” localization of these peptides (Fig. 6B). The intensity of the phosphate-diester stretching band (R–O–P–O–R′) also decreased as observed for 1 and 2 in liposome systems (Fig. S7A†). Regarding the amide I band of peptide 3 (Fig. 6C) in the presence of DOPC/DOPG liposomes a broadening of the band was observed with a small shoulder at 1620 cm−1 which may refer to some sheet-like secondary structure. A similar broad band was also seen for 4 in the presence of liposomes with a shoulder around 1665 cm−1 which may be allocated to intramolecular hydrogen bonding. When comparing the water-soluble RS compounds, the highest change in peptide secondary structure was presented by peptide 5 upon interaction with the lipid bilayer (Fig. S6D–F†). The relative amount of random coil structure in the peptide 5–liposome system decreased, while the appearance of the band component at ∼1675 cm−1 strongly indicates an increase in intermolecular H-bonding.

In overall, these observations from FTIR spectra confirmed that the peptide 5 with increased amount of intermolecular H-bonds is situated below the head group region. This effect was observed neither for peptides 1 and 2, nor for 3 and 4, as these were located mainly near to or embedded into the lipid head group regions.
Further on, the presence of DOPC/DOPG liposomes caused a substantial blue shift in the CD spectrum of peptide 3 (Fig. 6D). Both the zero-crossing point and the λmin were displaced to lower wavelengths by 6 and 4–5 nm, respectively, suggesting a stronger effect of the membrane on peptide conformation. The negative band of 4 in the presence of liposomes totally overlaps with that obtained in water and its minimum shows only a small, 2 nm red shift (Fig. 6D). Such a slight alteration of the CD profile does not indicate gross conformational change in the peptide. When compared to CD spectra of peptide 5 in water, the zero-crossing point remains at 203 nm also in the lipid system, whereas the positive band exhibits a moderate red shift. The negative band shows two minima at 210 and 217 nm, which cannot be seen in aqueous solution. These changes may correlate with the increase of strand-like conformation content induced by the membrane interaction. However, the lower signal intensity due to the presence of liposomes makes direct comparison of the aqueous and membrane phases difficult. Thus, conformational variations of foldamer 5 were further investigated in solvents with different polarity (Fig. S23†). The minimum of the negative band detected at 215 nm in water is blue shifted to 207 nm in methanol, and the same shift can be observed for the zero-crossing point as well. On the other hand, this blue shift in octanol is smaller, resulting in a shallow negative minimum at ∼212 nm. Octanol is widely used to mimic membrane environment and based on the slight difference observed in the shape of CD curves for water and octanol, it is proposed that the peptide conformations in these solvents lie close to each other. The most significant shift in methanol suggests larger structural changes, which may indicate appearance of monomers. To support this, MD calculations were also performed using methanol as solvent. Eight individual peptide molecules were placed in random orientation using the same setting parameters as in water simulations (for more details see the Materials and methods section of the ESI†). In contrast to the aqueous phase, oligomer formation did not occur in methanol over the first μs of the simulation time. Note that formation of dynamic assemblies in water occurred already at the first 100 ns of the simulation time. To further confirm monomer state, the simulation was continued until a full 2 μs of simulation time, under which no assembly formation was observed as demonstrated by the lower number of intermolecular hydrogen bonds (Fig. S20†).
To gain molecular level insight to membrane interactions, 5 was subjected to further MD simulations in the presence of a lipid bilayer. The aim of these simulations was to investigate how peptide 5 could be embedded in the lipid chain region as suggested by experimental results detailed above. Accordingly, simulations were started with peptide 5 centered into the middle of the bilayer. The applied atomic restraints during the minimization and equilibration process are summarized in Table S3.† To estimate the optimal extent of self-assembly, three parallel simulations were performed, with a monomeric, tetrameric and an octameric form as starting structure. Considering the hydrophobic surrounding, the oligomeric form was assumed to be similar to the water-soluble two-layered assemblies but having the leucine on the exterior side and the charged residues on the interior side. For the monomeric and tetrameric state a rapid, spontaneous reorientation occurred in the first ∼100 ns (Fig. 7A), after which both forms are situated mainly in the vicinity of the bilayer surface, where they are stabilized over the remaining simulation time (900 ns). In contrast, the octameric form remained in the hydrophobic interior of the lipid acyl chains (Fig. 7A), with very little variation throughout the entire simulation time. This foldamer oligomer also retains connection with both ends of the lipid bilayer, by having several neighboring lipids bent from the original membrane surface. An overall thinning of the membrane from both sides is caused in the vicinity of the oligomer (Fig. 7B). The experimental and simulation limitations prevent the quantification of the number of peptides present in aqueous or lipid bilayer inserted oligomers. Nevertheless, based on the above it is concluded that an octameric association of peptide 5 is already large enough to properly shield the charged residues in the depth of the lipid bilayers. The overall structure resembles to the water-soluble one, namely it is also a two-layered assembly with several H-bonds between the individual peptides in each layer. Conformational analysis of the amino acid residues indicates similar torsional angle distributions for the aqueous and bilayer forms, though with a somewhat altered ratio (Fig. S18†). However, in contrast to the water soluble form, the angle between the plane of layer I and layer II shows much less variation in the lipid bilayer, its value stays ∼60°–80° throughout the entire simulation time (Fig. S21†). The lipid bilayer structure also contains numerous salt bridges formed by the lysine and glutamate side chains in the interior of the assembly. Note, that when comparing relative average positions of the monomeric, tetrameric and octameric forms, a qualitative mechanism on membrane insertion can be outlined (Fig. 7A). Accordingly, as the number of peptides in the oligomers increase, the position of peptide 5 gets closer to the center of the lipid bilayer. In the case of the octameric form, the observed stability in the depth of the membrane will also be supported by the apolar butyryl groups on the N-terminal of peptide 5. Furthermore, the large, ∼60°–80° angle between the two layers allows for such an orientation where all apolar N-terminals are placed towards the hydrophobic core of the bilayer (Fig. 7B). The presence of oligomers inside the lipid bilayer is also in line with the sequential build-up of the molecule. Since the alternating sequence prevents helix formation, the charged residues would fall on the same side in a more elongated conformation of a monomer, where Glu and Lys are likely to form a salt bridge in the hydrophobic membrane environment. However, a single Glu–Lys salt bridge will be still a rather polar region and the monomer state cannot shield that from the lipid bilayer. Thus, based on the IR and DSC results, the MD simulations and the sequence of peptide 5, it can be concluded that monomeric form inside the bilayer is unlikely.

Based on the experiments and MD simulations, the oligomers formed by the water-soluble foldamers 3–5 have a hydrophobic core in aqueous solution with intermolecular H-bonds in a more elongated backbone conformation. To better validate their characteristics, as well as to investigate their potential applicability, binding measurements were employed using markers of mainly hydrophobic features. The fluorescence probes ThT (Thioflavin-T) and ANS (8-anilinonaphthalene-1-sulfonic acid) are typically used for sheet-rich associates, such as amyloids100–102 and hydrophobic membranes103 or micelles.104 Furthermore, considering that these assemblies may provide unique opportunities as artificial cargo delivering biomolecular scaffolds, their hosting capacities for pyrene, a planar, water-insoluble molecule, were also tested.
In aqueous solutions, ANS, a well-known hydrophobic probe, shows a weak fluorescence signal with an emission maximum at ∼530 nm, which is blue shifted in hydrophobic environment to below 500 nm.105 Model membranes or micelles with net positive or neutral head-groups bind ANS buried at the hydrophobic–hydrophilic edge, as reflected by the emission maximum of 480 nm for the PC liposomes (Fig. 8A). Similarly, proteins with exposed hydrophobic patches with a nearby positively charged moiety where ANS fits well, exhibit remarkably blue-shifted emission maxima, too. When tested with the assemblies of 3–5, a dose-dependent shift of the maximum in the 465–470 nm range were detected upon consecutive addition of the peptides. This suggests the presence of an optimal ANS binding site where the probe is exposed to the solvent/buried from the solvent to a comparable extent, as in the case of lipid bilayers or suitable protein patches.

In contrast to the minor changes in emission maximum, larger differences were detected for the signal intensities as observed for peptide 5 in comparison to peptides 3 and 4 (Fig. 8B). Considering the sequential differences of peptides, this can presumably be attributed to the replacement of Arg in 3 and 4 to Lys in 5. This change might manifest in the effect observed as the positively charged peptide side chains could contribute to the binding via electrostatic attraction to the ANS sulfonyl group, or via cation–π interaction formed between cationic side chains and ANS aromatic rings.106 Alternatively, the higher capacity of Arg over Lys to form multiple thus stronger H-bond networks might result in higher oligomerization tendency, leading to the formation of bigger, less structured aggregates with enhanced ANS binding properties. In addition, the oligomeric state might also affect ANS binding, resulting in intensity variations, supported by the enhanced signal background arising from light scattering of larger particles observed in TEM images. Moreover, peptide assemblies and proteins in partially unfolded states, decorated with exposed hydrophobic patches, are characterized by remarkably enhanced ANS fluorescence, while well-folded structure with a more buried hydrophobic interior exhibit less pronounced ANS signals.107 Based on these considerations, the high signal intensities detected for 3 and 4 are likely indicative of a less folded, loosely packed overall structure, whereas the relatively low ANS binding intensity observed for 5 suggests more organized, folded associates, in agreement with the IR and TEM results. Considering the signal enhancement, titration data showed a nearly linear dose-dependence on foldamer concentration (Fig. 8B). This data may also suggest a critical association concentration (cac) below 20 μM as ANS binding was detected even at concentrations as low as 20–50 μM, indicating presence of foldamer assemblies capable of effectively hosting guest molecules at low peptide levels.
Host–guest interaction was further tested using the probe ThT, which is sensitive for the rigidity of the microenvironment.100 ThT alone also exhibited self-fluorescence,108 the intensity of which increased upon addition of peptides 3–5 (Fig. 8C). In consideration with the structure and morphology of the assemblies derived from our MD simulations and TEM images, ThT could bind to the side cavity formed between the two layers. This arrangement could provide a favourable surface for the dye to align in an extended conformation, which is the supposed prerequisite for the induced fluorescence. Further on, this is in line with the preferred ThT binding site, which was concluded earlier to be enriched in hydrophobic and aromatic side chains, but lacking many charged groups.100 Analysing the ThT signal, the induced intensity was the lowest for 4 and the highest for 3 which can be explained in terms of slightly different morphology of the assemblies, or sequential environment. For amyloid structures, the induced signal was proven to be proportional to the concentration, allowing fibril quantification based on the intensity, at least up to 10 μM amyloid.108 In comparison, the dose dependence curves show saturation type behaviour at higher foldamer levels, indicating a dissociation constant of the interaction in the micromolar range (Fig. 8C). Altogether, fluorescence-based findings suggest similar host capacity for 3–5 indicating the formation of assemblies. Fluorescent probes differing in hydrophobicity and charge reported variations in binding capacities, which are consistent with sequential and spatial preferences of the particular dye. The rather linear dependence of the probe interactions indicates no critical association concentration for the foldamers in the 20–250 μM range suggesting that the observed assemblies are formed readily at low concentrations.
Finally, to test these assemblies toward potential applicability as hosts or transporters of hydrophobic biomolecules or drugs, we employed peptide 5 and the planar small molecule, pyrene, which readily inserts into lipid membranes due to its hydrophobic nature.109–111 UV-Vis measurements indicated that the pyrene molecule is inserted into the foldamer assembly similarly as it inserts into lipid bilayers (Fig. 9A). The broad band with a maximum at 360 nm assigned to pyrene aggregates present in water,112 almost vanished upon titration with peptide 5 and the shape of the spectra in the lower (300–350 nm) wavelength range resembled that of monomeric pyrene inserted in a lipid bilayer.113,114 The reduction of the absorption band intensity at 360 nm has already initiated with the very first addition of 5 with as low as 5 μM concentration (Fig. 9A inset). This observation suggests that oligomerization of compound 5 takes place already in the low micromolar range. This is also supported by the signal enhancement results during ANS titration for 5 (Fig. 8B). Thus, qualitatively both measurements point towards a critical association concentration (cac) to be in this range, estimated to be ∼5–20 μM. However, taking into consideration that our peptides show some similarity to sheet-like amyloid peptide systems, which are known to have complex oligomerization processes,115–117 we cannot exclude that multiple aggregation states, starting potentially well below micromolar concentrations, occur also for the currently presented compounds.

Insertion of pyrene into the bundles formed by 5 has also been investigated by MD simulations. A single pyrene molecule was simulated with the solvated octameric oligomer of 5. According to the simulation results, the pyrene molecule interacts closely with peptide 5 in most of the total 1 μs simulation time. For a more in-depth analysis, the oligomer was sectioned into four regions (as detailed in the ESI†) and the position of the guest molecule was followed by measuring pair distances with each section. Accordingly, the fully hydrophobic pyrene is located either in the internal leucine core, providing an optimal hydrophobic environment, or near the core but closer to the butyryl groups of the N-terminus, partially exposed to the solution (Fig. 9B and S22†). In this way a small bundle is capable of hosting water-insoluble compound and no significant change in the oligomeric state and peptide conformation was found during the simulation. Only some re-ordering of the chains was observed whereas the number of intermolecular hydrogen bonds remained nearly the same. These suggest that the two-layered assemblies can be sufficiently dynamic to allow rearrangements upon incoming guest molecules and decrease or negate potentially arising steric strains within the assembly.
Out of the tested foldamers, the water-soluble ones (3–5) showed the highest affinity for self-assembly, according to NMR, IR, fluorescence spectroscopy, TEM, DLS and MD simulations. These β3-peptides have an affinity to assemble primarily into oligomers composed of only a small number of molecules, where self-assembly can take place even at relatively low, ∼5 μM peptide concentrations. The MD simulations indicate that these bundles have two opposite sides, where in each side intermolecular hydrogen bonds can be formed and more elongated peptide conformations dominate. Similar pattern of intermolecular hydrogen bonds between β-peptides were earlier predicted by ab initio quantum chemical investigations88,89 and were also observed for infinite fibrils of foldamers with pentacyclic β-amino acids.69 Note, that although a proportion of these bundles can further associate into larger finite rod-like morphologies with ∼80 nm length, our employed acyclic residues with the combination of homochiral and alternating heterochiral regions in the sequence makes a unique construct which is flexible enough to result in formation of finite oligomers. The fluorescence measurements show that the water-soluble forms of the oligomers have characteristics of both sheet-like assemblies and a membrane-like hydrophobic core. Furthermore, their most interesting property, namely solubility both in water and in lipid bilayers, is also likely due to the special structural characteristics as observed for oligomers of 5. Based on the IR and the MD results, there are 2–3 intermolecular H-bonds between each molecule in one layer, stabilizing the two separate sides of these oligomers. However, there are no H-bonds observed between the two layers forming the hydrophobic core. Consequently, there is likely no high enthalpic barrier for switching from water soluble form to a membrane soluble form in the presence of a lipid bilayer, where the latter could insert into the hydrophobic regions of the membrane as shown by IR and DSC results and by the MD membrane simulations. The lack of the H-bonds connecting the two opposing sides is likely crucial for such a Janus-faced character because when compared to β-barrel proteins, or to the more resembling natural examples, i.e. the cylindrin structure of toxic amyloid oligomers,118 the intermolecular H-bonds in the water-soluble state of those structures probably contributes to their stability, preventing them to enter in the membranes.118 Note, that despite several of the designed foldamers show both water soluble and membrane soluble forms, further studies will clearly be required to identify potential stages and kinetics of related self-assembly and membrane insertion processes, to characterize how sequential changes will modulate formation of small oligomers and their affinity to form larger assemblies, and also to further test their capacity to adopt to both hydrophilic and hydrophobic environments. Finally, the ability to readily host hydrophobic small molecules as demonstrated by using pyrene suggests that similar but more specific functions in the future could be reached for these assemblies. The designed chirality pattern and the resulting small scaffold is hoped to provide a platform from which numerous applications could be developed by subtle variation of the original sequence, without altering much the formed bundles. Moreover, the similarity of these assemblies to natural small amyloid oligomers is also hoped to widen our understanding on the complex morphological stages prior fibril formation, which may provide a better molecular insight to related processes resulting in pathological conditions.
The sequences (1–5) were synthesized by solid phase technique and Fmoc chemistry in continuous-flow apparatus119 using two equivalents of amino-acid and HATU as coupling reagent in DMF and four equivalents of DIPEA. More details in peptide synthesis and purification are reported in ESI.†
NMR measurements for peptides (3–5) in water (H2O : D2O = 90 : 10 v/v) solution were carried out on a Bruker Avance III 500 MHz spectrometer equipped with a cryo probe head. FTIR spectra of thin dry films were collected on a Varian 2000 Spectrometer fitted with a diamond attenuated total reflection cell by slowly evaporating the solvent under ambient conditions. Fluorescence measurements were performed using a Jobin Yvon Fluoromax-3 spectrofluorimeter and ANS and ThT probes were titrated upon consecutive addition of peptides (3–5). Mass spectrometric measurements were performed on a high resolution and high mass accuracy Thermo Scientific Q Exactive Focus hybrid quadrupole-orbitrap mass spectrometer coupled to a Dionex 3000 UHPLC system.
An extended Method section is reported in ESI.†
Molecular dynamics simulations were done using version 2018.6 of the GROMACS software,120 and the CHARMM36m force field extended with torsional potential parameters for β-amino acids developed recently.86 Simulations on peptides (3–5) were carried out in aqueous solution and lipid bilayer at 300 K temperature and 1 bar pressure with 2 fs timesteps, in explicit water (CHARMM-modified TIP3P model) with 150 mM NaCl added. More details in MD simulations are reported in ESI.†
T. B. S., I. M. and I. Cs. Sz. conceived the project. I. Cs. Sz. designed and performed CD, ATR-FTIR experiments and data analysis. J. M. contributed to ATR-FTIR data evaluation and discussion. A. V. and G. K. performed molecular dynamics simulations. D. B. and I. M. performed NMR experiments and data analysis. G. S. performed high resolution mass spectrometric measurements. T. J. performed fluorescence measurements, data analysis and discussion. F. Zs. contributed to CD data interpretation and discussion. A. B. and Z. V. were involved in the design and execution of TEM measurements and data interpretation. V. U. and F. F. were involved in discussion. I. Cs. Sz. and T. B. S. wrote the manuscript.
There are no conflicts to declare.
This work was funded by the Momentum Programme (LP2016-2 (T. B.-S.) and LP2017-8 (I. M.)) of the Hungarian Academy of Sciences, by the National Competitiveness and Excellence Program (NVKP_16-1-2016-0007) of the National Research, Development and Innovation Office NKFIH, Hungary, and by the GINOP grant (BIONANO_GINOP-2.3.2-15-2016-00017). We also thank for the National Research, Development and Innovation Office NKFIH, Hungary (grants OTKA PD-121326 and PD-124451), and for Marie Curie Fellowship for T. B.-S. (MSCA-IF BARREL 660030). The János Bolyai Research Scholarship (Z. V.) and the MTA Premium Post-Doctorate Research Program (G. S.) of the Hungarian Academy of Sciences are greatly acknowledged. These studies were also supported by grant VEKOP-2.3.3-15-2017-00020 from the European Union and the State of Hungary, co-financed by the European Regional Development Fund. The financial support of HunProtEx 2018-1.2.1-NKP-2018-00005 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the 2018-1.2.1-NKP funding scheme. The ELTE Thematic Excellence Programme (Szint+) supported by the Hungarian Ministry for Innovation and Technology is hereby acknowledged. The authors thank Terész Kiss for performing TEM measurements.
This journal is © The Royal Society of Chemistry 2020