
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomerization-induced enhancement of luminescence in Au28(SR)20 nanoclusters†
Yuxiang
Chen
 ,
Meng
Zhou
,
Qi
Li
,
Harrison
Gronlund
and
Rongchao
Jin
*
,
Meng
Zhou
,
Qi
Li
,
Harrison
Gronlund
and
Rongchao
Jin
*
Department of Chemistry, Carnegie Mellon University, Pennsylvania 15213, USA. E-mail: rongchao@andrew.cmu.edu
First published on 17th July 2020
Abstract
Understanding the origin and structural basis of the photoluminescence (PL) phenomenon in thiolate-protected metal nanoclusters is of paramount importance for both fundamental science and practical applications. It remains a major challenge to correlate the PL properties with the atomic-level structure due to the complex interplay of the metal core (i.e. the inner kernel) and the exterior shell (i.e. surface Au(I)-thiolate staple motifs). Decoupling these two intertwined structural factors is critical in order to understand the PL origin. Herein, we utilize two Au28(SR)20 nanoclusters with different –R groups, which possess the same core but different shell structures and thus provide an ideal system for the PL study. We discover that the Au28(CHT)20 (CHT: cyclohexanethiolate) nanocluster exhibits a more than 15-fold higher PL quantum yield than the Au28(TBBT)20 nanocluster (TBBT: p-tert-butylbenzenethiolate). Such an enhancement is found to originate from the different structural arrangement of the staple motifs in the shell, which modifies the electron relaxation dynamics in the inner core to different extents for the two nanoclusters. The emergence of a long PL lifetime component in the more emissive Au28(CHT)20 nanocluster reveals that its PL is enhanced by suppressing the nonradiative pathway. The presence of long, interlocked staple motifs is further identified as a key structural parameter that favors the luminescence. Overall, this work offers structural insights into the PL origin in Au28(SR)20 nanoclusters and provides some guidelines for designing luminescent metal nanoclusters for sensing and optoelectronic applications.
Introduction
Atomically precise metal nanoclusters have emerged as a new class of nanomaterials in recent years and hold promise in many applications such as chemical sensing, biological imaging, and catalysis.1–12 Luminescent metal nanoclusters are of particular interest owing to their unique properties, including their high stability, low toxicity, large Stokes shift, and long luminescence lifetime, and thus such nanoclusters have attracted significant research interest in recent years.13–24A major effort in current research on luminescent nanoclusters protected by thiolates is focused on the development of effective strategies to enhance their luminescence. To improve the quantum yield of photoluminescence (PL), it is of paramount importance to understand the PL mechanism and, in particular, the structural basis.18 Toward this goal, significant efforts have been made in recent years.25–37 Wu et al. identified that the presence of electron-rich atoms (e.g., oxygen and nitrogen) or functional groups (e.g., the carboxyl or amine group) in the thiolate ligands could enhance the PL of Au25(SR)18 nanoclusters via ligand to metal core charge transfer.25 Xie et al. reported an aggregation induced emission (AIE) mechanism in gold nanoclusters.28–30 Lee et al. reported that rigidifying the gold shell of the nanocluster can efficiently enhance PL.31 Foreign metal doping and alloying is also an important strategy for boosting the PL in metal nanoclusters.37–40 Theoretical insights into PL have been obtained by Aikens et al.26 Despite such progress, atomic-level understanding of the origin and structural basis for the PL mechanism in metal nanoclusters still remains a challenge.
Intense research on gold nanoclusters have created a series of atomically precise nanoclusters with diverse and tunable structures.1,41–49 For example, by utilizing ligand-based synthetic strategies, atomic-level control has been achieved in manipulating the cluster size,50 shape51 (such as hexagonal prism shaped Au40vs. tetragonal rod shaped Au52), surface structure,43,52 and core structure.53–55 With the valuable structural information, it has become possible to precisely correlate the PL properties of gold nanoclusters with the atomic structure.33
For thiolate-protected Aun(SR)m nanoclusters, both the inner gold kernel (hereafter referred to as core) and surface Aux(SR)x+1 motifs (hereafter referred to as shell) are considered as important contributors to their PL properties.28,56 For the well-studied icosahedral Au25(SR)18 nanocluster, the core-to-shell relaxation was observed in ultrafast electron dynamics and the PL mechanism was interpreted,57–61 including the theoretical insights.35 However, the PL origin and mechanism for other Aun(SR)m nanoclusters with non-icosahedral structures largely remain unclear, albeit all possess the core–shell structures. In consideration of the coupling between the core and the shell, the individual roles of these two intertwined structural parameters have not been elucidated. Thus, an important step toward atomic-level understanding of PL is to decouple these two structural parts by selectively changing one while retaining the other and then investigate the effect to identify the mechanistic pathway. The 28-gold-atom nanocluster,52i.e., Au28(CHT)20 (CHT = cyclohexanethiolate) and Au28(TBBT)20 (TBBT = p-tert-butylbenzenethiolate), which share the same core but different shell structures, can serve as an ideal system for such a purpose.
Herein, the luminescence origin of the two Au28(SR)20 nanoclusters is investigated. Interestingly, the Au28(CHT)20 nanocluster exhibits more than 15-fold higher PL quantum yield than Au28(TBBT)20. Time-resolved PL measurements identify a long lifetime component in Au28(CHT)20, indicating that the nonradiative pathway is suppressed compared to that in Au28(TBBT)20. To understand the enhancement of PL, ultrafast transient absorption spectroscopy is employed to probe the electron dynamics of these two correlated nanoclusters. Based on all the results, we present a shell-mediated core emission mechanism to explain the structural origin of the PL in Au28(SR)20 nanoclusters. In such a luminescence pathway, the emission solely arises from the metal core while the electron dynamics of the core is distinctly influenced by the shell.
Results and discussion
The synthesis of the Au28(TBBT)20 nanocluster followed a previously reported approach,49 and the Au28(CHT)20 was obtained by a ligand-induced isomerization reaction of Au28(TBBT)20 with excess cyclohexanethiol at 80 °C.52 Both Au28(SR)20 nanoclusters are thermally robust and their total structures were determined by X-ray crystallography49,52 as shown in Fig. 1. The Au28(CHT)20 and Au28(TBBT)20 nanoclusters have the same inner Au14 core structure (Fig. 1a and d) but distinctly different shell structures (i.e., surface staple motifs, Fig. 1b and e). It is worth noting that, for face-centered cubic (FCC) gold nanoclusters, there are several alternative ways to categorize the core and shell gold atoms.62 Here we consider the gold atoms with the shortest Au–Au bond lengths (i.e., average 2.83 Å in Au28(CHT)20 and 2.84 Å in Au28(TBBT)20) as the core (Fig. 1a and d). In this view, both Au28(SR)20 isomers have the same Au14 core, which consists of a dimer of bitetrahedral Au7. The Au7 unit can further be dissected into two Au4 units that share one common vertex (see the ESI, Fig. S1†). This view of the core structure is supported by X-ray absorption spectroscopy,63 Bader charge analysis,64 and other theoretical analyses.65–67 Of note, other views (e.g., cuboctahedron or a cubic box)52,62 do not affect the fact that the two Au28(SR)20 nanoclusters have the same gold core but different shells.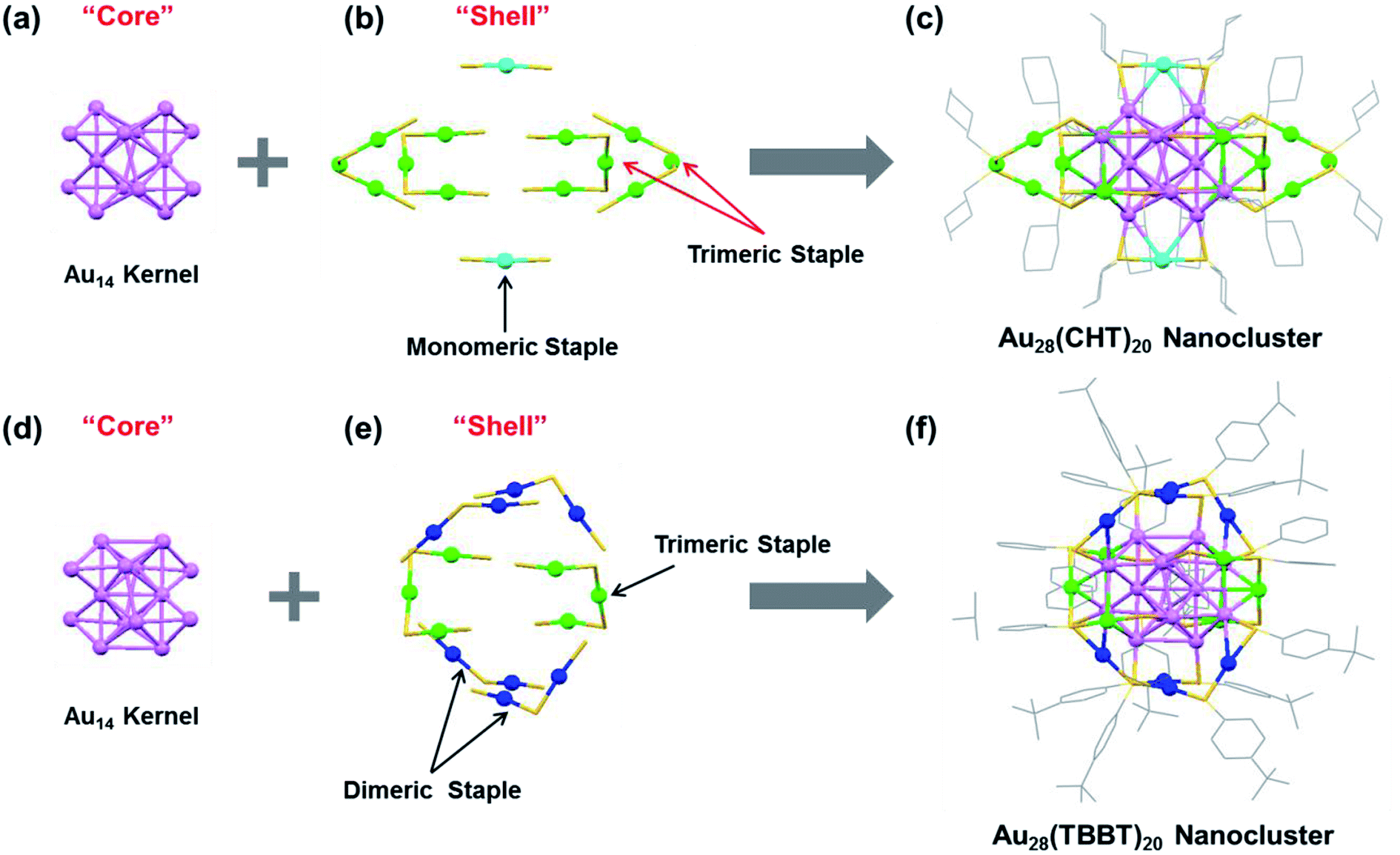 | ||
| Fig. 1 X-ray structures of (a–c) Au28(CHT)20 and (d–f) Au28(TBBT)20 featuring the “core” and “shell” components. Color labels: purple = core Au atoms; other colors = shell Au atoms. The carbon tails in (b and e) are omitted for clarity. Redrawn from ref. 49 and 52. | ||
The shell structure exhibits completely different arrangements between the two Au28(SR)20 nanoclusters. In the Au28(CHT)20 nanocluster, the Au14 core is surrounded by two monomeric staple motifs (SR-Au-SR) and four trimeric staple motifs (SR-Au-SR-Au-SR-Au-SR), Fig. 1b. The trimeric motifs form two pairs of interlocked configuration (highlighted with red arrows in Fig. 1b). In contrast, the shell of Au28(TBBT)20 contains four dimeric staples (SR-Au-SR-Au-SR) and two trimeric staples (SR-Au-SR-Au-SR-Au-SR), Fig. 1e. The total structures of Au28(CHT)20 and Au28(TBBT)20 are shown in Fig. 1c and f. The two Au28(SR)20 nanoclusters motivate us to investigate their PL properties in the current work.
The optical absorption spectra of the Au28(CHT)20 and Au28(TBBT)20 nanoclusters are shown in Fig. 2a and c, respectively. Both Au28(SR)20 nanoclusters exhibit very similar UV-vis optical features despite the blue shifting of the absorption bands for Au28(CHT)20 compared with those of Au28(TBBT)20. It is worth noting that these two Au28(SR)20 isomers also exhibit a similar HOMO–LUMO band gap around 1.7 eV as determined from their optical spectra and differential pulse voltammogram.68 The closely resembling optical absorption spectra indicate that the core structure dictates the absorption spectra.
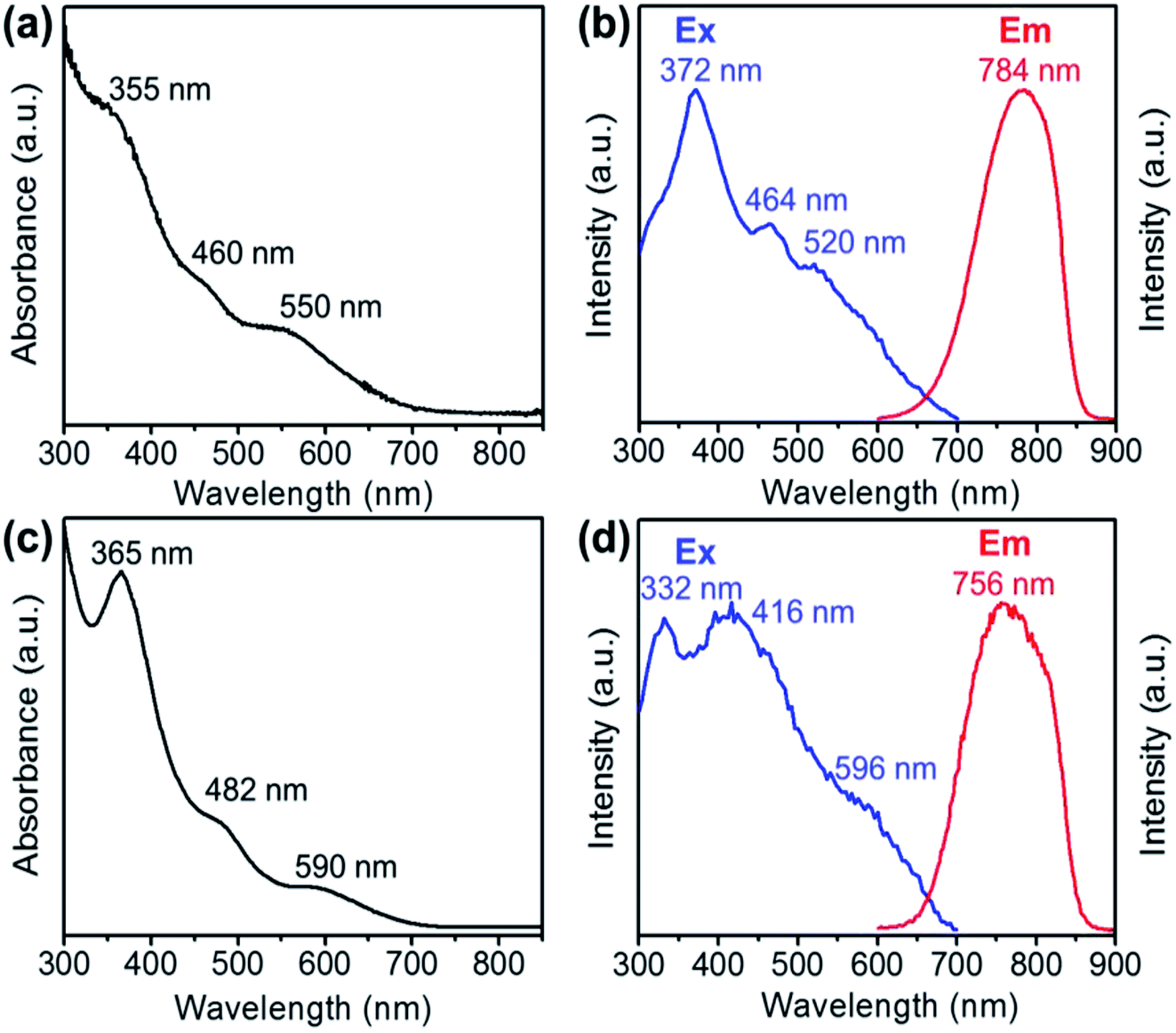 | ||
| Fig. 2 UV-vis absorption spectra of (a) Au28(CHT)20 and (c) Au28(TBBT)20. Photoluminescence excitation (Ex, monitored emission at the maximum intensity) and emission (Em, excited at 372 nm) spectra of (b) Au28(CHT)20 and (d) Au28(TBBT)20. | ||
The photo excitation and emission spectra of Au28(CHT)20 and Au28(TBBT)20 are shown in Fig. 2b and d, respectively. The PL emission spectrum of Au28(CHT)20 is centered at ∼784 nm (Fig. 2b, red profile). The PL excitation spectrum exhibits three peaks with the highest peak at 372 nm (Fig. 2b, blue profile). For Au28(TBBT)20, the PL spectrum is centered at 756 nm which is blue shifted compared with Au28(CHT)20 (Fig. 2d). The PL excitation spectrum of Au28(TBBT)20 also shows three bands but less distinct, in comparison to Au28(CHT)20 (Fig. 2d). Interestingly, the PL intensity of Au28(CHT)20 is significantly higher than that of Au28(TBBT)20 as revealed in the excitation/emission contour maps (Fig. 3a and b). The PL excitation was scanned from 300 to 700 nm and the emission profile was recorded from 600 to 900 nm. As shown in Fig. 3, although both Au28(SR)20 nanoclusters emit in the 725 to 825 nm region, the quantum yield (QY) of Au28(CHT)20 is more than 15-fold higher than that of Au28(TBBT)20, i.e. 1.6% vs. 0.1%. Herein, the QY measurements used Au25(SG)18 (QY = 0.2%)25 as the reference. We note that gold nanoclusters protected by hydrophobic thiolates are often weak in luminescence (QY < 1%). Only a few gold nanoclusters, i.e., the present Au28(CHT)20 and the previously reported Au24(SR)20 (R = CH2Ph-tBu, CH2Ph, C2H4Ph) have QYs greater than 1%.15,16,27
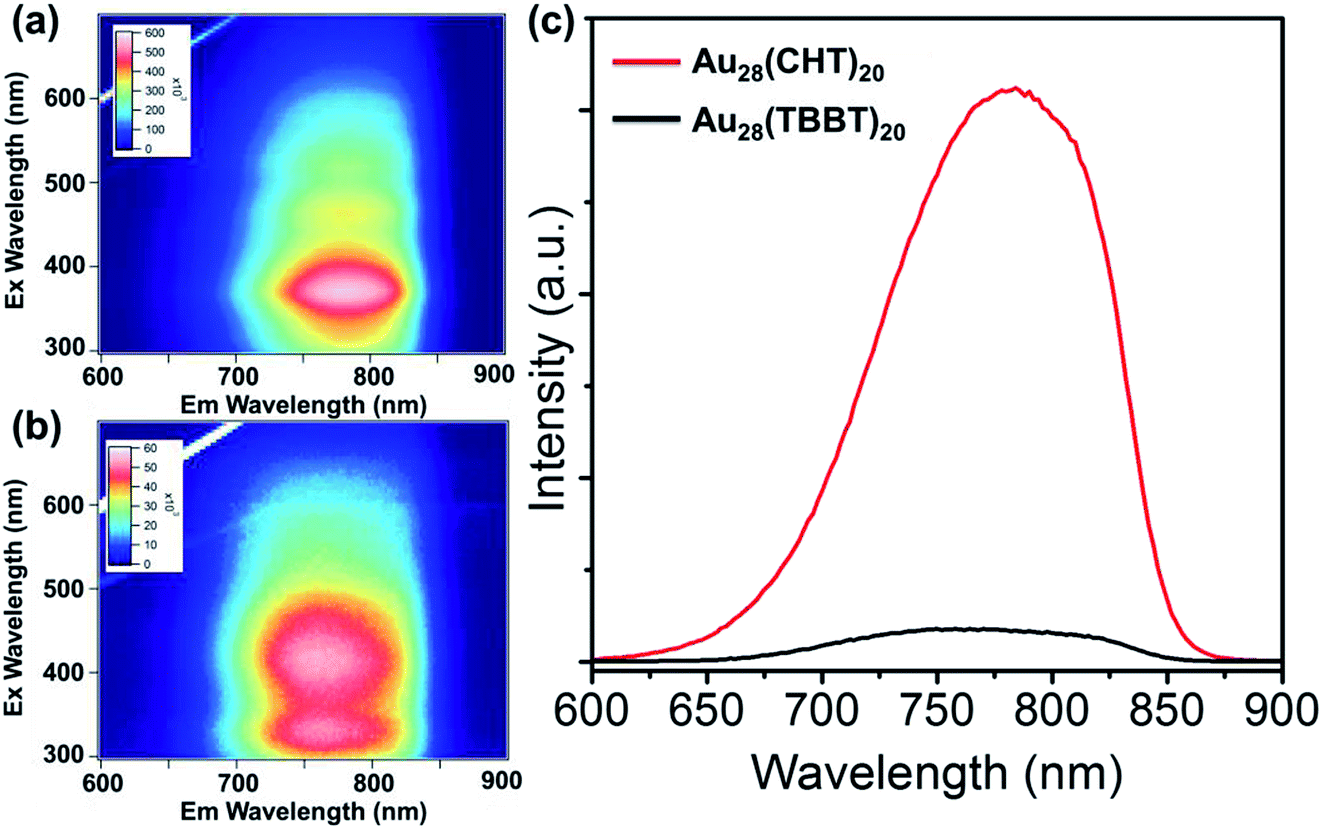 | ||
| Fig. 3 Excitation/emission contour maps of (a) Au28(CHT)20 and (b) Au28(TBBT)20. (c) Comparison of the emission spectra of Au28(CHT)20 and Au28(TBBT)20 excited at 372 nm. | ||
To gain insight into the luminescence behavior of the two nanoclusters, the PL lifetime was measured by time-correlated single photon counting (TCSPC). As shown in Fig. 4, multi-exponential fitting of the PL decay profiles gives rise to two lifetime components for each nanocluster, i.e., τ1 = 264 ns (25%) and τ2 = 1.70 μs (75%) for Au28(CHT)20, and τ1 = 59 ns (56%) and τ2 = 285 ns (44%) for Au28(TBBT)20. It is clear that Au28(CHT)20 exhibits a significantly longer PL lifetime than Au28(TBBT)20 (Fig. 4). The long-lived component explains the higher QY of Au28(CHT)20. The average lifetime (τ*) and QY are related by eqn (1),
| QY = τ*kR | (1) |
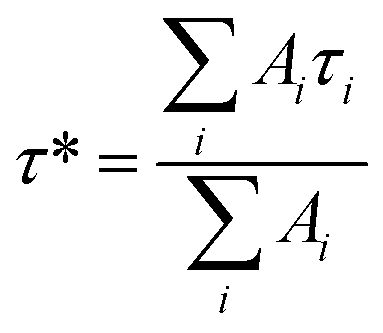 | (2) |
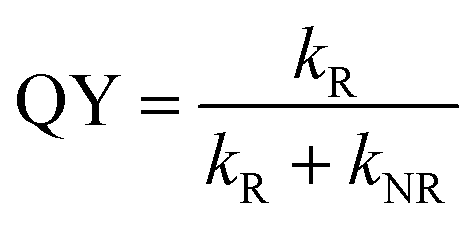 | (3) |
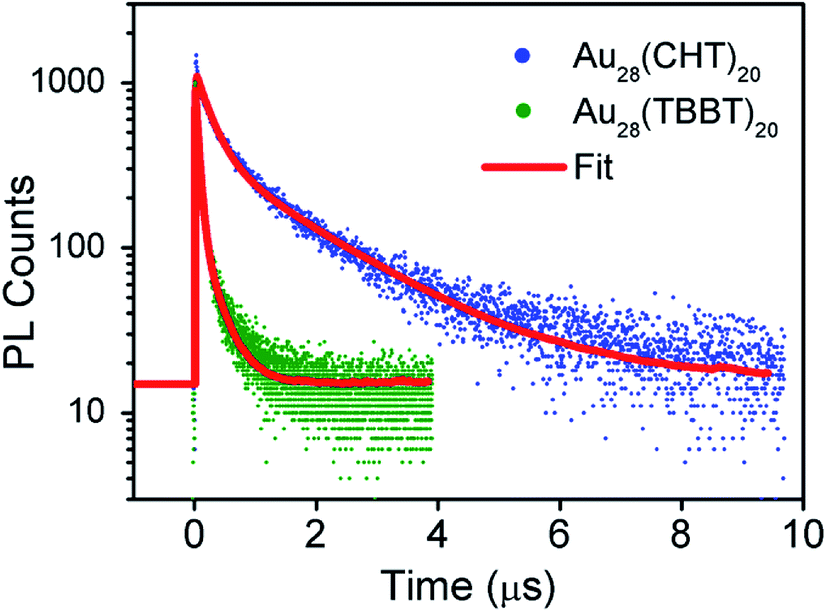 | ||
| Fig. 4 Comparison of the photoluminescence decays of Au28(CHT)20 and Au28(TBBT)20 nanoclusters at the emission maxima measured by TCSPC. | ||
The measured and calculated parameters are summarized in Table 1.
| QY (%) | τ* | k R (s−1) | k NR (s−1) | |
|---|---|---|---|---|
| a QY: quantum yield, τ*: average lifetime, kR: radiative decay rate, kNR: non-radiative decay rate. | ||||
| Au28(CHT)20 | 1.6 | 1341 ns | 1.19 × 104 | 7.32 × 105 |
| Au28(TBBT)20 | 0.1 | 158 ns | 6.33 × 103 | 6.32 × 106 |
Generally, PL can be increased by (i) enhancing the radiative rate (accordingly, the PL lifetime becomes shorter), and/or (ii) suppressing the nonradiative rate (accordingly, the PL lifetime becomes longer). In our system, apparently the PL enhancement in Au28(CHT)20 falls in the second category since the lifetime becomes much longer. The mechanism for suppressing the non-radiative decay rate (kNR) in the Au28(CHT)20 nanocluster is further discussed below.
To further understand the origin of the difference in the PL intensity and lifetime for the two Au28(SR)20 nanoclusters, femtosecond and nanosecond transient absorption (TA) spectroscopy measurements were performed to probe the detailed excited state relaxation and dynamics. As shown in Fig. 5a and b, the two Au28(SR)20 isomers exhibit very similar TA spectra at Δt = 1 ns. The excited state absorption (ESA) overlaps with ground state bleaching (GSB) in both Au28(SR)20 isomers. All the GSB and ESA peaks of Au28(CHT)20 are blue-shifted compared with those of Au28(TBBT)20, which agrees well with the trend in their steady-state absorption spectra (Fig. 2a and c). Significantly, despite the two nanoclusters having similar TA spectra, drastic differences were observed in their excited state lifetimes (Fig. 5c and d), that is, the ESA of Au28(TBBT)20 decays to zero within ∼300 ns while the decay of ESA in Au28(CHT)20 at a similar probe wavelength takes a significantly longer time (∼10 μs). Global fitting shows that the excited state lifetime of Au28(CHT)20 has three components (8 ns, 105 ns and 1.7 μs), while Au28(TBBT)20 shows two components (58 ns and 200 ns). The significantly longer excited state lifetime of Au28(CHT)20 from TA measurements is in line with the PL lifetime results measured by TCSPC, which further supports that the long-lived excited state is responsible for the stronger PL in the Au28(CHT)20 nanocluster, that is, the PL enhancement mechanism is via suppressing the nonradiative rate, as opposed to enhancing the radiative rate. The resemblance of the excited state lifetimes measured by nanosecond TA spectroscopy and the TCSPC explicitly indicates that the TA and PL signals have a similar geometrical and electronic origin.
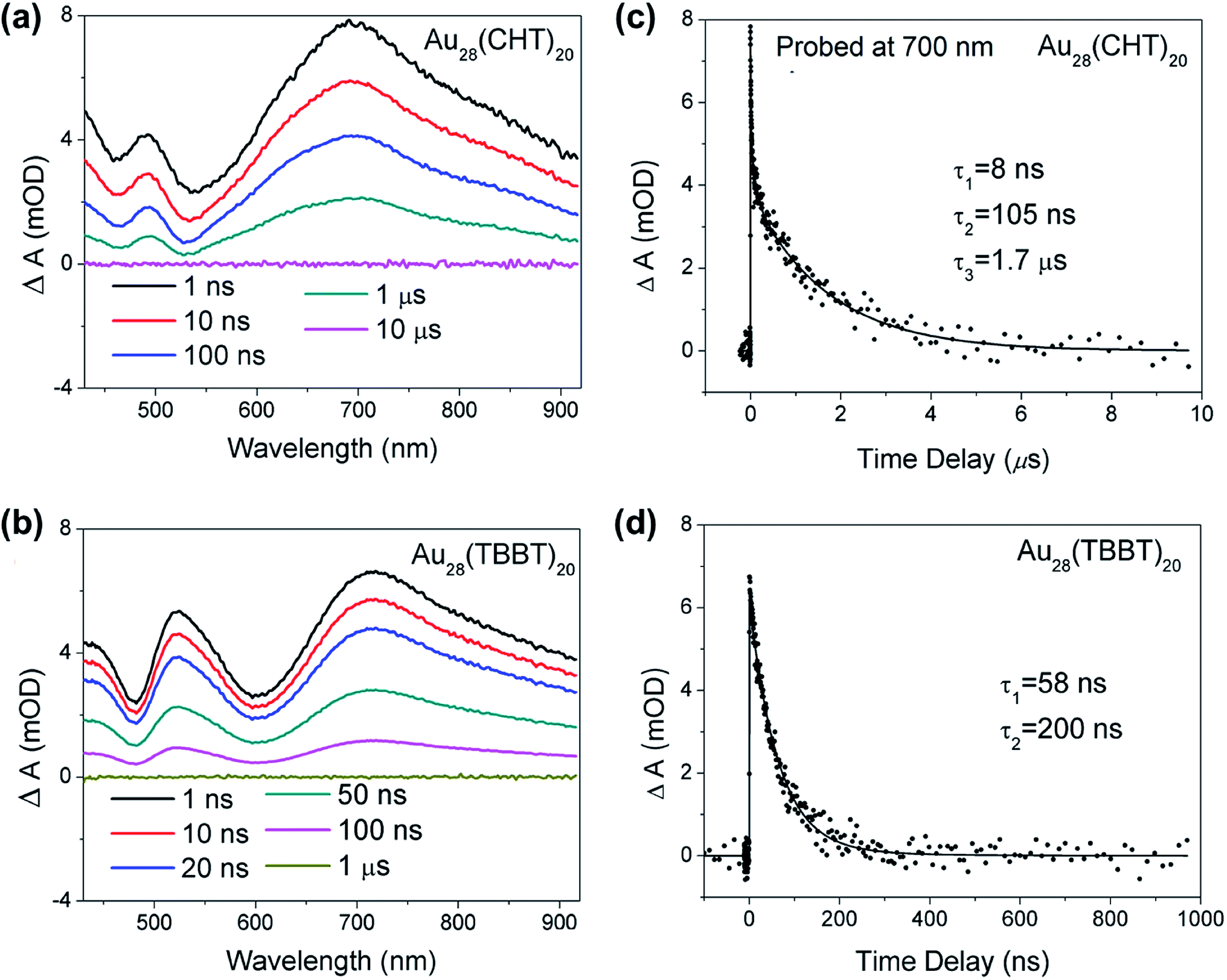 | ||
| Fig. 5 Nanosecond transient absorption spectra of (a) Au28(CHT)20 and (b) Au28(TBBT)20 at selected time delays pumped at 360 nm. Kinetic traces probed at 700 nm for (c) Au28(CHT)20 and (d) Au28(TBBT)20. The corresponding fits are obtained from global fitting. | ||
In addition to the above nanosecond dynamics, femtosecond transient absorption measurements with a pump at 600 nm (near-bandgap excitation) were also carried out to probe the ultrafast electron dynamics of the two Au28(SR)20 nanoclusters. Interestingly, both Au28(SR)20 nanoclusters did not exhibit any ∼1 ps component that corresponds to the core–shell relaxation observed in a Au25(SR)18 nanocluster.57–59 Since no additional spectral features with near-bandgap excitation was observed (Fig. 6), we conclude that there is no core-to-shell electron transfer (or core–shell relaxation) in both Au28(SR)20 nanoclusters. As shown in Fig. 6, both Au28(SR)20 nanoclusters exhibit picosecond and nanosecond decaying components with similar evolution associated spectral (EAS) features. Therefore, the picosecond relaxation can be attributed to the structural relaxation.69 These results are distinctly different from the core–shell relaxation mode previously observed in Au25(SR)18 and Au38(SR)24 nanoclusters.57–59,69
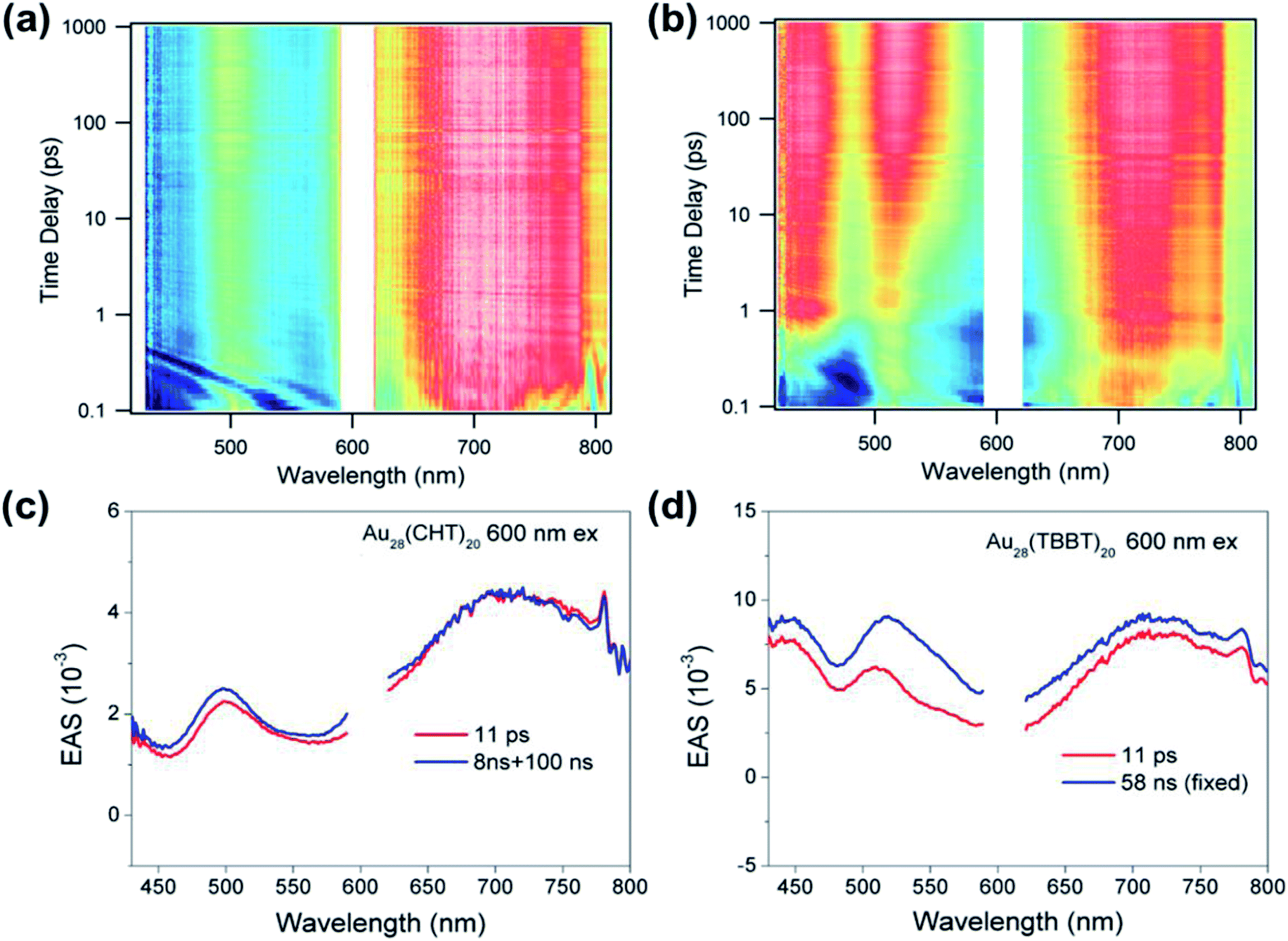 | ||
| Fig. 6 Femtosecond transient absorption data map of (a) Au28(TBBT)20 and (b) Au28(CHT)20 pumped at 600 nm. Evolution associated spectra (EAS) obtained from global fitting of (c) Au28(TBBT)20 and (d) Au28(CHT)20. Note: scattering around 600 nm due to the pump pulse was cut off in both Au28(SR)20 nanoclusters. | ||
Based on the femtosecond and nanosecond transient absorption measurements, the relaxation pathway for both Au28(SR)20 isomers is summarized in Scheme 1 with respective time constants. Such a relaxation pathway is completely different from the previously observed two-state relaxation for Au25(SR)18 and Au38(SR)24 nanoclusters.59,60,69 The absence of core–shell electronic relaxation in both Au28(SR)20 nanoclusters suggests that the electrons are mostly localized in the metal core where the TA signals originated. Therefore, the inner metal core is the key and constitutes the structural origin for the distinct differences in the PL intensity and excited state lifetime between the two Au28(SR)20 nanoclusters. As both Au28(SR)20 nanoclusters have the same Au14 core (note: the Au–Au bond length difference between the two Au14 cores is less than 0.4%), we conclude that the shell structures heavily influence or even dictate the electronic relaxation and dynamics of the metal core, which indirectly affects the PL properties. These results suggest that the PL originated from the metal core, instead of the shell.
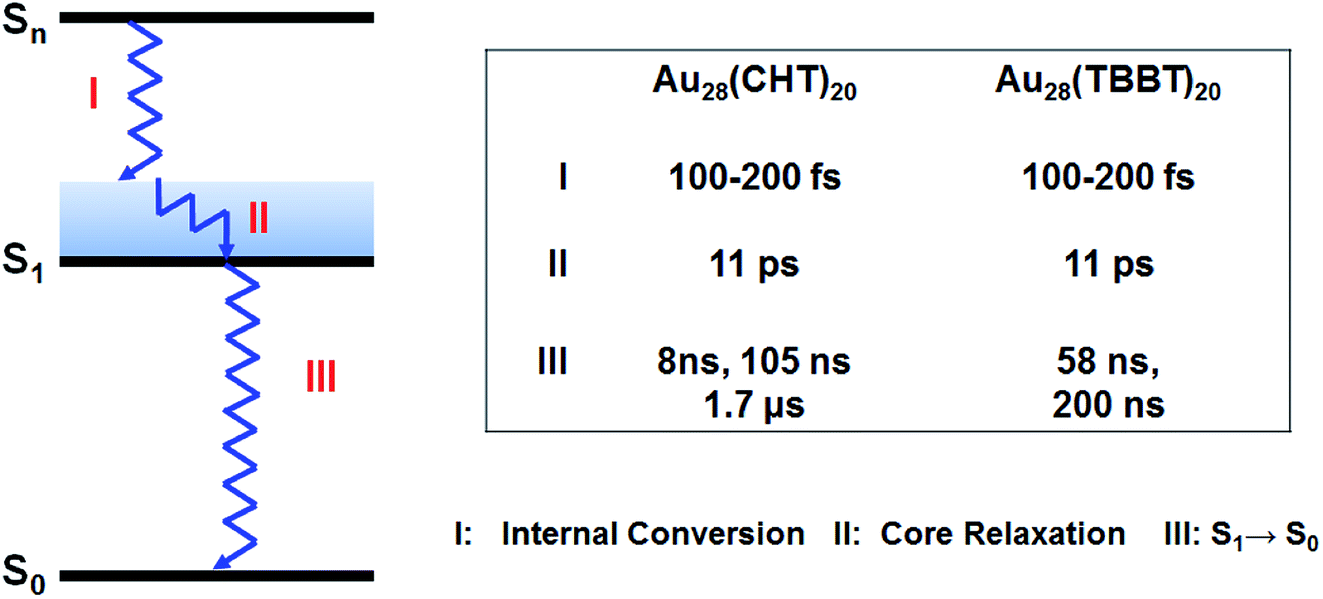 | ||
| Scheme 1 Relaxation diagram of Au28(SR)20 nanoclusters and time constants obtained from transient absorption measurements. | ||
Based on the above results, we propose a shell-mediated core emission mechanism to explain the structural origin of the PL in the Au28(SR)20 nanoclusters. In such a luminescence pathway, the emission solely arises from the metal core while the electron dynamics of the core is modulated by the shell. This core-originated PL mechanism in Au28(SR)20 nanoclusters is quite unique, and it is distinctly different from the previously observed surface-initiated emission behavior for Au25(SR)18 and other water soluble glutathione (SG)-protected gold nanoclusters.19,25 Such a different origin of PL (i.e., core vs. surface) is attributed to the unique Au4-assembled or FCC core structure in Au28(SR)20 nanoclusters as opposed to the icosahedron-based structures in Au25(SR)18 and other related nanoclusters. This core-originated emission may also exist in other FCC based gold nanoclusters. Other than the core-originated PL, the 15-fold enhancement in Au28(CHT)20 compared to Au28(TBBT)20 implies that the shell structure also plays an important role, which is due to the electronic interactions between the core and the shell.
Finally, it is worth noting that Au(I)-SR complexes70 and polymers71 could be luminescent. Thus, we prepared72 and tested both Au(I)CHT and Au(I)TBBT for PL, but no appreciable PL was found (see the ESI, Fig. S2†). Upon correlating with the X-ray structures of Au28(CHT)20 and Au28(TBBT)20, one can find that, in Au28(CHT)20, the staples are quite dangling out, which leads to less interactions with the kernel and thus slower excitation energy dumping through the surface (to solvent), whereas the Au28(TBBT)20 has a tighter interaction between staples and the kernel, which leads to faster dumping of electronic excitation energy. The interlocking of staple motifs also enhances the PL owing to surface rigidification. In summary, given the structure-dependence of PL, the origins of PL in metal nanoclusters are most probably multi-fold (e.g. FCC-related origin vs. icosahedral structure origin), and future work is expected to map out more mechanisms through structure-PL correlations.
Conclusion
Using the pair of Au28(CHT)20 and Au28(TBBT)20 nanoclusters as a model system, we have investigated the photoluminescence origin. Interestingly, we find that the QY of the Au28(CHT)20 nanocluster is more than 15-fold higher than that of Au28(TBBT)20. Correlating the PL and the excited state electron dynamics with the atomic structures of the two Au28(SR)20 nanoclusters reveals a shell-mediated core emission mechanism. The discovery of the core-originated emission mechanism in FCC based Au28(SR)20 nanoclusters is significant since it provides new insights into the intriguing PL phenomenon in non-icosahedral gold nanoclusters. The present study offers important guidelines for rationally enhancing the luminescence of metal nanoclusters for various applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Yongbo Song for assistance with the preparation and PL measurements of Au(I)CHT and Au(I)TBBT complexes. R. J. acknowledges financial support by the Air Force Office of Scientific Research and the U.S. National Science Foundation (DMR-1808675).References
- M. Zhou, T. Higaki, G. Hu, M. Y. Sfeir, Y. Chen, D. Jiang and R. Jin, Science, 2019, 364, 279–282 CrossRef CAS.
- Z. J. Guan, F. Hu, S. F. Yuan, A. A. Nan, Y. M Lin and Q.-M. Wang, Chem. Sci., 2019, 10, 3360–3365 RSC.
- M. Yu, J. Zhou, B. Du, X. Ning, C. Authement, L. Gandee, P. Kapur, J. T. Hsieh and J. Zheng, Angew. Chem., Int. Ed., 2016, 55, 2787–2791 CrossRef CAS PubMed.
- Y. Z. Li, R. Ganguly, K. Y. Hong, Y. Li, M. E. Tessensohn, R. Webster and W. K. Leong, Chem. Sci., 2018, 9, 8723–8730 RSC.
- X. Yuan, Z. Luo, Y. Yu, Q. Yao and J. Xie, Chem.–Asian J., 2013, 8, 858–871 CrossRef CAS PubMed.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS.
- A. W. Cook and T. W. Hayton, Acc. Chem. Res., 2018, 51, 2456–2464 CrossRef CAS.
- Y. Chen, M. L. Phipps, J. H. Werner, S. Chakraborty and J. S. Martinez, Acc. Chem. Res., 2018, 51, 2756–2763 CrossRef CAS PubMed.
- K. K. Chakrahari, R. P. B. Silalahi, J. H. Liao, S. Kahlal, Y. C. Liu, J. F. Lee, M. H. Chiang, J. Y. Saillard and C. W. Liu, Chem. Sci., 2018, 9, 6785–6795 RSC.
- Y. M. Su, Z. Wang, G. L. Zhuang, Q. Q. Zhao, Z. P. Wang, C. H. Tung and D. Sun, Chem. Sci., 2019, 10, 564–568 RSC.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 14767 CrossRef CAS PubMed.
- X.-K. Wan, W. W. Xu, S.-F. Yuan, Y. Gao, X.-C. Zeng and Q.-M. Wang, Angew. Chem., Int. Ed., 2015, 127, 9819–9822 CrossRef.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- X. Kang, S. Wang and M. Zhu, Chem. Sci., 2018, 9, 3062–3068 RSC.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- C. Yao, S. Tian, L. Liao, X. Liu, N. Xia, N. Yan, Z. Gan and Z. Wu, Nanoscale, 2015, 7, 16200–16203 RSC.
- Z. Tang, T. Ahuja, S. Wang and G. Wang, Nanoscale, 2012, 4, 4119–4124 RSC.
- J. Liu, P. N. Duchesne, M. Yu, X. Jiang, X. Ning, R. D. Vinluan III, P. Zhang and J. Zheng, Angew. Chem., Int. Ed., 2016, 55, 8894–8898 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- D. M. Chevrier, V. D. Thanthirige, Z. Luo, S. Driscoll, P. Cho, M. A. MacDonald, Q. Yao, R. Guda, J. Xie, E. R. Johnson, A. Chatt, N. Zheng and P. Zhang, Chem. Sci., 2018, 9, 2782–2790 RSC.
- K. Pyo, V. D. Thanthirige, S. Y. Yoon, G. Ramakrishna and D. Lee, Nanoscale, 2016, 8, 20008–20016 RSC.
- L. L. M. Zhang, G. Zhou, G. Zhou, H. K. Lee, N. Zhao, O. V. Prezhdo and T. C. W. Mak, Chem. Sci., 2019, 10, 10122–10128 RSC.
- J. Jiang, C. V. Conroy, M. M. Kvetny, G. J. Lake, J. W. Padelford, T. Ahuja and G. Wang, J. Phys. Chem. C, 2014, 118, 20680–20687 CrossRef CAS.
- Y.-T. Tseng, R. Cherng, S. G. Harroun, Z. Yuan, T.-Y. Lin, C.-W. Wu, H.-T. Chang and C.-C. Huang, Nanoscale, 2016, 8, 9771–9779 RSC.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1802751 CrossRef.
- X. Dou, X. Yuan, Y. Yu, Z. Luo, Q. Yao, D. T. Leong and J. Xie, Nanoscale, 2014, 6, 157–161 RSC.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS.
- K. L. D. M. Weerawardene, P. Pandeya, M. Zhou, Y. Chen, R. Jin and C. M. Aikens, J. Am. Chem. Soc., 2019, 141, 18715–18726 CrossRef CAS.
- X. Kang, S. Wang, Y. Song, S. Jin, G. Sun, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3611–3614 CrossRef CAS PubMed.
- K. L. D. M. Weerawardene and C. M. Aikens, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS.
- K. G. Stamplecoskie, Y.-S. Chen and P. V. Kamat, J. Phys. Chem. C, 2014, 118, 1370–1376 CrossRef CAS.
- X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18834–18840 CrossRef CAS PubMed.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS PubMed.
- T. Dainese, M. Agrachev, S. Antonello, D. Badocco, D. M. Black, A. Fortunelli, J. A. Gascon, M. Stener, A. Venzo, R. L. Whetten and F. Maran, Chem. Sci., 2018, 9, 8796–8805 RSC.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef CAS PubMed.
- S. Zhuang, L. Liao, Y. Zhao, J. Yuan, C. Yao, X. Liu, J. Li, H. Deng, J. Yang and Z. Wu, Chem. Sci., 2018, 9, 2437–2442 RSC.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 8710–8713 CrossRef CAS.
- Z.-J. Guan, J.-L. Zeng, Z.-A. Nan, X.-K. Wan, Y.-M. Lin and Q.-M. Wang, Sci. Adv., 2016, 2, e1600323 CrossRef.
- K. Konishi, M. Iwasaki, M. Sugiuchi and Y. Shichibu, J. Phys. Chem. Lett., 2016, 7, 4267–4274 CrossRef CAS.
- Y. Niihori, S. Hossain, S. Sharma, B. Kumar, W. Kurashige and Y. Negishi, Chem. Rec., 2017, 17, 473–484 CrossRef CAS.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS.
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS.
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076–10079 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef.
- A. Dass, S. Theivendran, P. R. Nimmala, C. Kumara, V. R. Jupally, A. Fortunelli, L. Sementa, G. Barcaro, X. Zuo and B. C. Noll, J. Am. Chem. Soc., 2015, 137, 4610–4613 CrossRef CAS.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC.
- M. S. Devadas, J. Kim, E. Sinn, D. Lee, T. Goodson III and G. Ramakrishna, J. Phys. Chem. C, 2010, 114, 22417–22423 CrossRef CAS.
- M. S. Devadas, V. D. Thanthirige, S. Bairu, E. Sinn and G. Ramakrishna, J. Phys. Chem. C, 2013, 117, 23155–23161 CrossRef CAS.
- H. Qian, M. Y. Sfeir and R. Jin, J. Phys. Chem. C, 2010, 114, 19935–19940 CrossRef CAS.
- T. D. Green and K. L. Knappenberger, Nanoscale, 2012, 4, 4111–4118 RSC.
- T. D. Green, C. Yi, C. Zeng, R. Jin, S. McGill and K. L. Knappenberger, J. Phys. Chem. A, 2014, 118, 10611–10621 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- D. M. Chevrier, C. Zeng, R. Jin, A. Chatt and P. Zhang, J. Phys. Chem. C, 2015, 119, 1217–1223 CrossRef CAS.
- S. Knoppe, S. Malola, L. Lehtovaara, T. Bürgi and H. Häkkinen, J. Phys. Chem. A, 2013, 117, 10526–10533 CrossRef CAS.
- W. W. Xu, B. Zhu, X. C. Zeng and Y. Gao, Nat. Commun., 2016, 7, 13574 CrossRef CAS.
- Z. Ma, P. Wang and Y. Pei, Nanoscale, 2016, 8, 17044–17054 RSC.
- L. Liu, J. Yuan, L. Cheng and J. Yang, Nanoscale, 2017, 9, 856–861 RSC.
- T. Higaki, C. Liu, Y. Chen, S. Zhao, C. Zeng, R. Jin, S. Wang, N. L. Rosi and R. Jin, J. Phys. Chem. Lett., 2017, 8, 866–870 CrossRef CAS.
- M. Zhou, S. Tian, C. Zeng, M. Y. Sfeir, Z. Wu and R. Jin, J. Phys. Chem. C, 2017, 121, 10686–10693 CrossRef CAS.
- V. W. Yam, V. K. Au and S. Y. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- C. Lavenn, N. Guillou, M. Monge, D. Podbevšek, G. Ledoux, A. Fateevae and A. Demessence, Chem. Commun., 2016, 52, 9063–9066 RSC.
- C. Zeng, M. Zhou, C. Gayathri, R. R. Gil, M. Y. Sfeir and R. Jin, Chin. J. Chem. Phys., 2018, 31, 555 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01270j |
| This journal is © The Royal Society of Chemistry 2020 |