
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of the photoelectrochemical behavior of Au nanocluster–TiO2 electrode by doping†
Malenahalli H.
Naveen‡
 a,
Rizwan
Khan‡
b,
Muhammad A.
Abbas‡
a,
Eunbyol
Cho
c,
Geun Jun
Lee
b,
Hahkjoon
Kim
d,
Eunji
Sim
*c and
Jin Ho
Bang
*abe
a,
Rizwan
Khan‡
b,
Muhammad A.
Abbas‡
a,
Eunbyol
Cho
c,
Geun Jun
Lee
b,
Hahkjoon
Kim
d,
Eunji
Sim
*c and
Jin Ho
Bang
*abe
aNanosensor Research Institute, Hanyang University, 55 Hanyangdaehak-ro, Sangnok-gu, Ansan, Gyeonggi-do 15588, Republic of Korea. E-mail: jbang@hanyang.ac.kr
bDepartment of Bionano Technology, Hanyang University, Republic of Korea
cDepartment of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: esim@yonsei.ac.kr
dDepartment of Chemistry, Duksung Women's University, Seoul 01369, Republic of Korea
eDepartment of Chemical and Molecular Engineering, Department of Applied Chemistry, Hanyang University, Republic of Korea
First published on 2nd June 2020
Abstract
Despite the successful debut of gold nanoclusters (Au NCs) in solar cell applications, Au NCs, compared to dyes and quantum dots, have several drawbacks, such as lower extinction coefficients. Any modulation of the physical properties of NCs can have a significant influence on the delicate control of absorbance, energy levels, and charge separation, which are essential to ensure high power conversion efficiency. To this end, we systematically alter the optoelectronic structure of Au18(SR)14 by Ag doping and explain its influence on solar cell performance. Our in-depth spectroscopic and electrochemical characterization combined with computational study reveals that the performance-dictating factors respond in different manners to the Ag doping level, and we determine that the best compromise is the incorporation of a single Ag atom into an Au NC. This new insight highlights the unique aspect of NCs—susceptibility to atomic level doping—and helps establish a new design principle for efficient NC-based solar cells.
Introduction
Optically- and optoelectronically-active metal nanoclusters (NCs) are an attractive alternative to quantum dots (QDs) and dyes in light energy conversion applications.1 In this regard, gold (Au) NCs are of particular interest due to their excellent stability in the corrosive redox environment present in energy conversion devices. Long excited-state lifetimes and energy levels appropriate for charge transfer render Au NCs the best candidate to produce efficient solar cell devices.2,3 The feasibility of using thiolated NCs as a photosensitizer in solar cell applications has been demonstrated, and a newly-coined term—metal-nanocluster-sensitized solar cells (MCSSCs)—has been well-described.4–7 Early breakthroughs by the Kamat group and others, including recent reports by our group, are setting the stage for the development of efficient MCSSCs by revealing the effects of various performance-dictating factors.7–12 For instance, the absorption capability of an NC is usually a function of its size. Unfortunately, an increase in size has been shown to accompany increased interfacial recombination.6 Therefore, the best power conversion efficiency (PCE) in MCSSCs is achieved using only Au NCs that have optimum absorption and recombination characteristics.Despite significant strides in the PCE of MCSSCs since their inception, it still lags far behind that of competitors (dye- or QD-sensitized solar cells).13,14 Therefore, to further improve PCE, it is necessary to understand the shortcomings of Au NCs and to devise a strategy to overcome these limitations. In general, the properties of NCs can be tuned using three key strategies: size variation,3,15,16 ligand engineering,17–19 and hetero-atomic doping.20–23 As demonstrated in our recent work,6 Au18(SR)14 are the most useful of the various-sized NCs because of a well-balanced interplay of interfacial events. Regarding ligands, reduced glutathione (GSH)-protected NCs have been demonstrated to show the highest stability and efficiency for light-harvesting applications. In particular, the presence of electron-rich functional groups in the GSH ligand makes it a unique candidate by ensuring long excited state lifetimes.24 Given these points, one can speculate that doping GSH-protected Au18(SR)14 NCs would be the next logical step in further modulating the physical properties that could make them more efficient sensitizers for MCSSCs. In fact, the doping effect is not trivial in Au NCs because, with substitution of only a single Au atom, the optoelectronic properties of Au NCs can be significantly altered—often in unpredictable ways—as demonstrated previously.25–27 Therefore, investigation of the doping effect is an important addition to establishing an NC design principle for light energy conversion applications.
One critical parameter of Au NC inferiority as sensitizers compared to dyes or QDs is the low molar extinction coefficient (∼103 M−1 cm−1),28 which is more than an order of magnitude lower than those of dyes and QDs.29,30 Therefore, doping Au18(SR)14 NCs with other metal atoms that can supplement their light absorption capability could increase the PCE of MCSSCs. Silver (Ag) is a candidate for this purpose because Ag NCs tend to have higher extinction coefficients for visible and UV radiation because they exhibit stronger quantum confinement effect31,32 and lack of damping due to d electrons.33,34 Furthermore, Ag doping could be beneficial for enhancing excited-state lifetimes. In fact, time-resolved spectroscopic measurements reveal that Ag-doped Au NCs have a longer excited-state lifetime than their undoped parents.35 Also, Schneider and co-workers enhanced the photoluminescence (PL) of Au NCs using Ag doping (a long lifetime > 200 ns) and achieved better photo-stability.36 However, despite this understanding of the effects of Ag doping on improved optical properties, these enhancements have yet to be realized in a working light harvesting device.
Given the results reported in the literature, there is no doubt that Ag can significantly augment the optical properties of Au NCs. However, Ag can negatively affect the stability of Au NCs due to their inherently low stability.37 Furthermore, inappropriate doping levels could change their energy levels in such a way that is detrimental to the charge transfer to TiO2. Therefore, careful, systematic control over doping levels is required to amplify the sensitization potential of Au18(SR)14. To this end, we precisely modulated the optoelectronic structure of Au18(SR)14 using Ag doping and investigated the impact on the PCE of MCSSCs. As synthesis protocols are widely known to significantly affect the properties of Au NCs, we adopted a strategy developed by the Xie group that can provide a simple way to incorporate 1–4 Ag atoms into Au18(SR)14 NCs as Au18−xAgx(SR)14 by controlling the ratio of Ag/Au precursors.37 A computational study based on density functional theory (DFT) was performed to investigate variations in the electronic structure of Au18−xAgx(SR)14 NCs. In addition, time-resolved spectroscopy combined with electrochemical impedance spectroscopy (EIS) shed light on various interfacial processes occurring in the MCSSCs sensitized with Au18−xAgx(SR)14. In addition to attaining comprehensive insights into the optoelectronic and photoelectrochemical behavior of Au18−xAgx(SR)14 NCs, we were able to further increase the PCE of MCSSCs to a new high of 4.22%.
Experimental
Materials
All chemicals were used as received. Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.99%), reduced L-glutathione (GSH, ≥98%), sodium hydroxide (NaOH, 99.99% trace metal basis), tetraoctylammonium bromide (≥98%), acetonitrile (anhydrous, 99.8%), and tetrabutylammonium hexafluorophosphate (for electrochemical analysis, 99%) were purchased from Sigma-Aldrich. Acetonitrile (reagent grade) for NC precipitation and toluene were purchased from Daejung Chemical & Metals. AgNO3 was obtained from INUISHO Precious Metals. Co., Ltd. TiCl4 was purchased from Junsei Chemical Co., Ltd. The TiO2 pastes used for the mesoporous layer (Ti-nanoxide, T/SP) and the scattering layer (18NR-AO) were purchased from Solaronix and GreatCellSolar, respectively. The I−/I3− electrolyte (Electrolyte EL-HPE) was purchased from GreatCellSolar.Synthesis of Au18(SR)14 and Ag-doped Au18−xAgx(SR)14 NCs
Au18(SR)14 NCs and Ag-doped Au18−xAgx(SR)14 NCs were synthesized by following the previously reported CO-reduction method.37 For the synthesis of Au18(SR)14 NCs, 12.5 mL aqueous solution of HAuCl4 (20 mM) and 10 mL of GSH (50 mM) were added to a 500 mL round-bottom flask containing 227.5 mL of deionized water. No Ag precursor was introduced into this reaction mixture. After stirring for 2 min, the pH of the solution was adjusted to 10 using a freshly prepared 1 M NaOH solution. The reaction solution was then purged with carbon monoxide for 4 min at a flow rate of 10 cm3 min−1. The resultant solution was stirred for 24 h. For the synthesis of Au18−xAgx(SR)14 NCs, Au (HAuCl4, 20 mM) and Ag (AgNO3, 20 mM) precursors were used in certain molar ratios. The samples NC-2, NC-3, and NC-4 were prepared with molar ratios of Ag and Au (R[Ag]/R[Au]) precursors set to 0.05, 0.10, and 0.20, respectively. These precursors were added into three different 500 mL round-bottom flasks containing 227.5 mL of deionized water, followed by adding 10 mL of reduced L-glutathione (50 mM). Then, the remainder of the procedure was the same as for NC-1. After 24 h, the as-synthesized NC solutions were precipitated by mixing with acetonitrile at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The resultant precipitates were collected by centrifugation at 9000 rpm for 10 min. The collected precipitates were resuspended in water with pH adjusted to 4. The resultant solution was used for the sensitizing of TiO2 films.
:3. The resultant precipitates were collected by centrifugation at 9000 rpm for 10 min. The collected precipitates were resuspended in water with pH adjusted to 4. The resultant solution was used for the sensitizing of TiO2 films.
Fabrication of solar cells
Double-layered TiO2 electrodes were prepared by screen printing on TiCl4-treated F-doped SnO2 (FTO) glass. The first layer of a 10 μm-thick mesoporous TiO2 film was deposited, followed by a 10 μm-thick TiO2 scattering film. The prepared films were sintered according to the previously reported method.6 The TiCl4 post-treatment was performed for all double-layered electrodes prior to dipping in sensitizing solutions. Photoelectrodes were prepared by immersing the TiO2 film in each sensitizing solution for 24 h in the dark. Prior to this, the sensitization solution was prepared by resuspending the purified NCs in deionized water and adjusting the pH to 4 using dilute HCl. Since the isoelectric point of anatase TiO2 is 6.89, the pH of the sensitization solution needed to be adjusted below the isoelectric point to facilitate the electrostatic interaction between the negatively charged carboxylic groups of GSH and the positively charged TiO2. After adsorption of the NCs, the sensitized TiO2 films were rinsed with deionized water and ethanol sequentially to remove any unattached Au NCs. The loading amount of NCs on TiO2 film was calculated by desorbing the attached NCs in 1 M NaOH solution. The absorbance of the resulting solution was measured by UV-vis absorption spectroscopy and the Beer–Lambert law was used to calculate the concentration of NCs. Prior to cell fabrication, an iodine electrolyte was dropped onto Pt-sputtered FTO glass containing a hot-melt ionomer (Surlyn) and melted at 90 °C for 5 min on the counter electrode. The counter electrode and the photoanode were then carefully coupled using steel clips to complete the MCSSC device.Characterization
UV-vis absorption spectra of the synthesized NC solutions and the NC-adsorbed TiO2 films were measured using a UV-vis spectrophotometer (SCINCO S-3100) equipped with a diffuse reflector (for film absorbance measurements). Compositional formula of the synthesized NCs (NC-1, NC-2, NC-3, and NC-4) were analyzed by electrospray ionization mass spectrometry (ESI-MS) in negative ion mode, with the spray voltage of 3 kV and the capillary temperature at 250 °C. NCs samples were prepared at a concentration of 4 mg mL−1 in 50% methanol. The photocurrent density–photovoltage (J–V) curve characteristics were measured using a Keithley 2400 source meter, while the MCSSCs (active area: 0.188 cm2) were illuminated under a solar simulator (HAL-320, Asahi Spectra). A standard silicon diode (CS-20, Asahi Spectra) was used to adjust the light intensity of the solar simulator to 1 sun (Air Mass 1.5G). The incident photon-to-current efficiency (IPCE) was measured in the wavelength range of 300–800 nm using a PV Measurements Inc. system (QEX7) and a Keithley 2400 source. PL lifetime measurements were performed using a custom-built inverted fluorescence microscope (Olympus, IX-70) equipped with a picosecond pulsed diode laser (375 nm, 100 μW) and a detector (GaAs photomultiplier tube, Hamamatsu, H7422-40). The EIS measurements were carried out in the dark (0.1 Hz to 100 kHz, AC amplitude of 10 mV) using an electrochemical workstation (REF600) from Gamry Instruments. Echem Analyst software was used for fitting.Square wave voltammetry (SWV) measurements
Water-soluble Au18−xAgx(SR)14 NCs were phase-transferred into an organic phase by following the previously reported immiscible biphasic procedure for Au25 NCs.38,39 Briefly, 20 mg of each Au NC was dissolved in 100 mL of deionized water. Then, 6 mL of 10 mM tetraoctylammonium bromide in toluene was added to the water-soluble Au NCs, and an immiscible layer was formed above the water phase. The water–toluene mixture containing the Au NCs was stirred vigorously, and one drop of 1 M NaOH solution was added. After 2 min, stirring was stopped, and the phase-transfer reaction took place within 30 s, as observed by the color change of the toluene. The aqueous phase was then replaced with fresh deionized water, and this process was repeated 3–4 times to remove water-soluble impurities. Toluene was evaporated out using a rotary evaporator, and the NCs were re-dissolved in acetonitrile for SWV measurements. Electrochemical experiments were performed with an electrochemical workstation (CHI 660D, CH Instruments) using acetonitrile as the solvent and 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte in a three-electrode configuration. Polished Pt disk electrode (0.04 mm) was used as the working electrode, Pt wire as the counter electrode, and a nonaqueous Ag/Ag+ quasi reversible electrode (Ag QRE) as the reference electrode. SWV was conducted at a peak amplitude of 30 mV, 2 mV of increment, and a frequency of 30 Hz. Prior to each measurement, the electrolyte was thoroughly degassed with N2 gas.Computational methods
DFT calculations, including geometry optimization, were carried out using the TURBOMOLE package version 7.0.2 at the B3LYP/def2-SVP level.40–42 We used 1.0d-8 and 1.0d-6 a.u. (i.e., scfconv 8 and denconv 1.0d-6 in TURBOMOLE) for energy and density convergence criteria, respectively. To consider the scalar relativistic effect of Au and Ag atoms, the effective core potential was used via def2-ecp.43 The atomic contribution to the molecular orbital was calculated using the Mulliken population method in the Multiwfn program.44,45 Depending on the doping sites, many isomers can exist for each Au18−xAgx(SR)14, and hence five major isomers were chosen for each cluster according to their thermodynamic stability. Detailed procedures and data are provided in the ESI S1 and S2.†Results and discussion
Optical properties and electronic structure of Au18−xAgx(SR)14 NCs
Au18(SR)14 and Au18−xAgx(SR)14 NCs were synthesized by mixing appropriate ratios of Au and Ag precursors (no Ag precursor was introduced to prepare pure Au18(SR)14) using a method discussed previously.37 Formation of NCs was confirmed by ESI-MS (Figs. S1 and S2†) and the absorption spectra of the as-prepared NCs matched well with those reported for Au18−xAgx(SR)14 NCs (Fig. 1 and S3†). The characteristic peak of Au18(SR)14 appeared at 562 nm. However, with increased concentration of Ag precursor, this peak shifted to a shorter wavelength. The first excitonic peak appeared at a wavelength of 522 nm when the Ag and Au precursors were introduced in a ratio of 0.20 (R[Ag]/[Au]). The change in UV-vis absorption spectra was also visible as a change in the color of each NC solution. The greenish-brown color of Au18(SR)14 changed gradually to dark red with increased Ag doping (inset of Fig. 1). It has been shown theoretically and experimentally that doping Au18(SR)14 with Ag causes a blue shift in the UV-vis absorbance spectra.27,37,46,47 Therefore, the changes in color and spectral features of the as-synthesized NCs were a clear indication of successful Ag doping. Varying levels of Ag doping were achieved using Ag and Au precursors in R[Ag]/[Au] ratios of 0, 0.05, 0.10, and 0.20. Pure Au18(SR)14 NCs were obtained when no Ag precursor was introduced during synthesis. These Au18(SR)14 NCs will be referred to as NC-1. The Au18−xAgx(SR)14 NCs synthesized with an R[Ag]/[Au] ratio of 0.05 produced NCs with x ≤ 2. This NC solution contained three distinct NCs—Au18(SR)14, AgAu17(SR)14, and Ag2Au16(SR)14—and AgAu17(SR)14 was the exclusively dominant species.37 These NCs will be referred to as NC-2. The Au18−xAgx(SR)14 NCs synthesized with R[Ag]/[Au] ratios of 0.10 and 0.20 will be denoted as NC-3 (x ≤ 3) and NC-4 (1 ≤ x ≤ 4), respectively. All four NC solutions showed excellent stability in the solution phase. It is noteworthy to mention that highly pure Au18−xAgx(SR)14 with a single x value would be ideal to study the effect of Ag doping. However, from a practical point of view, it is extremely difficult to separate a mixture of Au18−xAgx(SR)14 NCs since the difference in their molecular weight is very small. As shown in Fig. 1, in addition to the blue shift in the characteristic peak of Au18(SR)14, the absorption capability of Au18−xAgx(SR)14 increased substantially at wavelengths between 400 and 600 nm for all levels of Ag doping. Furthermore, the increase in absorbance was exponentially proportional to the doping level (Fig. S4†). These changes in absorption spectra indicate that Ag doping had a significant influence on the electronic structure of Au18−xAgx(SR)14.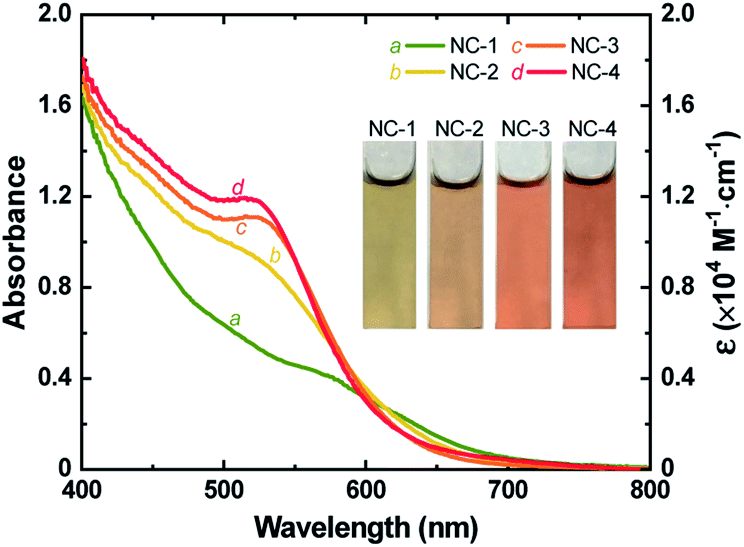 | ||
| Fig. 1 UV-vis absorption spectra (left) and molar extinction coefficient (right) of as-synthesized NC-1, NC-2, NC-3, and NC-4. Inset shows digital photographs of an aqueous solution of each NC. | ||
The HOMO–LUMO (HL) gaps of all four NCs were determined experimentally using SWV (Fig. 2A). Voltammetry measurements have been a very powerful technique in unraveling the electronic structures of NCs.48,49 This measurement not only verifies assertions about changes in the HL gap with Ag doping but can also determine their suitability as a sensitizer for TiO2 in construction of MCSSCs (Fig. 2B). The HL gap of all the NCs was determined after subtracting the charging energy (O2–O1) from the gap between first oxidation (O1) and reduction (R1) potentials.49 The HOMO levels of all four NCs were fairly constant at −5.30 eV; this measurement showed an energy difference of 0.48 eV between the HOMO and the redox potential of the I−/I3− electrolyte, which implied a sufficient driving force for efficient regeneration of all NCs. The open-circuit potentials of each NC solution showed negative potential (around −0.5 to −0.6 V), indicating the NCs existed in anionic form. The LUMO level increased from −3.35 to −3.17 eV in the samples from NC-1 to NC-4, which indicates that Ag doping of Au18(SR)14 proceeds mostly with a shift in the LUMO level of the NCs. The LUMO level of all the NCs was found to be sufficiently high with respect to the TiO2 conduction band (−4.26 eV), suggesting that, from the thermodynamic point of view, charge transfer from each NC to TiO2 could take place efficiently enough to drive photocurrent.
 | ||
| Fig. 2 Square wave voltammograms of (A) NC-1, NC-2, NC-3, and NC-4 in acetonitrile containing 0.1 M of Bu4NPF6 electrolyte. (B) Energy level diagram in vacuum scale constructed from the HOMO and LUMO levels as determined by SWV measurements. In part (A), for clarity, the intensity of reduction peaks in the SWV curves is magnified to three times for NC-1, NC-3, and NC-4 and two times for NC-2 with respect to those of respective oxidation peaks. | ||
In order to better understand how the modulation of the electronic structure of Au18−xAgx(SR)14 is driven by the Ag-doping level and to study its implications on solar cell performance, we carried out DFT calculations. Au18(SR)14 contains an Au9 metallic core composed of two octahedrons that are connected and share a triangular facet (Fig. 3A and B).50–54 This metallic core is covered by five Au-ligand motifs—three monomers, one dimer, and one tetramer motif. We generated all the possible isomers of Au18−xAgx(SR)14 with x ≤ 4 in the quest to find the most stable isomers. Our calculations concluded that the most likely structures were produced when doping of Au18(SR)14 with Ag was proceeded by substituting Ag for the Au atoms in the Au9 core (Fig. 3C, S5 and Table S1†). These results were consistent with previously reported theoretical and experimental studies.53,54 Since the energy difference among various isomers was quite small, we considered the Boltzmann distribution of isomers to look at relative populations (Table S2 and Fig. S6†). This analysis further confirmed that almost the entire population of the Au18−xAgx(SR)14 consisted of NCs where Ag substituted Au in the Au9 core. The presence of Ag in the core can also have important implications on the stability. As Ag NCs suffer from severe inherent instability especially in the corrosive redox environment, the presence of Ag in the core could prevent their direct exposure to the redox couple and provide stability to the solar cell.
 | ||
| Fig. 3 (A) Optimized structure of Au18(SCH3)14 NC. (B) The Au9 core of the Au18(SCH3)14. Each core atom is tagged to indicate the doping positions for further discussion. (C) Energy difference (ΔE) of the most probable isomers from Au18(SCH3)14 when up to four Au atoms were replaced with Ag atoms. The five lowest energy isomers (lowest to higher) are indicated with specific symbols. The exact location of substitution sites for the lowest energy isomers is given in Fig. S5.† (D) DFT calculated HOMO–LUMO energy positions of Au18−xAgx(SCH3)14 NCs with 1 ≤ x ≤ 4. The upper and lower edges of the boxes are the weighted average of energy positions and bars on each side show the total range of the energy positions of the five lowest energy isomers in each category. | ||
While the s orbitals of both Ag and Au are singly occupied, their optical properties are substantially different. This results from the stronger relativistic effect in Au that holds the electrons tightly in the Au atomic orbitals.47 As a consequence of the contracted electron cloud in Au, the 6s and 5d orbitals lie close to each other. In contrast, the 4d and 5s orbitals in Ag are farther apart.55 Hence, the doping of Au NCs with Ag is usually accompanied by a blue shift of the first excitonic peak that results in an increase in the HL gap.27,37,56 Indeed, our calculations showed an increase in the HL gap when the Ag content of Au18−xAgx(SR)14 increased from 0 to 4 (Fig. 3C), which was consistent with the experimental evidence from SWV and UV-vis absorption spectra (Fig. 1 and 2). Furthermore, the electronic structure of Au18−xAgx(SR)14 showed that Ag doping did not change the HOMO level significantly and that the change in HL gap was mostly driven by variation in LUMO level (Fig. 3D, S7–S27, and Table S3†), which was also well-consistent with the experimental observations (Fig. 1 and 2). According to a previous report, the bonding molecular orbital (MO) of Au–Ag clusters is found to resemble that of Au2 clusters, whereas their anti-bonding MO is on par with that of Ag2 clusters.57 Therefore, it is inferred that LUMO levels are more susceptible to the Ag doping, but HOMO levels tend to be relatively insensitive to the doping. The absence of Landau damping from 4d electrons due to positions relative to 5s electrons produces a stronger quantum confinement effect and induces optical properties that are superior to those of Au NCs.5,55 Therefore, doping of Au NCs with Ag can increase the extinction coefficients of bimetallic NCs, making them more efficient light harvesters than pure Au NCs. Indeed, the extinction coefficients of Ag-doped Au18(SR)14 were significantly higher than those of their pure counterparts (Fig. 1).
Solar cell performance
Mesoporous TiO2 films on FTO were sensitized with NC-1, NC-2, NC-3, and NC-4 to prepare photoanodes for the fabrication of solar cell devices. All the samples showed a similar amount of NC loading (Table S4†). The UV-vis absorption spectra of these photoanodes are given in Fig. S28.† The Ag doping clearly enhanced absorption capability compared to that of Au18(SR)14 over the whole observed wavelength range (400–800 nm). Increasing the doping level was directly correlated with an increase in the light harvesting ability of the photoanodes. These results were consistent with the absorption spectra of NC-1, NC-2, NC-3, and NC-4 in the solution phase. Working solar cell devices were fabricated using the NC-sensitized TiO2 films as the photoanodes, Pt-sputtered FTO glass as the counter electrode, and an I−/I3− redox couple as the electrolyte. They were then evaluated under 1 sun illumination to explore their photovoltaic characteristics. The solar cells sensitized with NC-1, NC-2, NC-3, and NC-4 will hereafter be referred to as NC-1-SC, NC-2-SC, NC-3-SC, and NC-4-SC, respectively. The PCE of NC-1-SC was 3.79%, with a short-circuit current (JSC) of 8.07 mA cm−2, an open-circuit voltage (VOC) of 0.683 V, and a fill factor (FF) of 0.688 (Fig. 4A and Table 1). This result was consistent with a previous report of Au18(SR)14-sensitized MCSSCs.6 Doping Au18(SR)14 with only a single Ag atom increased the PCE to 4.22%, which is 11% higher than the previous best PCE. The increase in PCE of NC-2-SC resulted from an increase in all three key solar cell parameters (i.e., JSC, VOC, and FF). This result suggests that the enhanced light absorption capacity might not be the only effect of Ag doping. Indeed, in contrast to our initial expectations, the PCEs of NC-3-SC and NC-4-SC were only 3.32% and 2.38%, which were even lower than that of NC-1-SC. Despite the better FF values seen in NC-3-SC and NC-4-SC, the inferior PCEs mostly resulted from lower values of JSC and VOC. Therefore, it was inferred that regardless of the positive impact of light absorbing ability of NCs, Ag doping with more than one atom must have deteriorated the delicate charge separation process in the working MCSSCs. | ||
| Fig. 4 (A) J–V curves and (B) IPCE spectra and integrated JSC of NC-1-SC, NC-2-SC, NC-3-SC, and NC-4-SC. Integrated JSC was calculated from the IPCE spectra. | ||
| Photo-electrode | J SC (mA cm−2) | V OC (V) | FF | PCE (%) |
|---|---|---|---|---|
| a All values are an average ± standard deviation of parameters from J–V curves of 5 devices. | ||||
| NC-1-SC | 8.07 ± 0.08 | 0.683 ± 0.001 | 0.688 ± 0.001 | 3.79 ± 0.01 |
| NC-2-SC | 8.45 ± 0.15 | 0.694 ± 0.009 | 0.719 ± 0.012 | 4.22 ± 0.07 |
| NC-3-SC | 6.68 ± 0.24 | 0.671 ± 0.008 | 0.740 ± 0.015 | 3.32 ± 0.08 |
| NC-4-SC | 5.06 ± 0.05 | 0.637 ± 0.015 | 0.739 ± 0.005 | 2.38 ± 0.06 |
The J–V measurements revealed that the increase in doping level, which did enhance the optical properties, did not correlate directly with JSC of the MCSSC. Therefore, we analyzed the IPCE of the MCSSCs, which provides information about the contribution of various spectral regions to JSC of each solar cell (Fig. 4B). While NC-2-SC exhibited lower IPCE values than NC-1-SC in the long wavelength region due to its wider HL gap, it outperformed NC-1-SC in the wavelength region of 400–600 nm, which contributed to its higher JSC. Despite better absorbance of the photoelectrodes in NC-3-SC and NC-4-SC, their IPCEs were lower than those of NC-1-SC over the entire observed spectra. Therefore, it was necessary to deconvolve the IPCE spectra to identify the factors that deteriorated JSC of NC-3-SC and NC-4-SC. IPCE spectra consist of light harvesting, charge separation, and charge collection components and details about the IPCE deconvolution process are given in ESI S1.†
Charge collection was assumed to be 100% in all devices in this study. This was a well-considered assumption based on our previous report on Au18(SR)14-sensitized MCSSCs6 and EIS measurements performed in the current work. More insights into this will be provided in a later part of this paper. Because charge collection was assumed to be 100%, therefore, absorption normalized IPCE, which is referred to as absorbed photon-to-current efficiency (APCE), would only reflect the variance in the charge separation among the MCSSCs under study (Fig. S29B†). The APCE spectra of NC-1-SC and NC-2-SC were very similar except in the 400–550 nm region, where the APCE values of NC-2-SC were slightly better than those of NC-1-SC. This could be due to better charge injection or lower recombination in NC-2-SC. However, it can be inferred that most of the improvement in NC-2-SC compared to NC-1-SC originated from the improved absorbance by NC-2 due to Ag doping. The APCE values of NC-3-SC and NC-4-SC were significantly lower than those of their counterparts; hence, we performed further analyses to elucidate this discrepancy.
Charge injection kinetics and interfacial recombination
Charge separation efficiency, ηsep, is composed of two components: charge injection and sensitizer regeneration efficiency. Charge injection from fluorescent sensitizers to other species can easily be explored through PL lifetime measurements.58,59 Materials with a very wide bandgap (e.g., ZrO2) have a conduction band that lies higher than the LUMO level of NCs, and hence they cannot accept excited electrons from NCs. Thus, the PL lifetime of NCs adsorbed onto these materials can serve as an indicator of their intrinsic lifetime. On the other hand, the PL lifetime of NCs adsorbed onto TiO2, which has the potential to accept excited electrons from NCs, will be greatly shortened due to the creation of additional recombination pathways. Therefore, the PL lifetime measurements of NCs on ZrO2 and TiO2 films can be used to determine the apparent rate constant (kc) for charge injection to the conduction band of TiO2 from photo-excited NCs (Fig. 5). The PL lifetimes and kc values of all four NCs are given in Table S5.† NC-1 and NC-2 exhibited similar kc values, hinting at the presence of comparable charge injection kinetics in the two electrodes. However, the kc value of NC-3 was half an order of magnitude and that of NC-4 was more than an order of magnitude lower than that of NC-2 (Fig. 5B). The dramatically lower charge injection rates in NC-3-SC and NC-4-SC were the key factors responsible for the low JSC in these MCSSCs. | ||
| Fig. 5 Photoluminescence decay curves of NC-sensitized TiO2 and ZrO2 films. (A) NC-1, NC-2, NC-3, and NC-4 adsorbed on TiO2 and ZrO2 films. The solid lines represent tri-exponential kinetic fits (A1e−kτ1 + A2e−kτ2 + A3e−kτ3). (B) Apparent rate constants (kc) as calculated by kc = 1/τTiO2 − 1/τZrO2. | ||
Recombination resistance (Rr) is directly related to VOC in the solar cells.5,6 As given in Fig. 6A, NC-2-SC, which exhibited the highest VOC, had the highest Rr, and the Rr values of all MCSSCs followed the same trend as did VOC (NC-2-SC > NC-1-SC > NC-3-SC > NC-4-SC). Therefore, we concluded that an adequate level of Ag doping in Au18(SR)14 not only improves its absorption capability but can also be beneficial in reducing interfacial recombination. However, higher levels of Ag-doping in NC-3 and NC-4 had detrimental consequences for interfacial recombination that led to lower VOC in NC-3-SC and NC-4-SC. The electron lifetime trend (τn = RrCμ) was in accord with the observed VOC of the MCSSCs (Fig. 6B).60 The electron conductivities (σn) of all NC-sensitized TiO2 electrodes were very similar except for a slight improvement in Ag-doped NCs in the high bias region (Fig. 6C). More importantly, the ratio of small perturbation diffusion length (Ln) to the thickness of the TiO2 film (d) was greater than two over the whole bias range in all MCSSCs (Fig. 6D and S32†). As illustrated in earlier reports using the diffusion equation, for well-performing DSSCs or MCSSCs, the Ln/d ratio must be higher than two to achieve a 100% charge collection.6,61 This validates the assumption in the IPCE analysis that ηcoll was 100% for all MCSSCs investigated in this work and implies that none of the solar cell devices investigated were limited by the charge collection process.
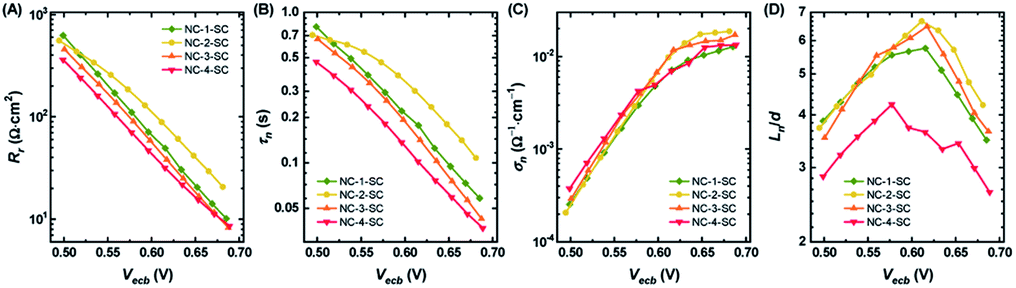 | ||
| Fig. 6 Physical parameters extracted by fitting equivalent circuit to EIS spectra. (A) Recombination resistance (Rr), (B) electron lifetime (τn), (C) electron conductivity in TiO2 (σn), and (D) ratio of small perturbation diffusion length (Ln) to TiO2 thickness (d) as a function of equivalent conduction band voltage (Vecb). | ||
Stability
Since Ag NCs are usually linked to low stability, it is important to study the stability of MCSSCs with increased Ag-doping levels.62 Photocurrents of the solar cells were measured under continuous illumination at 1 sun conditions as a function of time. As seen in Fig. 7, the photocurrent of NC-1-SC, NC-2-SC, and NC-3-SC degraded to ∼90% of the initial value after 1 h of continuous illumination despite the corrosive nature of the iodide species toward Au and Ag. This similarity in degradation rate indicates that doping of Au18(SR)14 with up to three Ag atoms does not significantly affect the stability of Au18(SR)14 for solar cell applications. The stability of NC-2-SC and NC-3-SC could be due to the locations of doped Ag atoms. Unlike the metal–ligand motifs, which were present on the surface of Au NCs and hence were directly exposed to the redox couple, Ag atoms were located in the inner metallic core of the NCs in our study.37 The lack of direct contact between Ag atoms and electrolyte may have circumvented their inherent low stability. However, incorporation of a fourth Ag atom into Au18(SR)14 did indeed lead to faster degradation of photocurrent compared to other NCs. Nonetheless, this stability is unprecedented compared to MCSSCs based on pure Ag NCs.5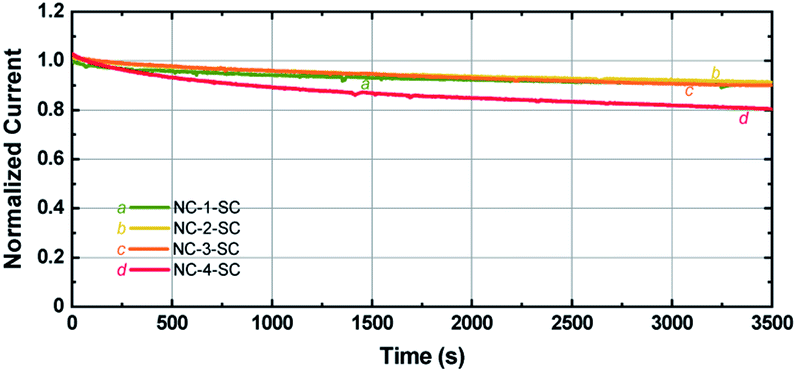 | ||
| Fig. 7 Photocurrent stability of NC-1-SC, NC-2-SC, NC-3-SC, and NC-4-SC under continuous illumination at 1 sun conditions. | ||
Conclusions
Given the significant influence of the physical properties of a photosensitizer on its photoelectrochemical behavior, it is of great importance to establish a property–performance relationship for the next generation of metal-nanocluster-sensitized solar cells. Along with the size and ligand structure, doping is a crucial element that dictates the physical properties of Au NCs. In this work, Au18(SR)14, the best-known NC sensitizer, was systematically modified by Ag doping, and the optoelectronic and photoelectrochemical properties of the resulting Ag-doped Au18(SR)14 were thoroughly investigated. The Ag doping significantly increased the extinction coefficient of Au18(SR)14. With the aid of DFT calculations, it was also found that the substituted Ag atoms contributed preferentially to the LUMO level over the HOMO level, resulting in an increase in HL gap. Time-resolved PL lifetime measurements and EIS analysis shed light on various interfacial charge transfer and recombination events, showing that solar cell performance is complicatedly dictated by the subtle changes in the electronic structure of Ag-doped Au18(SR)14. The best compromise for achieving a new PCE record of 4.22% was obtained when a single Ag atom was incorporated into Au18(SR)14. Interestingly, doping Au18(SR)14 with up to three Ag atoms did not cause any significant loss in photoelectrochemical stability, which could be attributed to the locations of the doped Ag atoms in the Au9 core. With this new comprehensive understanding, it is expected that various synthesis strategies for manipulating the physical properties of Au NCs will be devised to enhance the light energy conversion efficiency of MCSSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2019R1A2C1003429 and NRF-2017R1A2B2003552) and by the Ministry of Education (NRF-2018R1A6A1A03024231).Notes and references
- M. A. Abbas, P. V. Kamat and J. H. Bang, ACS Energy Lett., 2018, 3, 840–854 CrossRef CAS.
- K. G. Stamplecoskie, Y.-S. Chen and P. V. Kamat, J. Phys. Chem. C, 2014, 118, 1370–1376 CrossRef CAS.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS PubMed.
- M. S. Kim, M. A. Abbas and J. H. Bang, Bull. Korean Chem. Soc., 2016, 37, 791–792 CrossRef CAS.
- M. A. Abbas, S. J. Yoon, H. Kim, J. Lee, P. V. Kamat and J. H. Bang, ACS Appl. Mater. Interfaces, 2019, 11, 12492–12503 CrossRef CAS PubMed.
- M. A. Abbas, T.-Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2016, 138, 390–401 CrossRef CAS PubMed.
- Y.-S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822–8825 CrossRef CAS PubMed.
- N. Sakai and T. Tatsuma, Adv. Mater., 2010, 22, 3185–3188 CrossRef CAS PubMed.
- K. G. Stamplecoskie and A. Swint, J. Mater. Chem. A, 2016, 4, 2075–2081 RSC.
- M. A. Abbas, S. J. Yoon, R. Khan, J. Lee and J. H. Bang, J. Phys. Chem. C, 2019, 123, 14921–14927 CrossRef CAS.
- M. A. Abbas, R. Thota, K. Pyo, D. Lee and J. H. Bang, ACS Energy Lett., 2020, 5, 1404–1406 CrossRef CAS.
- M. A. Abbas, R. Khan, S. J. Yoon and J. H. Bang, ACS Appl. Mater. Interfaces, 2020, 12, 16566–16575 CrossRef CAS PubMed.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS.
- K. Zhao, Z. Pan and X. Zhong, J. Phys. Chem. Lett., 2016, 7, 406–417 CrossRef CAS PubMed.
- Y. Yu, X. Chen, Q. Yao, Y. Yu, N. Yan and J. Xie, Chem. Mater., 2013, 25, 946–952 CrossRef CAS.
- S. Link, M. A. El-Sayed, T. Gregory Schaaff and R. L. Whetten, Chem. Phys. Lett., 2002, 356, 240–246 CrossRef CAS.
- J. Li, R. R. Nasaruddin, Y. Feng, J. Yang, N. Yan and J. Xie, Chem.–Eur. J., 2016, 22, 14816–14820 CrossRef PubMed.
- X. Yuan, N. Goswami, W. Chen, Q. Yao and J. Xie, Chem. Commun., 2016, 52, 5234–5237 RSC.
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS PubMed.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- H. Qian, D.-e. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-e. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS PubMed.
- K. Kwak, Q. Tang, M. Kim, D.-e. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- Y. Sun, A. C. Onicha, M. Myahkostupov and F. N. Castellano, ACS Appl. Mater. Interfaces, 2010, 2, 2039–2045 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- S. Kumar, M. D. Bolan and T. P. Bigioni, J. Am. Chem. Soc., 2010, 132, 13141–13143 CrossRef CAS PubMed.
- S. Link, Z. L. Wang and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 3529–3533 CrossRef CAS.
- S. H. Yau, B. A. Ashenfelter, A. Desireddy, A. P. Ashwell, O. Varnavski, G. C. Schatz, T. P. Bigioni and T. Goodson, J. Phys. Chem. C, 2017, 121, 1349–1361 CrossRef CAS.
- B. A. Ashenfelter, A. Desireddy, S. H. Yau, T. Goodson and T. P. Bigioni, J. Phys. Chem. C, 2015, 119, 20728–20734 CrossRef CAS.
- R. Ho-Wu, P. K. Sahu, N. Wu, T. K. Chen, C. Yu, J. Xie and T. Goodson, J. Phys. Chem. C, 2018, 122, 24368–24379 CrossRef CAS.
- X. Le Guével, V. Trouillet, C. Spies, K. Li, T. Laaksonen, D. Auerbach, G. Jung and M. Schneider, Nanoscale, 2012, 4, 7624–7631 RSC.
- Y. Yu, Q. Yao, T. Chen, G. X. Lim and J. Xie, J. Phys. Chem. C, 2016, 120, 22096–22102 CrossRef CAS.
- M. A. Habeeb Muhammed and T. Pradeep, J. Cluster Sci., 2009, 20, 365–373 CrossRef CAS.
- K. Kwak and D. Lee, J. Phys. Chem. Lett., 2012, 3, 2476–2481 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- TURBOMOLE V7.0 2015, A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH. Since 2007, available from http://www.turbomole.com2015 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1841–1846 CrossRef CAS.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- E. B. Guidez, V. Mäkinen, H. Häkkinen and C. M. Aikens, J. Phys. Chem. C, 2012, 116, 20617–20624 CrossRef CAS.
- J. F. Hicks, A. C. Templeton, S. Chen, K. M. Sheran, R. Jasti, R. W. Murray, J. Debord, T. G. Schaaff and R. L. Whetten, Anal. Chem., 1999, 71, 3703–3711 CrossRef CAS PubMed.
- D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- A. Tlahuice and I. L. Garzón, Phys. Chem. Chem. Phys., 2012, 14, 3737–3740 RSC.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS PubMed.
- B. Molina and A. Tlahuice-Flores, Phys. Chem. Chem. Phys., 2016, 18, 1397–1403 RSC.
- J. Xiang, P. Li, Y. Song, X. Liu, H. Chong, S. Jin, Y. Pei, X. Yuan and M. Zhu, Nanoscale, 2015, 7, 18278–18283 RSC.
- N. Bartlett, Gold Bull., 1998, 31, 22–25 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. C, 2008, 112, 19797–19800 CrossRef CAS.
- H. M. Lee, M. Ge, B. R. Sahu, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. B, 2003, 107, 9994–10005 CrossRef CAS.
- J. H. Bang and P. V. Kamat, ACS Nano, 2009, 3, 1467–1476 CrossRef CAS PubMed.
- J. H. Bang and P. V. Kamat, ACS Nano, 2011, 5, 9421–9427 CrossRef CAS PubMed.
- W. H. Leng, P. R. F. Barnes, M. Juozapavicius, B. C. O'Regan and J. R. Durrant, J. Phys. Chem. Lett., 2010, 1, 967–972 CrossRef CAS.
- L. Peter, Acc. Chem. Res., 2009, 42, 1839–1847 CrossRef CAS PubMed.
- N. Shahzad, F. Chen, L. He, W. Li and H. Wang, J. Power Sources, 2015, 294, 609–619 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental computational and experimental methods, more characterization results and electrochemical analysis, and coordinates for the structures of Au18(SCH3)14. See DOI: 10.1039/d0sc01220c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |