
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fine-tuning the electronic structure of heavy-atom-free BODIPY photosensitizers for fluorescence imaging and mitochondria-targeted photodynamic therapy†
Sujie
Qi
 a,
Nahyun
Kwon
a,
Yubin
Yim
a,
Van-Nghia
Nguyen
*ab and
Juyoung
Yoon
*a
a,
Nahyun
Kwon
a,
Yubin
Yim
a,
Van-Nghia
Nguyen
*ab and
Juyoung
Yoon
*a
aDepartment of Chemistry and Nanoscience, Ewha Womans University, Seoul 03760, Republic of Korea. E-mail: jyoon@ewha.ac.kr
bInstitute of Research and Development, Duy Tan University, Da Nang 550000, Vietnam. E-mail: nguyenvannghia8@duytan.edu.vn
First published on 17th March 2020
Abstract
Theranostics that combines both diagnosis and therapy into a single platform has recently emerged as a promising biomedical approach for cancer treatment; however, the development of efficient theranostic agents with excellent optical properties remains a challenge. Here, we report novel mitochondria-targeting BODIPY photosensitizers (R-BODs) that possess considerable singlet oxygen generation capabilities and good fluorescence properties for imaging-guided photodynamic therapy (PDT). The incorporation of sulfur atoms into the π-conjugated skeleton of BODIPY along with the introduction of different functional groups at the meso-position of the BODIPY core is essential for tuning the photophysical and photosensitizing properties. Notably, the MeOPh-substituted thiophene-fused BODIPY (MeO-BOD, R = p-methoxyphenyl) displayed the highest singlet oxygen generation capability (ΦΔ ≈ 0.85 in air-saturated acetonitrile) and a moderate fluorescence quantum yield (Φf = 17.11). Furthermore, MeO-BOD showed good biocompatibility, low dark toxicity and superior fluorescence imaging properties in living cells. More importantly, the PDT efficacy of mitochondria-specific anchoring of MeO-BOD was remarkably amplified with an extremely low half-maximal inhibitory concentration (IC50) value of 95 nM. We believe that the incorporation of an electron-donating group at the meso-position of the thiophene-fused BODIPY platform may be an effective approach for developing theranostic agents for precision cancer therapy.
Introduction
Photodynamic therapy (PDT) is an effective clinical treatment strategy for malignant tumors.1 In the PDT process, a photosensitizer (PS) is activated under light irradiation, and the excited PS subsequently interacts with molecular oxygen to generate cytotoxic reactive oxygen species (ROS), which can oxidize biomolecules resulting in cancer cell death.2,3 In particular, theranostics has recently been recognized as a promising medical technology that combines diagnostic and therapeutic capabilities in one dose to achieve the real-time and precise monitoring of the therapeutic effect of the drug.4 Moreover, PDT combined with image-guided diagnosis has attracted considerable attention owing to its prominent advantages such as high spatiotemporal selectivity, noninvasiveness, and fewer side effects.5–7 Therefore, the design of novel PSs that can effectively produce both fluorescence and ROS is in high demand.To date, numerous organic dyes, such as porphyrins, phthalocyanines, cyanine, squaraine, diketopyrrolopyrrole (DPP), and boron dipyrromethane (BODIPY) derivatives, have been developed as theranostic agents.7–13 Among these, BODIPY dyes have attracted great interest as theranostic agents in photodynamic cancer therapy due to their excellent photochemical stability, good biocompatibility, high molar extinction coefficients, high quantum efficiencies of fluorescence and facile modification.11–17 At present, for obtaining remarkable PDT efficiency of BODIPY dyes, the most popular approach is the introduction of heavy halogen atoms (Br and I) to promote spin–orbit coupling (SOC), which enhances intersystem crossing (ISC) and improves the singlet oxygen (1O2) generation capability.18–24 However, the incorporation of heavy atoms increases the dark-toxicity and quench fluorescence.25 Thus, BODIPY PSs without heavy halogen atoms are preferred as theranostic agents. Recently, several approaches to enhance the ISC, such as the use of double excited states,26,27 spin converters,28 and photoinduced electron transfer (PET),29–31 have been implemented in the development of PSs without heavy atoms; however, the search for novel PSs is still required for PDT to reach its full potential.
Intriguingly, one alternative strategy to strengthen the ISC with an efficient triplet population is to increase the SOC while decreasing the singlet–triplet energy gaps; to do this, a thiophene moiety was introduced into the π-conjugated system of BODIPY,32–35 but the oncological applications of this approach have not been fully studied.35 Furthermore, the introduction of electron-donating groups at the meso-site of the BODIPY platform is also an emerging strategy for achieving efficient PSs.36,37 The abovementioned factors inspired us to consider designing PSs with different functional substituents incorporated at the meso-position of thiophene-fused BODIPY to achieve compounds suitable for clinical use. To the best of our knowledge, there have been no attempts to design efficient theranostic agents by taking advantage of the above two strategies.
Herein, we designed and synthesized a series of heavy-atom-free thiophene-BODIPY derivatives by incorporating different functional groups (pyridinyl (PY), phenyl (PH), p-methoxyphenyl (MeO) and N,N-dimethylaminophenyl (DMA)) at the meso-position of the thiophene-BODIPY platform (Scheme 1). The experimental results revealed that the 1O2 generation ability of these thiophene-BODIPY derivatives through a type II process gradually increased with increasing electron-donating ability of the substituent (ΦΔ: PY-BOD < PH-BOD < MeO-BOD). Fortunately, MeO-BOD had prominent dual functions, showing both a high 1O2 quantum yield and moderate fluorescence intensity. Furthermore, the cellular experimental results showed that MeO-BOD could be utilized as a mitochondria-specific diagnostic agent to reinforce the PDT effect.
 | ||
| Scheme 1 Synthetic route to the thiophene-fused BODIPY derivatives. | ||
Results and discussion
Synthesis and photophysical properties
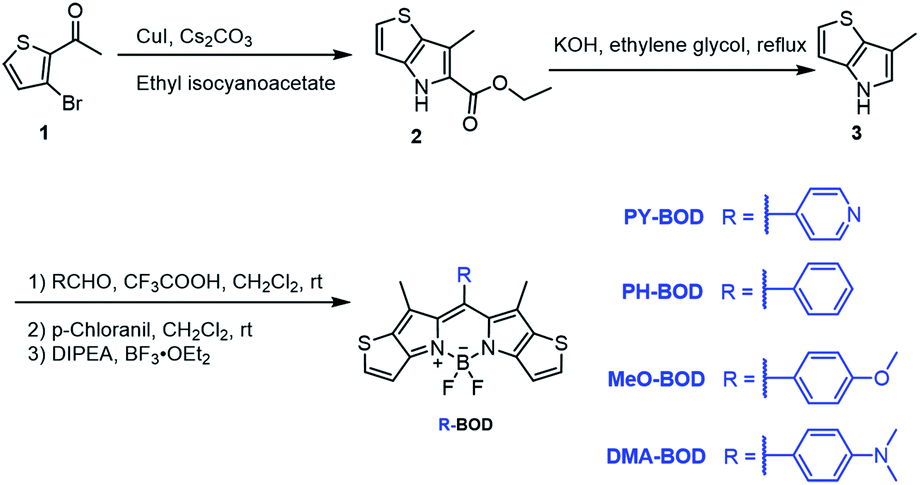
The different thiophene-fused BODIPY derivatives were synthesized through the trifluoroacetic acid-catalyzed condensation of thiophene-fused pyrroles with different benzaldehydes, followed by oxidation and complexation (Scheme 1). Detailed experimental procedures are provided in the ESI.† The structures of all these thiophene-fused BODIPY derivatives were fully confirmed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (Fig. S1–S14†). UV-vis absorption and fluorescence spectra of the new thiophene-BODIPY derivatives were acquired (Fig. 1, S15 and S16†). These key photophysical property data are summarized in Table 1.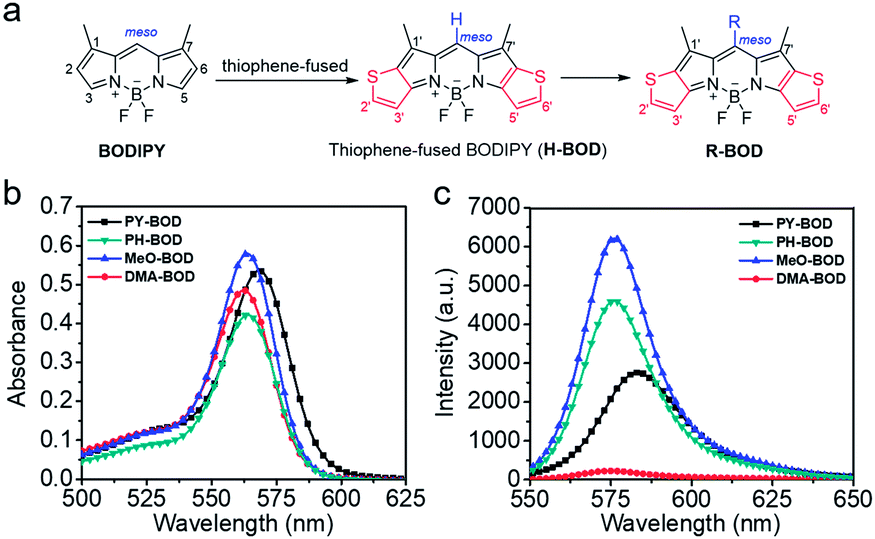 | ||
| Fig. 1 (a) Design of thiophene-fused BODIPY derivatives, (b) UV-Vis absorption and (c) emission spectra of PY-BOD, PH-BOD, MeO-BOD and DMA-BOD in toluene at c = 5.0 μM and λex = 540 nm. | ||
| Compounds | λ abs [nm] | ε × 10−3a [M−1 cm−1] | λ em [nm] | Stokes shift [nm] | Φ f (%) | Φ Δ |
|---|---|---|---|---|---|---|
| a In toluene (5.0 × 10−6 M). b Fluorescence quantum yield estimated relative to rhodamine 101 as the standard (Φf = 1.0 in methanol). c Singlet oxygen quantum yield was determined with respect to rose bengal (ΦΔ (RB) = 0.54 in ACN). d Literature value.19 | ||||||
| BODIPY | 502 | 120.00 | 508 | 6 | 70.0 | ≈0 |
| PY-BOD | 568 | 107.09 | 585 | 17 | 9.82 | 0.52 |
| PH-BOD | 564 | 111.60 | 576 | 12 | 18.83 | 0.69 |
| MeO-BOD | 564 | 115.78 | 579 | 15 | 17.11 | 0.85 |
| DMA-BOD | 562 | 96.99 | 577 | 15 | 2.50 | 0.04 |
The absorption and fluorescence spectra of all the thiophene-fused BODIPY derivatives were similar in shape to those of H-BOD, indicating that the R substituents had little effect on the HOMO–LUMO energy gap of the thiophene-fused BODIPY chromophore because they are nearly orthogonal to the thiophene-fused BODIPY core (Fig. 1a).32 All the studied compounds retained high absorption molar extinction coefficients (ε ∼ 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), and the maximum absorptions were shifted to longer wavelengths (λabs ∼ 565 nm) compared with that of unmodified BODIPY. The absorption spectra of all the thiophene-BODIPY derivatives showed a slight blueshift with increasing solvent polarity. This phenomenon indicates that the dipole moments of the ground state of these thiophene-BODIPY derivatives might be larger than those of the first singlet excited states.32 On the other hand, PY-BOD, PH-BOD and MeO-BOD exhibited moderate fluorescence intensities, while DMA-BOD was poorly emissive due to a PET mechanism.38,39 The fluorescence quantum yields of all the thiophene-BODIPY derivatives were evaluated in toluene (Table 1), and PY-BOD, PH-BOD and MeO-BOD were shown to be excellent candidates for fluorescence imaging.
000 M−1 cm−1), and the maximum absorptions were shifted to longer wavelengths (λabs ∼ 565 nm) compared with that of unmodified BODIPY. The absorption spectra of all the thiophene-BODIPY derivatives showed a slight blueshift with increasing solvent polarity. This phenomenon indicates that the dipole moments of the ground state of these thiophene-BODIPY derivatives might be larger than those of the first singlet excited states.32 On the other hand, PY-BOD, PH-BOD and MeO-BOD exhibited moderate fluorescence intensities, while DMA-BOD was poorly emissive due to a PET mechanism.38,39 The fluorescence quantum yields of all the thiophene-BODIPY derivatives were evaluated in toluene (Table 1), and PY-BOD, PH-BOD and MeO-BOD were shown to be excellent candidates for fluorescence imaging.
1O2 generation ability
Subsequently, the singlet oxygen generation capability of PY-BOD, PH-BOD, MeO-BOD and DMA-BOD was assessed in air-saturated acetonitrile (ACN) under 560 nm irradiation. A commercial 1O2 probe, 1,3-diphenylisobenzofuran (DPBF), was used as an indicator, and rose bengal (RB, ΦΔ = 0.54 in ACN) was used as the reference.40 As shown in Fig. 2a and S17,† the absorbance at 410 nm of DPBF decreased gradually in the presence of the thiophene-BODIPY derivatives under continuous light irradiation. According to the linear relationship of the decay curves (Fig. 2b), the 1O2 quantum yields of PY-BOD, PH-BOD, MeO-BOD and DMA-BOD were calculated to be 0.52, 0.69, 0.85 and 0.04, respectively (Table 1). The strongest 1O2 generation ability of MeO-BOD among all the R-BODs suggested the electron donating group (MeOPh–) in the thiophene-fused BODIPY derivatives played an important role in enhancing the 1O2 generation. Based on these results, we propose a plausible mechanism for MeO-BOD. The introduction of the donating group probably favors 1O2 generation first by increasing the formation of the charge transfer (CT) state via photoinduced charge transfer (PCT) (1BOD-Donor → BOD[δ−]-Donor[δ+]). The charge recombination of the CT state further triggers the production of T1 of the thiophene-fused BODIPY derivatives (BOD[δ−]-Donor[δ+] → 3BOD-Donor). Finally, 1O2 is generated by energy transfer from T1 to molecular oxygen (3BOD-Donor + O2 → BOD-Donor + 1O2) (Scheme S1†).29–31,37,41,42 Unfortunately, DMA-BOD showed almost no 1O2 generating ability in ACN, which implied that the electron-donating group could also lead to a PET process, which triggered the quenching of fluorescence and forbidding of non-radiative transitions such as ISC, thereby prohibiting the 1O2 generation.43 Thus, the 1O2 formation efficiency could be controlled by finely tuning the electronic properties of the meso-substituent on the thiophene-BODIPY platform, especially by the introduction of a suitable electron-donating group. Taken together, these results indicate that MeO-BOD has potential as a theranostic agent for cancer treatment.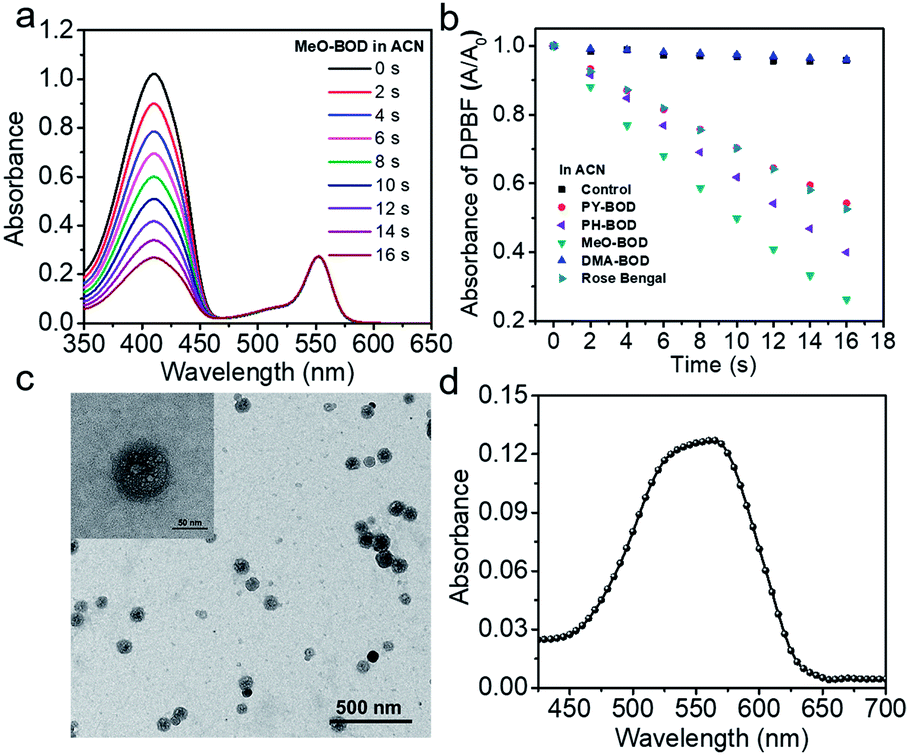 | ||
| Fig. 2 (a) Time-dependent photodegradation of DPBF with MeO-BOD; (b) the DPBF degradation rate curves with PY-BOD, PH-BOD, MeO-BOD, DMA-BOD and rose bengal in ACN; (c) a TEM image of MeO-BOD NPs (inset: high magnification TEM image); and (d) UV-Vis absorption spectrum of MeO-BOD (5 μM) in deionized water. | ||
Cellular fluorescence imaging and subcellular colocalization
Given the above inspiring results, we attempted to investigate the fluorescence (FL) imaging and PDT efficiency of MeO-BOD in living cells. To this end, MeO-BOD nanoparticles (NPs) were prepared by adding the DMSO stock solution of MeO-BOD into water.44 The transmission electron microscopy (TEM) image in Fig. 2c indicated that the MeO-BOD NPs had a regular spherical morphology with a diameter of approximately 72 nm. The size distribution of the nanoparticles was determined using dynamic light scattering (DLS), which showed that the average size was 68 ± 7 nm (Fig. S18†). These particles are suitably sized for passive targeting through the enhanced permeability and retention (EPR) effect.45 In addition, the evident decrease in absorbance and the broad absorption spectrum and the redshift of the fluorescence spectrum of MeO-BOD in deionized water (DW) suggested that the formation of MeO-BOD NPs might be due to J-aggregation (Fig. 2d and S19b†).46 The MeO-BOD NPs could be disassembled and the instinctive fluorescence peak of MeO-BOD could be restored in the presence of FBS (10%) in DW (Fig. S19†).Then, we further explored the cellular uptake of MeO-BOD in HeLa cells by using confocal laser scanning microscopy. As illustrated in Fig. 3a, MeO-BOD could be rapidly internalized by living cells, and the FL images showed strong emission in the cell cytoplasm. Therefore, MeO-BOD could be employed as an imaging-guided PDT agent. Furthermore, to test the main organelle locations of MeO-BOD, we co-stained HeLa cells with MeO-BOD and commercial MitoTracker Green (MTG) or LysoTracker Green (LTG). The colocalization experiments indicated that MeO-BOD were mainly localized in mitochondria, as indicated by the high Pearson's coefficient (0.98), instead of the lysosomes (Pearson's coefficient 0.62) (Fig. 3b–e). In addition, the time-dependent colocalization fluorescence imaging of MeO-BOD with MTG in HeLa cells was performed. As shown in Fig. S20,†MeO-BOD almost internalize into the mitochondria of HeLa cells after 1 h. Recent reports have demonstrated that the BODIPY dye itself could localize into the mitochondria of cells due to its low electron density character.47 The unmodified BODIPY platform has a low electron density (+δ) character. Also, the incorporation of sulfur atoms into the π-conjugated skeleton of BODIPY perhaps led to more reduction of electron density of the BODIPY core,48 which might facilitate the mitochondrial accumulation of MeO-BOD. Subcellular organelles are indispensable in maintaining cellular biological function.49 In particular, the generation of 1O2 in mitochondria can induce direct dysfunction and trigger cell apoptosis. Thus, mitochondria-targeted theranostic agents could maximize cancer treatment efficiency.
 | ||
| Fig. 3 (a) Confocal laser scanning microscopy images of HeLa cells incubated with MeO-BOD (1.0 μM) for different times (0, 15, 30 and 60 min); scale bar: 30 μm. Confocal laser scanning microscopy colocalization fluorescence images of MeO-BOD (1.0 μM) with (b) LTG and (c) MTG (500 nM) in HeLa cells, respectively. The fluorescence intensity correlation of MeO-BOD with (d) LTG and (e) MTG, respectively. Rp is Pearson's coefficient; scale bar: 20 μm. | ||
In vitro PDT efficacy evaluation
To demonstrate the PDT efficacy of MeO-BOD in living cells, we first tested its cytotoxicity by the methyl thiazolyltetrazolium (MTT) assay in HeLa cells. As indicated in Fig. 4a, MeO-BOD had negligible cytotoxicity in the dark, revealing its excellent biocompatibility in vitro. Under 560 nm light irradiation (0.1 W cm−2, 5 or 10 min), the viability of HeLa cells gradually decreased with the increasing concentration of MeO-BOD, and the growth inhibition ratio reached ∼88% even at a very low concentration of 0.15 μM (Fig. 4b). The half-maximal inhibitory concentration (IC50) of MeO-BOD for HeLa cells was as low as 95 nM under 560 nm light irradiation (0.1 W cm−2, 10 min). The extremely low IC50 of MeO-BOD could be attributed to the high 1O2 quantum yield and efficient mitochondria-specific ROS generation upon light irradiation. To clarify the cytotoxicity in the PDT process, we tracked the morphological variations of HeLa cells in the presence of MeO-BOD under 559 nm laser irradiation using confocal laser scanning microscopy. As described in Fig. S21,† with increasing irradiation time (0–10 min), the morphology of the HeLa cells preincubated with MeO-BOD obviously changed; gradual thinning of the cell membrane and the formation of numerous blebs (red line) were observed. In contrast, the cells not exposed to MeO-BOD exhibited no appreciable morphological changes under the same laser irradiation.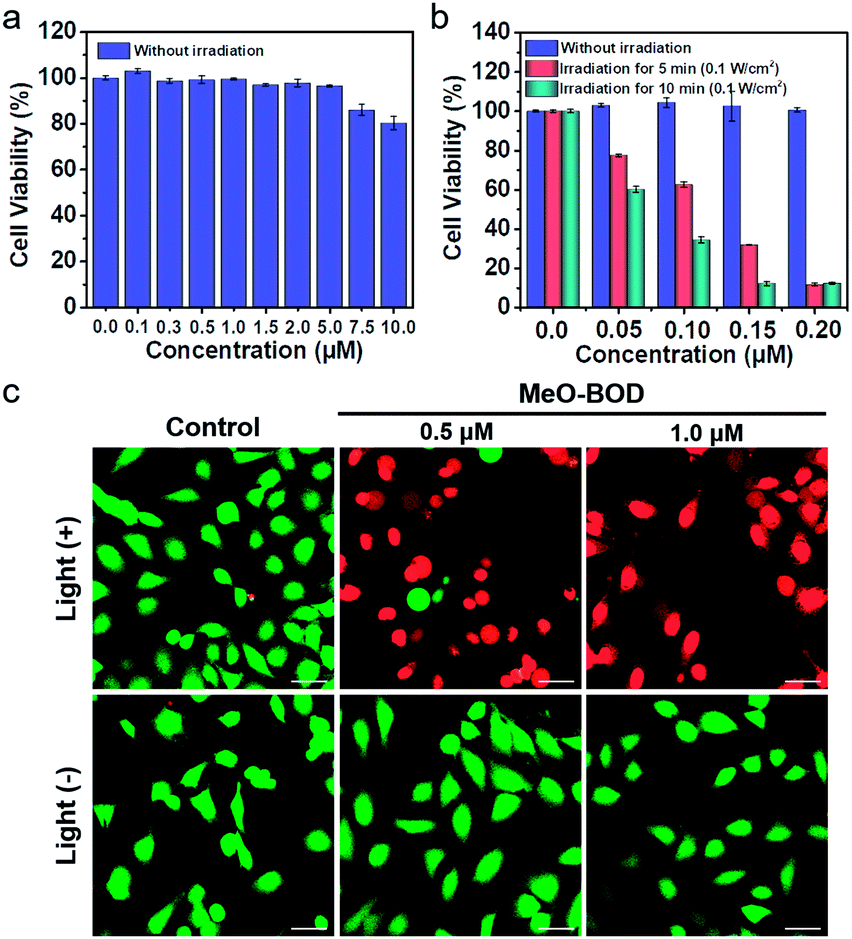 | ||
| Fig. 4 Concentration-dependent changes in the cell viability of HeLa cells treated with MeO-BOD using a typical MTT assay (a) in the dark and (b) under light irradiation. Cells were irradiated with 560 nm light (0.1 W cm−2, 5 and 10 min). (c) Fluorescence images of calcein AM/PI-stained HeLa cells after preincubation with MeO-BOD (0.5 and 1.0 μM) for 1 h and irradiation with 560 nm light (0.1 W cm−2, 10 min). Scale bar: 40 μm. | ||
Additionally, to intuitively establish the PDT efficacy of MeO-BOD (0.5 and 1.0 μM), live/dead cell co-staining of HeLa cells with calcein AM (green, live cells) and PI (red, dead cells) was performed to visualize the cell viability (Fig. 4c). The control group showed strong green fluorescence for live cells, which also verified the low dark toxicity and good biocompatibility of MeO-BOD. In comparison, almost all the HeLa cells treated with MeO-BOD were killed, and intense red fluorescence was observed. All these results confirmed that MeO-BOD could be employed as a theranostic agent for cancer treatment in vitro.
Apoptosis mechanism of MeO-BOD-mediated PDT
Finally, to elucidate the potential therapeutic mechanism at the cellular level, we further evaluated the cellular 1O2 generation capability of MeO-BOD by using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) as the 1O2 indicator in HeLa cells (Fig. 5a). The HeLa cells treated with only DCFH-DA and only MeO-BOD showed almost no fluorescence, whereas the group pretreated with MeO-BOD before light irradiation and then incubated with DCFH-DA showed apparent green fluorescence from oxidized 2,7-dichlorofluorescein (DCF). These experimental results validated the 1O2 generation ability of MeO-BOD in living cells. As mentioned above, the production of 1O2 in mitochondria causes mitochondrial destruction, resulting in cell apoptosis. The reduction of mitochondrial membrane potential (MMP) is a crucial signal of mitochondrial damage. Hence, the MMP changes were monitored by using confocal fluorescence images using JC-1 dye, as its fluorescence color changed between its aggregates (red, high MMP) and monomers (green, low MMP). The untreated control group under 560 nm light irradiation (0.1 W cm−2 for 10 min) displayed strong red emission and weak green emission, indicating that the cells were healthy with a high MMP. By comparison, the cells treated with MeO-BOD suffered from depolarization of the mitochondrial membrane, as demonstrated by the increase of green fluorescence intensity (Fig. 5b). These observations further suggested that the cell death was induced by a mitochondria-associated pathway.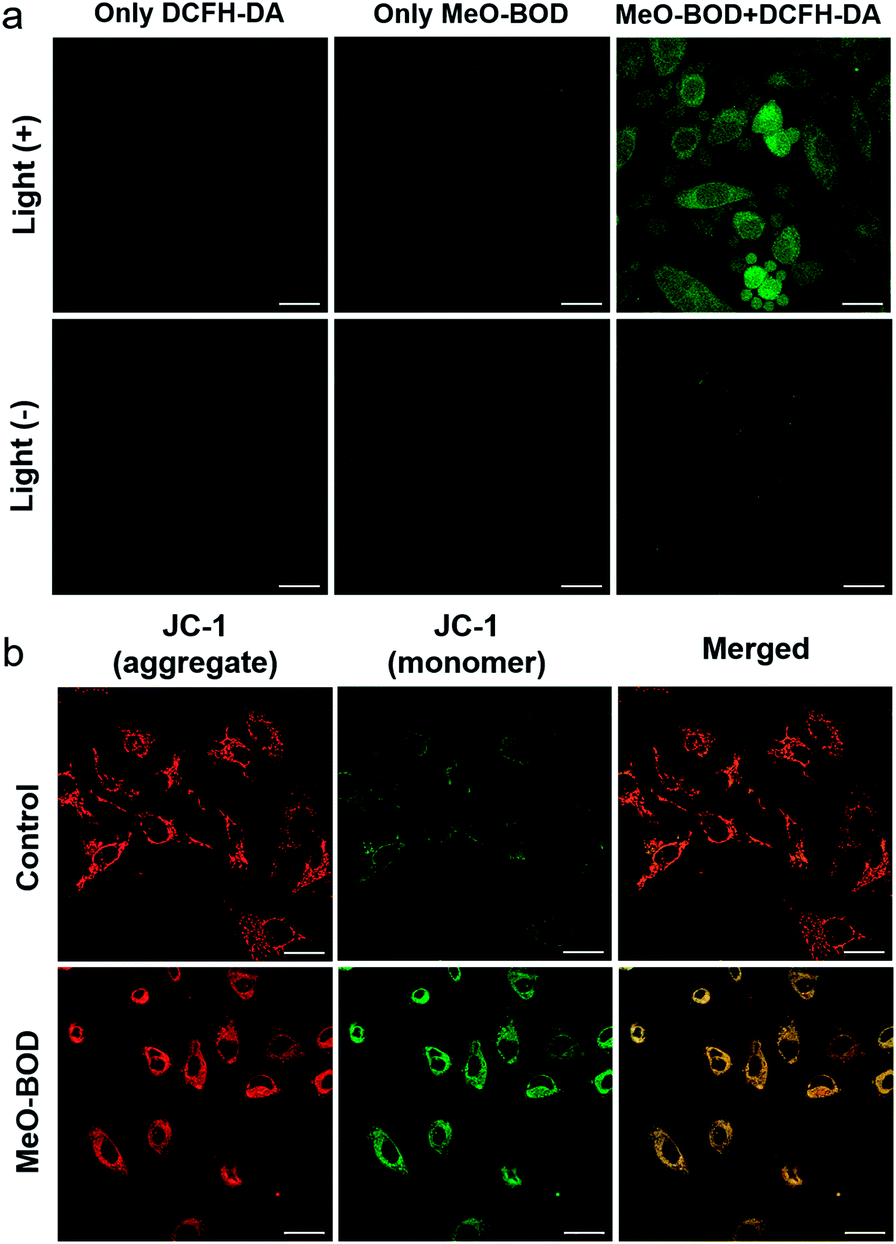 | ||
| Fig. 5 (a) Fluorescence images of ROS generation in HeLa cells after incubation with MeO-BOD (1.0 μM) for 1 h using DCFH-DA as an indicator (10 μM). The green fluorescence indicates that DCFH-DA is oxidized to DCF. (b) Fluorescence images of the mitochondrial membrane potential of HeLa cells after incubation without or with MeO-BOD (1.0 μM) using JC-1 as the indicator (2 μM), λex = 473 nm, red channel for aggregates (healthy cells): 575–675 nm, and green channel for monomers (apoptotic cells): 490–540 nm. Samples were irradiated with 560 nm light (0.1 W cm−2, 10 min). Scale bar: 30 μm. | ||
Conclusions
We synthesized thiophene-fused BODIPY analogues with different functional groups at the meso-position of the BODIPY core as mitochondria-targeted theranostic agents for imaging and PDT. The varying R-substituents had little effect on the absorption and emission maxima. PY-BOD, PH-BOD and MeO-BOD exhibited moderate fluorescence quantum yields, while that of DMA-BOD was much lower due to a PET process. Among the analogues, MeO-BOD exhibited excellent dual functionality, both considerable 1O2 generation ability and high brightness. The cell experiments manifested that MeO-BOD offered many advantages: good biocompatibility, mitochondria-specific fluorescence imaging, and a very low IC50 value (≈95 nM). Therefore, MeO-BOD could be employed as an imaging-guided PDT agent for cancer treatment. In addition, the apoptosis mechanism of light-induced PDT might be a result of ROS-induced damage to mitochondria, which was demonstrated by detecting the changes of the mitochondrial membrane potential. Briefly, finely tuning the electronic structure of the substituent at the meso-site of a heavy-atom-free thiophene-fused BODIPY core is a promising strategy for developing highly efficient theranostic agents with minimal side effects to accomplish the integration of diagnosis and therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2012R1A3A2048814 for J. Y.). Mass spectral data were obtained from the Korea Basic Science Institute (Daegu) on a Jeol JMS 700 high resolution mass spectrometer.Notes and references
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- J. Zhang, L. Ning, J. Huang, C. Zhang and K. Pu, Chem. Sci., 2020, 11, 618–630 RSC.
- J. Dai, Y. Li, Z. Long, R. Jiang, Z. Zhuang, Z. Wang, Z. Zhao, X. Lou, F. Xia and B. Z. Tang, ACS Nano, 2020, 14, 854–866 CrossRef CAS PubMed.
- Y. Yuan, C.-J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- Y. Cai, W. Si, W. Huang, P. Chen, J. Shao and X. Dong, Small, 2018, 14, 1704247 CrossRef PubMed.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- X. Yi, F. Wang, W. Qin, X. Yang and J. Yuan, Int. J. Nanomed., 2014, 9, 1347–1365 CrossRef PubMed.
- X. Li, C. y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS PubMed.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820–15826 CrossRef CAS PubMed.
- R. Wang, K. Dong, G. Xu, B. Shi, T. Zhu, P. Shi, Z. Guo, W.-H. Zhu and C. Zhao, Chem. Sci., 2019, 10, 2785–2790 RSC.
- H. Wang, W. Zhao, X. Liu, S. Wang and Y. Wang, ACS Appl. Bio Mater., 2020, 3, 593–601 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- W. Sun, X. Zhao, J. Fan, J. Du and X. Peng, Small, 2019, 15, 1804927 CrossRef PubMed.
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- J. Tian, J. Zhou, Z. Shen, L. Ding, J.-S. Yu and H. Ju, Chem. Sci., 2015, 6, 5969–5977 RSC.
- Q.-J. Hu, Y.-C. Lu, C.-X. Yang and X.-P. Yan, Chem. Commun., 2016, 52, 5470–5473 RSC.
- L. Huang, Z. Li, Y. Zhao, Y. Zhang, S. Wu, J. Zhao and G. Han, J. Am. Chem. Soc., 2016, 138, 14586–14591 CrossRef CAS PubMed.
- H. S. Jung, J. Han, H. Shi, S. Koo, H. Singh, H.-J. Kim, J. L. Sessler, J. Y. Lee, J.-H. Kim and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 7595–7602 CrossRef CAS PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
- S. Xu, W. Wu, X. Cai, C.-J. Zhang, Y. Yuan, J. Liang, G. Feng, P. Manghnani and B. Liu, Chem. Commun., 2017, 53, 8727–8730 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed.
- S. Kolemen, M. Işık, G. M. Kim, D. Kim, H. Geng, M. Buyuktemiz, T. Karatas, X.-F. Zhang, Y. Dede, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2015, 54, 5340–5344 CrossRef CAS PubMed.
- M. Üçüncü, E. Karakuş, E. Kurulgan Demirci, M. Sayar, S. Dartar and M. Emrullahoğlu, Org. Lett., 2017, 19, 2522–2525 CrossRef PubMed.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2019, 5, 138–155 CAS.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- X.-F. Zhang and N. Feng, Chem.–Asian J., 2017, 12, 2447–2456 CrossRef CAS PubMed.
- S. Ji, J. Ge, D. Escudero, Z. Wang, J. Zhao and D. Jacquemin, J. Org. Chem., 2015, 80, 5958–5963 CrossRef CAS PubMed.
- S. G. Awuah, S. K. Das, F. D'Souza and Y. You, Chem.–Asian J., 2013, 8, 3123–3132 CrossRef CAS PubMed.
- K. Tanaka, H. Yamane, R. Yoshii and Y. Chujo, Bioorg. Med. Chem., 2013, 21, 2715–2719 CrossRef CAS PubMed.
- R. L. Watley, S. G. Awuah, M. Bio, R. Cantu, H. B. Gobeze, V. N. Nesterov, S. K. Das, F. D'Souza and Y. You, Chem.–Asian J., 2015, 10, 1335–1343 CrossRef CAS PubMed.
- X. Xia and Y. Qian, Analyst, 2018, 143, 5218–5224 RSC.
- C. Wang and Y. Qian, Biomater. Sci., 2020, 8, 830–836 RSC.
- W. Lin, W. Zhang, S. Liu, Z. Li, X. Hu, Z. Xie, C. Duan and G. Han, ACS Appl. Mater. Interfaces, 2019, 11, 43928–43935 CrossRef CAS PubMed.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed.
- V. N. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C. Y. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202002843.
- W. Hu, Y. Lin, X.-F. Zhang, M. Feng, S. Zhao and J. Zhang, Dyes Pigm., 2019, 164, 139–147 CrossRef CAS.
- Y. Liu, C. Xu, L. Teng, H.-W. Liu, T.-B. Ren, S. Xu, X. Lou, H. Guo, L. Yuan and X.-B. Zhang, Chem. Commun., 2020, 56, 1956–1959 RSC.
- X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- C. Duan, Y. Zhou, G.-G. Shan, Y. Chen, W. Zhao, D. Yuan, L. Zeng, X. Huang and G. Niu, J. Mater. Chem. C, 2019, 7, 3471–3478 RSC.
- T. Gayathri, S. Karnewar, S. Kotamraju and S. P. Singh, ACS Med. Chem. Lett., 2018, 9, 618–622 CrossRef CAS PubMed.
- K. Tanaka, H. Yamane, R. Yoshii and Y. Chujo, Bioorg. Med. Chem., 2013, 21, 2715–2719 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 26529–26558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01171a |
| This journal is © The Royal Society of Chemistry 2020 |