
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective electrochemical reduction of carbon dioxide to ethanol via a relay catalytic platform†
Juan
Du
ab,
Shaopeng
Li
ab,
Shulin
Liu
ab,
Yu
Xin
ab,
Bingfeng
Chen
 a,
Huizhen
Liu
*ab and
Buxing
Han
ab
a,
Huizhen
Liu
*ab and
Buxing
Han
ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: liuhz@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 24th April 2020
Abstract
Efficient electroreduction of carbon dioxide (CO2) to ethanol is of great importance, but remains a challenge because it involves the transfer of multiple proton–electron pairs and carbon–carbon coupling. Herein, we report a CoO-anchored N-doped carbon material composed of mesoporous carbon (MC) and carbon nanotubes (CNT) as a catalyst for CO2 electroreduction. The faradaic efficiencies of ethanol and current density reached 60.1% and 5.1 mA cm−2, respectively. Moreover, the selectivity for ethanol products was extremely high among the products produced from CO2. A proposed mechanism is discussed in which the MC–CNT/Co catalyst provides a relay catalytic platform, where CoO catalyzes the formation of CO* intermediates which spill over to MC–CNT for carbon–carbon coupling to form ethanol. The high selectivity for ethanol is attributed mainly to the highly selective carbon–carbon coupling active sites on MC–CNT.
Introduction
The excessive use of fossil fuels and deforestation result in the continuous increase in carbon dioxide (CO2) concentration in the atmosphere and ocean, which is a crucial issue for human beings.1 The efficient conversion of CO2 to fuels and high-value-added chemicals can promote the development of the cyclic utilization of carbon resources and a reduction in CO2 emissions.2,3 Electrocatalytic reduction of CO2 is a promising route due to its mild operating conditions (ambient pressure and temperature) and modular reaction systems, which can be easily controlled by adjusting electrochemical parameters, making them easy to scale-up for applications.4–6 Ethanol is a widely used intermediate in chemical synthesis and a liquid fuel with a high energy density (26.8 MJ kg−1).7 Direct electroreduction of CO2 to ethanol is of great importance. However, it is challenging because the process involves not only the transfer of multiple proton–electron pairs but also carbon–carbon coupling with a high overpotential.Cu is documented to be a key component for multicarbon chemical production. Unfortunately, most Cu-based electrodes are more favorable for ethylene over ethanol. On plasma-activated Cu nanocube electrocatalysts, the faradaic efficiencies (FE) of ethylene and ethanol were 45% and 22%, respectively.8 Electrodeposited CuAg alloy films showed a good electroreduction performance with an FE of 60% for ethylene and 25% for ethanol.9 An oxide-derived Cu/carbon catalyst exhibited an FE of 24% for ethanol production.10 On bimetallic Ag/Cu electrocatalysts, ethanol could be formed with an FE of 41%.11 Hierarchically structured Cu dendrite electrodes with a superhydrophobic surface attained an FE of 56% for ethylene and 17% for ethanol production.12 An FE for ethanol of 41% on porphyrin-based Fe/Cu was reported and the ratio of ethanol to ethylene was 1.0 under optimized operating conditions.13 Copper–nitrogen-doped carbon materials achieved aqueous CO2 electroreduction to ethanol at an FE of 55% under optimized conditions.14 On copper-cuprous oxide electrodes, ethanol and acetic acid could be produced, while the main product was acetic acid.15
In addition to Cu-based electrocatalysts, Ag-, Fe- and carbon-material-based catalysts have also been reported for the electroreduction of CO2 to ethanol. Ag-anchored N-doped graphene/carbon foam could efficiently catalyze the conversion of CO2 to ethanol with an FE of 82.1–85.2%, but the current density was only 0.35 mA cm−2.16 Fe2P2S6 nanosheets were used as an efficient electrocatalyst for the highly selective electroreduction of CO2 to methanol and ethanol, with an FE of 23.1% for ethanol.17 An Ru(II) polypyridyl carbene complex immobilized on N-doped porous carbon displayed an FE of 27.5% for ethanol with very low current density.18 Carbon material electrodes can catalyze the conversion of CO2 to ethanol with high selectivity but their current density is very low. For example, an FE of 77% for ethanol was obtained on nitrogen-doped ordered cylindrical mesoporous carbon with a current density <1 mA cm−2. Moreover, the selectivity of ethanol among the products produced from CO2 was nearly 100%.19 Further study showed that medium micropores embedded in the channel walls of nitrogen-doped ordered mesoporous carbon could promote ethanol production from CO2 electroreduction, and the FE for ethanol reached 78%.20
A literature survey shows that the electrocatalysts that have been reported exhibit high current density and low selectivity for ethanol or high selectivity with low current density. It is obvious that more efforts should be made to develop highly efficient electrocatalysts for the electroreduction of CO2 to ethanol. CO is an important intermediate for the generation of C2+ products from CO2 electroreduction. It has been reported that high-concentration CO could be generated on the doping metal sites and spill over to the Cu sites for carbon–carbon coupling before further reduction to C2+ products on Cu-based bimetallic electrodes. Rich local CO intermediates can effectively promote the next carbon–carbon coupling process in CO2 electroreduction. Moreover, the highly selective carbon–carbon coupling active sites on a catalyst are crucial for the electroreduction of CO2 to ethanol with high selectivity.
Cobalt-based materials can be used as catalysts for the electroreduction of CO2 to CO. On semiconducting Co3O4 nanofibers, CO could be formed with an FE of 65%.21 Cobalt phthalocyanine (CoPc) molecules anchored on carbon nanotubes exhibit an FE of 95% for CO production.22 Inspired by the high selectivity by carbon materials for CO2 electroreduction to ethanol and the good performance of cobalt-based materials for CO2 electroreduction to CO, in this work, we designed a catalyst MC–CNT/Co, in which CoO was anchored in N-doped carbon material composed of mesoporous carbon (MC) and carbon nanotubes (CNT), for CO2 electroreduction, which provided a relay catalytic platform, where CoO catalyzes the formation of *CO intermediates that spill over to MC–CNT for carbon–carbon coupling to ethanol. The selectivity for ethanol and acetaldehyde could reach nearly 100% among the CO2 electroreduction products at −0.2 to −0.45 V and an FE of 60.1% was achieved for ethanol under a low overpotential (−0.32 V vs. RHE) with a current density of 5.1 mA cm−2. The Co species, ordered mesoporous structure and CNT had an excellent synergistic effect for promoting CO2 electroreduction to ethanol.
Experimental
Reagents
Toray carbon paper (CP, TGP-H-60, 19 × 19 cm), Nafion D-521 dispersion (5% w/w in water and 1-propanol, ≥0.92 meg per g exchange capacity) and Nafion N-117 membrane (0.180 mm thick, ≥0.90 meg per g exchange capacity) were purchased from Alfa Aesar China. 3-(Aminopropyl)trimethoxysilane (APTMS), 2-methylimidazole (purity >98%), tetraethyl orthosilicate (TEOS) (purity >99%), and cobalt nitrate hexahydrate (Co(NO3)2) were purchased from Beijing Innochem Company, Pluronic P123 (average Mn ∼ 5800) was obtained from Sigma. Hydrochloric acid, sodium hydroxide, and toluene (purity >99%) were provided by the Beijing Chemical Company.Synthesis of the electrode materials
Materials characterization
The morphologies of the electrodes were characterized using a S4800 scanning electron microscope (SEM) and a JEOL-2100F transmission electron microscope (TEM) operated at 200 kV. Powder X-ray diffraction (XRD) patterns were performed on an X-ray diffractometer (Model D/MAX2500, Rigaka) with Cu-Kα radiation. X-ray photoelectron spectroscopy (XPS) analysis was conducted on a Thermo Scientific ESCA Lab 250Xi using 200 W monochromatic Al Kα radiation. The content of Co in the catalysts was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Vista-MPX). The N2 adsorption/desorption isotherms were determined using a Micromeritics TriStar 3020 instrument at −196 °C. The Brunauer–Emmett–Teller (BET) method was employed to calculate the specific surface area, while the Barrett–Joyner–Halenda (BJH) method was applied to analyze the pore size distribution using the adsorption branch of the isotherm. The FTIR spectra were collected at a resolution of 4 cm−1 on a Bruker Vector 27 spectrophotometer in the 400–4000 cm−1 region. The IR spectra of the samples were measured by the conventional KBr pellet method. Thermogravimetric analysis (Pyris 1 TGA) was performed under an Ar flow from 20 to 800 °C at a heating rate of 10 °C min−1.Electrochemical measurements
An electrochemical workstation (CHI 660E, Shanghai CH Instruments Co., China) was used for all CO2 reduction experiments. Typically, a 10 mg sample and 20 μL of Nafion solution (5 wt%) were dispersed in 1 mL of deuterium acetonitrile by sonicating for 30 min to form a homogeneous ink. Then, the dispersion was loaded onto carbon paper with dimensions of 1 cm × 1 cm. For CO2 reduction experiments, cyclic voltammogram (CV) measurements were performed in an H-type cell using 0.5 M KHCO3 (60 mL, pH = 7.8) as the electrolyte with a typical three-electrode setup, which contained a working electrode, a platinum gauze auxiliary electrode, and an Ag/AgCl reference electrode. All the measured potentials in this work are cited with respect to the RHE using the following conversion: ERHE (V) = EAg/AgCl (V) + 0.197 V + (0.059 V × pH). Before electrolysis, the electrolyte was purged with Ar or CO2 gas for at least 30 min. The CV measurement in gas-saturated electrolyte was conducted at a sweep rate of 5 mV s−1 in the potential between −0.6 V and −1.4 V (vs. Ag/AgCl). The process was kept under constant magnetic stirring. The electrolysis experiments were conducted at 25 °C in a typical H-type cell. It consisted of a cathode, an anode (platinum gauze auxiliary electrode) and an Ag/AgCl reference electrode. In the experiments, the cathode and anode compartments were separated by a Nafion-117 proton exchange membrane. KHCO3 aqueous solution (0.5 M) was used as the anodic electrolyte. Then, potentiostatic electrochemical reduction of CO2 was carried out with CO2 bubbling (5 mL min−1). The reduction of acetaldehyde was also performed over an MC–CNT/Co electrode at −0.32 V (vs. RHE) in 0.5 M KHCO3 electrolysis solution with 0.05 M acetaldehyde. The electrolysis experiment was conducted at 25 °C in an H-type cell with a Nafion-117 proton exchange membrane and lasted for 2 h. The liquid products were analyzed by 1H NMR (Bruker Avance III 400 HD spectrometer) in acetonitrile-d3 with dimethyl sulfoxide (DMSO) as an internal standard. The gaseous product was collected using a gas bag and analyzed by a gas chromatograph (GC, HP 4890D), which was equipped with a TCD detector using helium as the internal standard. Each data point was an average of the measurements collected from at least three separate NMR or GC analyses. Each prepared catalyst was used only once for CO2 reduction at a chosen potential.Gaseous products.
| FE = moles of products per second/theoretical moles equivalent per second |
Liquid products. In NMR spectra, DMSO was used as the internal standard, and the relative peak area of CH3CH2OH could be calculated.
The number of electrons required to produce liquid product during the entire CO2 electroreduction reaction is:
| N = C × V × NA × ne |
We can obtain the Qtatal from the chronoamperogram and the total number of electrons by measuring:
| Ntotal = Qtotal/e |
The faradaic efficiency (FE) of the liquid product is:
| FE = N/Ntotal × 100% |
Results and discussion
Ordered MS was prepared using TEOS as the precursor and P123 as the template. Amino groups were grafted onto MS using APTMS as an amination reagent, which was proved by the N–H vibration peaks at 1560 and 3120 cm−1 in the FTIR spectra (Fig. S1†). The mesoporous structure of MS was retained after amino group grafting (Fig. S2†).24–26 MC–CNT/Co was synthesized in three steps, as shown in Fig. 1a. First, the cobalt species was adsorbed onto MS to obtain Co2+/MS owing to the electrostatic force between the amino and cobalt ion. Second, MC–CNT/Co/MS was obtained by thermal treatment of a mixture composed of Co2+/MS composite and powdered 2-methylimidazole under an Ar atmosphere, in which 2-methylimidazole was used as the carbon and nitrogen precursor. Subsequently, MC–CNT/Co/MS was treated with 10 wt% NaOH aqueous solution to remove silica, resulting in the MC–CNT/Co. SEM and TEM images show that MC–CNT/Co has a 3D network structure formed by CNT connected to rod-like mesoporous carbon (Fig. 1b and f). As shown in Fig. 1d and e, the graphene layers with a lattice distance of 0.34 nm could be clearly seen. It has been reported that Co could catalyze the formation of CNT.27 Ordered mesoporous structures observed from TEM images of MC–CNT/Co showed that the pores of the MS template were replicated (Fig. 1c). Additionally, cobalt nanoparticles (diameter: 3–5 nm) were also detected, and their lattice spacing was 0.25 nm, which was assigned to CoO (111) (Fig. 1d).28,29 The content of cobalt in MC–CNT/Co was 13.20 wt%, which was analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES). The corresponding elemental mapping reveals its elemental distribution, which shows a uniform N, Co distribution in the carbon framework (Fig. 1g). | ||
| Fig. 1 (a) Schematic illustration of the synthesis process, (b) SEM, (c and f) TEM, and (d and e) high-resolution TEM images, and (g) the corresponding elemental mapping of C, O, Co and N elements of MC–CNT/Co. | ||
Fig. 2a shows the XRD patterns of MC–CNT/Co. The broad diffraction peaks around 23.3° and 26.0° correspond to the amorphous and graphitized carbon, respectively. Three peaks at 36.8°, 43.1° and 63.4° are indexed to CoO (111), CoO (200) and CoO (220) (CoO-75-0418).30,31 As depicted in Fig. 2b, MC–CNT/Co exhibited a type IV adsorption–desorption isotherm and type H3 hysteresis loops, showing the existence of mesopores. The specific surface area calculated using the BET method was 303 m2 g−1. The BJH method was used to calculate the pore-size distribution from the adsorption branch of the nitrogen isotherm and the average pore size was 4.4 nm (Fig. 2c).
 | ||
| Fig. 2 (a) XRD patterns, (b) N2 adsorption and desorption curves, (c) pore-size distribution and (d) XPS spectrum of the Co 2p of MC–CNT/Co. | ||
The electronic states of different elements in MC–CNT/Co were investigated by XPS. As shown in Fig. S4,† Co, N, C and O elements were observed. The high-resolution Co 2p3/2 XPS spectrum of MC–CNT/Co (Fig. 2d) can be deconvoluted into three sets of peaks at 780.6 and 779.3 eV and a satellite at 785.5 eV, which are attributed to Co(II)Nx and Co(II)O.32–34 The O 1s signal (Fig. S5a†) was deconvoluted into three oxygen configurations, carboxyl oxygen (532 eV), hydroxyl oxygen (534 eV) in amorphous hydrogenated carbon and Co–O (529 eV).33 Deconvolution of the N 1s peak (Fig. S5b†) revealed two peaks that can be assigned to pyridinic-N (398.5 eV) and pyrrolic-N (400.0 eV), respectively.34
The CV tests over MC–CNT/Co were performed in Ar-saturated and CO2-saturated 0.5 M KHCO3 electrode aqueous solution (pH 7.8), respectively. In this work, the applied potential is referenced to the reversible hydrogen electrode (RHE) and the current density is calculated from the geometric surface area. As shown in Fig. 3a, an obvious reduction peak can be observed at ca. −0.3 V (vs. RHE) in a CO2-saturated electrolyte, which shows the reduction of CO2. Controlled-potential CO2 electrolysis experiments were performed in 0.5 M KHCO3 aqueous electrolyte solution using a typical H type cell. The onset potential of MC–CNT/Co was as low as −0.25 V (vs. RHE), which is more positive than that of many other carbon-based electrodes.19,35–38 More importantly, ethanol and acetaldehyde (trace) were the only liquid products detected by NMR spectroscopy. H2 was the only gaseous product in the range of −0.20 to −0.45 V (vs. RHE), showing that the selectivity for C2 liquid products from CO2 is 100% on the MC–CNT/Co catalyst. The current density and FE for ethanol and acetaldehyde were 5.1 mA cm−2 and 70.7% (60.1% for ethanol and 10.0% for acetaldehyde), respectively (Fig. 3b and c). Apart from ethanol, a small amount of CO was detected at −0.48 V and −0.52 V (versus RHE), which is a very important intermediate for C2 products, as in previously reported results.39 MC–CNT/Co exhibited a higher FE for ethanol than Cu-based catalysts and a higher current density than carbon-material-based electrodes. In addition, the MC–CNT/Co catalyst showed long-term stability in electrolysis, which was known from the fact that both current density and FE did not change considerably with electrolysis time over 20 h at −0.32 V (vs. RHE), as shown in Fig. S6.†
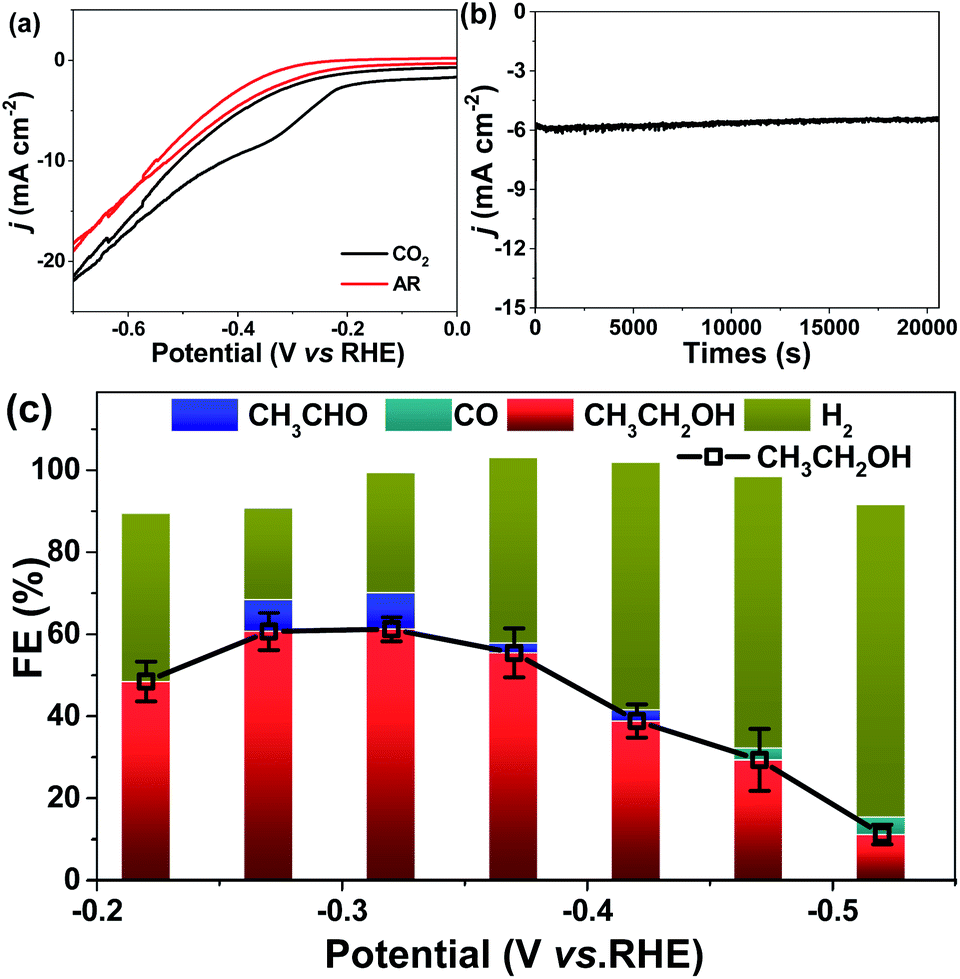 | ||
| Fig. 3 (a) Cyclic voltammogram curves in the potential range from 0 V to −0.7 V (vs. RHE) at a sweep rate of 5 mV s−1 in 0.5 M Ar-saturated and CO2-saturated 0.5 M KHCO3 on MC–CNT/Co electrodes, (b) total current density versus time in 0.5 M KHCO3 under a CO2 atmosphere on electrodes at −0.32 V (vs. RHE) and (c) FE for ethanol, aldehyde, carbon monoxide and hydrogen production obtained from the electrochemical reduction of CO2 for 6 h on MC–CNT/Co electrode at −0.2 to −0.6 V (vs. RHE). | ||
From the product distribution shown in Fig. 3c, we deduced that acetaldehyde was a possible intermediate in the formation of ethanol and this can be confirmed by the electroreduction of acetaldehyde. The reduction of acetaldehyde was performed over MC–CNT/Co at −0.32 V (vs. RHE) in 0.5 M KHCO3 electrolysis solution (Fig. S7†). It was found that ethanol was produced with 50% conversion and an FE of 98%.
To confirm that the product was derived from CO2 reduction, blank experiments using Ar to replace CO2 in the electrolysis at −0.3 V vs. RHE (Fig. S8†) were conducted and no product was formed after electrolysis for 6 h. The reaction in isotope-labeled 13CO2 and 12CO2 saturated 0.5 M KHCO3 electrolyte solution at −0.32 V vs. RHE was also studied over MC–CNT/Co. The product was analyzed by 1H NMR after electrolysis for 1 h. In the 1H NMR spectra in ESI Fig. S9,† we can only see the H signal of the 13CH3 group on ethanol, which is split into two peaks by coupling with the 13C atom. The results indicate that carbon atoms in the product were from CO2 rather than from other C-based chemicals in the reaction system.18,40
To unveil the reasons behind the good performance of MC–CNT/Co, MC–CNT and pure CoOx were checked as reference samples for the electroreduction of CO2. MC–CNT was prepared by etching CoO in MC–CNT/Co with hydrochloric acid solution (2 M) for 72 h, and characterized by TEM, XRD and XPS. Fig. S10† shows CNT and the mesopore structure of MC–CNT, but no detectable Co or CoO nanoparticles or subnano-clusters were observed. Moreover, no signals of Co or CoO were detected in the XRD or XPS spectra (Fig. S11†). These results indicate that Co or CoO was removed by HCl. A pure CoOx sample was also prepared by annealing Co(NO3)2 under an Ar atmosphere (Fig. S12†). CO2 electrolysis measurements were performed in CO2-saturated 0.5 M KHCO3 electrolyte solution using CoOx or MC–CNT as the catalysts and the electrolysis process was carried out in the range of −0.20 to −0.6 V (vs. RHE) for 2 h. On the CoOx electrode, CO was detected with a maximum FE of 5.6% at −0.46 V (vs. RHE), which is an important intermediate during CO2 electroreduction to C2 products (Fig. S13†). On the MC–CNT electrode, CO and ethanol as result of carbon–carbon coupling were produced with FE of 0.98% and 48.1% at −0.46 V (vs. RHE), respectively. The pyridinic-N and pyrrolic-N with high electron densities could facilitate the formation of ethanol, which is consistent with results reported in the literature.19,20 Furthermore, because of the excellent conductivity of CNT, the current density was measured to be 3.2 mA cm−2, which is higher than that reported for a carbon electrode. Recent studies have also proved the vital role of CNT in facilitating the electrocatalytic reduction of CO2.19,35,41,42 These experimental results suggest that MC–CNT/Co creates a relay catalytic platform where CoOx and MC–CNT cooperate very well to catalyze the formation of C2 products via CO.
Electrochemical impedance spectroscopy (EIS) was performed at −0.56 V vs. RHE in CO2-saturated 0.5 M KHCO3 solution to measure the charge transfer resistance (Rct) for MC–CNT/Co and MC–CNT. As shown in Fig. S14,† the Rct value of MC–CNT/Co was as low as 0.28 ohm, lower than that of MC–CNT (0.47 ohm), implying that electrons could be more easily transported on MC–CNT/Co.
According to the experimental data and reported results,35,43,44 a possible mechanistic pathway for the electrocatalytic production of ethanol over MC–CNT/Co is proposed and is shown in Scheme 1. One electron transfers to a CO2 molecule to form an intermediate CO2*− anion radical. Subsequently, the obtained CO2*− reacts with a proton–electron pair and forms CO* intermediates, which is mainly attributed to the CoO species. CO* is the key intermediate for the dimerization35 and it spills over to MC–CNT for carbon–carbon coupling to form *COCHO or *COCO intermediates. Pyrrolic-N and pyridinic-N in MC–CNT/Co and its mesoporous structure are both favorable for stabilizing CO* intermediates and promoting the carbon–carbon coupling reaction to ethanol.19,20 Furthermore, CoO species and MC–CNT are intimate in MC–CNT/Co because it is synthesized by one-pot pyrolysis, which is beneficial for the spillover of CO* intermediates to MC–CNT.
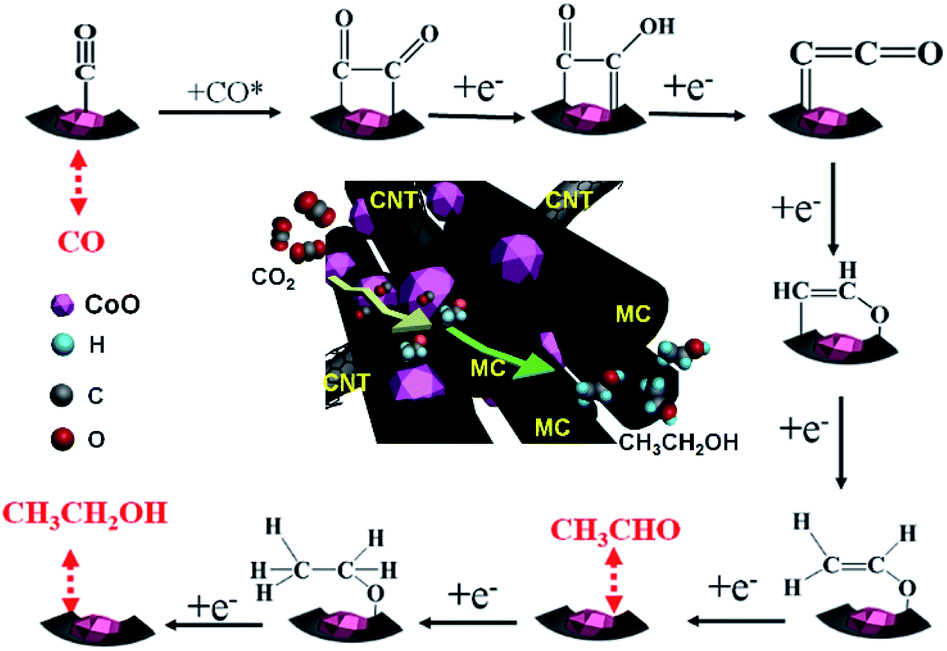 | ||
| Scheme 1 Proposed mechanism for the electrochemical reduction of CO2 to ethanol on an MC–CNT/Co electrode. | ||
Conclusions
In summary, the MC–CNT/Co catalyst designed in this work shows excellent performance for CO2 electroreduction to ethanol. The FE of ethanol could reach 60.1% at −0.32 V versus the reversible hydrogen electrode. The ordered mesoporous structure and CoO particles cooperate very well to promote the reaction. CoO catalyzes the formation of CO* intermediates that spill over to MC–CNT for carbon–carbon coupling to ethanol. Pyrrolic-N and pyridinic-N in MC–CNT/Co and its mesoporous structure are both favorable for stabilizing CO* intermediates and promoting the carbon–carbon coupling reaction. The high selectivity for ethanol was attributed mainly to the highly selective carbon–carbon coupling active sites on MC–CNT. We believe that a relay catalytical platform strategy can also be used for designing highly efficient catalysts for electroreduction of CO2 to C2+ products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017YFA0403003, 2017YFA0403101), the National Natural Science Foundation of China (21871277, 21603235 and 21403248), the Beijing Municipal Science & Technology Commission (Z191100007219009), and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed.
- A. Vasileff, X. Zhi, C. Xu, L. Ge, Y. Jiao, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 9411–9417 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS PubMed.
- H. Yang, Y. W. Hu, J. J. Chen, M. S. Balogun, P. P. Fang, S. Zhang, J. Chen and Y. Tong, Adv. Energy Mater., 2019, 9, 1901396 CrossRef.
- D. Gao, I. Sinev, F. Scholten, R. M. Arán-Ais, N. J. Divins, K. Kvashnina, J. Timoshenko and B. R. Cuenya, Angew. Chem., Int. Ed., 2019, 18, 17047–17053 CrossRef PubMed.
- H. Cui, Y. Guo, L. Guo, L. Wang, Z. Zhou and Z. Peng, J. Mater. Chem. A, 2018, 6, 18782–18793 RSC.
- H. Pramanik and S. Basu, Can. J. Chem. Eng., 2008, 85, 781–785 CrossRef.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. R. Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. d. Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. R. Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. G. Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- K. Lv, Y. Fan, Y. Zhu, Y. Yuan, J. Wang, Y. Zhu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 5025–5031 RSC.
- L. Ji, L. Chang, Y. Zhang, S. Mou, T. Wang, Y. Luo, Z. Wang and X. Sun, ACS Catal., 2019, 9, 9721–9725 CrossRef CAS.
- Y. Liu, X. Fan, A. Nayak, Y. Wang, B. Shan, X. Quan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26353–26358 CrossRef CAS PubMed.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS PubMed.
- Y. Song, S. Wang, W. Chen, S. Li, G. Feng, W. Wei and Y. Sun, ChemSusChem, 2020, 13, 293–297 CrossRef CAS PubMed.
- A. Aljabour, H. Coskun, D. A. Apaydin, F. Ozel, A. Hassel, P. Stadler, N. S. Sariciftci and M. Kus, Appl. Catal., A, 2018, 229, 163–170 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- A. Chen, Y. Yu, R. Wang, Y. Yu, W. Zang, P. Tang and D. Ma, Nanoscale, 2015, 7, 14686–14690 Search PubMed.
- S. H. Huang and D. H. Chen, J. Hazard. Mater., 2009, 163, 174–179 CrossRef CAS PubMed.
- S. Wang, K. Wang, C. Dai, H. Shi and J. Li, Chem. Eng. J., 2015, 262, 897–903 CrossRef CAS.
- L. You, F. Yuan and F. Ma, J. Phys. Chem. A, 2015, 89, 2298–2303 CAS.
- Z. Li, M. Shao, Q. Yang, Y. Tang, M. Wei, D. G. Evans and X. Duan, Nano Energy, 2017, 37, 98–107 CrossRef CAS.
- K. M. Nam, J. H. Shim, D.-W. Han, H. S. Kwon, Y.-M. Kang, Y. Li, H. Song, W. S. Seo and J. T. Park, Mater. Chem., 2010, 22, 4446–4454 CrossRef CAS.
- F. D. Wu and Y. Wang, J. Mater. Chem., 2011, 21, 6636 RSC.
- J. Zhao, Z. Shao and H. Zhang, Mater. Manuf. Processes, 2020, 35, 195–201 CrossRef CAS.
- F. Wu, S. Zhang, B. Xi, Z. Feng, D. Sun, X. Ma, J. Zhang, J. Feng and S. Xiong, Adv. Energy Mater., 2018, 8, 1703242 CrossRef.
- W. Hu, Q. Wang, S. Wu and Y. Huang, J. Mater. Chem. A, 2016, 4, 16920–16927 RSC.
- H.-C. Huang, I. Shown, S.-T. Chang, H.-C. Hsu, H.-Y. Du, M.-C. Kuo, K.-T. Wong, S.-F. Wang, C.-H. Wang, L.-C. Chen and K.-H. Chen, Adv. Funct. Mater., 2012, 22, 3500–3508 CrossRef CAS.
- H. Wang, X. Bo, A. Wang and L. Guo, Electrochem. Commun., 2013, 36, 75–79 CrossRef CAS.
- K. Lv, Y. Fan, Y. Zhu, Y. Yuan, J. Wang, Y. Zhu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 5025–5031 RSC.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio, J. H. Montoya, D. B. Trimarco, C. Roy, S. Meier, J. Wendland, J. K. Norskov, I. E. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2016, 55, 1450–1454 CrossRef CAS PubMed.
- W. M. Brooks, L. N. Moxon, J. Field, M. G. Irving and D. M. Doddrell, Biochem. Biophys. Res. Commun., 1985, 128, 107–112 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. Schouten, G. Mul and M. T. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- I. Ledezma-Yanez, E. P. Gallent, M. T. M. Koper and F. Calle-Vallejo, Catal. Today, 2016, 262, 90–94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01133a |
| This journal is © The Royal Society of Chemistry 2020 |