
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Autonomous intelligent agents for accelerated materials discovery
Joseph H.
Montoya
 a,
Kirsten T.
Winther
b,
Raul A.
Flores
b,
Thomas
Bligaard
bc,
Jens S.
Hummelshøj
a and
Muratahan
Aykol
*a
a,
Kirsten T.
Winther
b,
Raul A.
Flores
b,
Thomas
Bligaard
bc,
Jens S.
Hummelshøj
a and
Muratahan
Aykol
*a
aToyota Research Institute, Los Altos, CA 94022, USA. E-mail: murat.aykol@tri.global
bSLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
cDepartment of Energy Conversion and Storage, Technical University of Denmark, Lyngby, Denmark
First published on 30th July 2020
Abstract
We present an end-to-end computational system for autonomous materials discovery. The system aims for cost-effective optimization in large, high-dimensional search spaces of materials by adopting a sequential, agent-based approach to deciding which experiments to carry out. In choosing next experiments, agents can make use of past knowledge, surrogate models, logic, thermodynamic or other physical constructs, heuristic rules, and different exploration–exploitation strategies. We show a series of examples for (i) how the discovery campaigns for finding materials satisfying a relative stability objective can be simulated to design new agents, and (ii) how those agents can be deployed in real discovery campaigns to control experiments run externally, such as the cloud-based density functional theory simulations in this work. In a sample set of 16 campaigns covering a range of binary and ternary chemistries including metal oxides, phosphides, sulfides and alloys, this autonomous platform found 383 new stable or nearly stable materials with no intervention by the researchers.
1. Introduction
Scientific discovery has been synonymous with serendipity, and the desire to streamline it is not new. Principles governing autonomy of agents, search of hypothesis spaces, knowledge, sequential improvement, and statistical models in scientific discovery systems had already been articulated more than two decades ago.1–8 For materials, autonomous discovery is being fueled by two concurrent developments: the need for new technological materials and the progress in artificial intelligence (AI).9–11 Recent advances in rapid generation of materials data12–17 have created a new frontier where computational and experimental materials research is intersecting with AI.9,10 This new wave of AI in materials has a theme of acceleration, enabled either by bypassing tools or methods via surrogate models,18–23 or by identification of new materials via adaptive schemes that combine models with decision-making approaches.24–32A number of autonomous frameworks for materials discovery have been designed and demonstrated, such as optimization of carbon nanotube syntheses via a robotics interface;27 organic molecule synthesis robots33,34 for autonomously navigating complex chemical reaction networks with reagent-based decision-making; and composition-based autonomous search for low thermal hysteresis shape-memory alloys.35 Among other efforts,36–38 these demonstrate proof-of-concepts for autonomous discovery of materials for specific target properties or applications. In addition, open-source software packages such as ChemOS,39,40 which includes functionality for researcher interaction via natural-language-processing and robotics interfaces, and ESCALATE,41 which features a streamlined data capture and reporting framework, have demonstrated adaptable programming frameworks and ontologies for achieving autonomous discovery. However, there remains an opportunity for a framework which not only executes autonomous research, but facilitates the design of autonomous discovery procedures via a scientific method that tests the automated decision-making for their effectiveness in materials discovery.
This paper, concerned with both execution and design of autonomous discovery, introduces an end-to-end sequential framework that adopts an agent–experiment abstraction to solve complex discovery objectives in materials science. We present the framework in an application targeting discovery of stable inorganic compounds using ab initio calculations, a problem central to identifying new, useful candidate materials for technological applications.42–45 Using this framework, we show examples of (i) simulations of agents designed for stable materials discovery with existing data to gauge their performance, and (ii) active-deployment of these agents in real-world discovery campaigns of cloud-based density functional theory (DFT) calculations. We show that this autonomous platform can expand our knowledge of materials in target chemistries on user demand, and further, augment the data available in community databases like The Materials Project (MP),13 Open Quantum Materials Database (OQMD)15 or AFLOW12 with new, potentially useful materials. The framework is open-sourced for community development and use in other autonomous materials research settings.
2. Results and discussion
2.1 An AI framework for materials discovery
material* = argmin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Experiment(material) Experiment(material) |
f(X) could be solved with some systematic effort, hence discovery streamlined.
To be specific, let us focus on the problem of finding new inorganic materials in the most basic terms; i.e. previously unknown combinations of crystal structures and compositions that demonstrate a property, the most ubiquitous of which is thermodynamic stability. The effectiveness of design of experiments,49–51 or traditional optimization methods (e.g. gradient-based methods, off-the-shelf black-box or Bayesian optimizers, etc.) vanish quickly in such scenarios for a few reasons and we will invoke some of the early principles of discovery systems to suggest practical solutions for each. First, the experiments are often costly, and researchers have finite time and resources, such as a limited budget for running a set of experiments. The need for frugality can be satisfied by a sequential design that factors past attempts in informing its next stages. Nevertheless, rather than a fully sequential optimization framework where a single sample is run at a time, adopting a batch mode where multiple samples are run in parallel in each iteration is often desirable to minimize the total time-cost in applications where experiments take a long time.52,53 The advances in high-throughput computational and experimental workflows make batch mode further appealing in optimization of materials, offering a higher volume of knowledge per experiment duration, as well as potential failure mitigation by diversification of experimental parameters. Using batch mode, however, leads to some degree of loss in efficiency in using new information in decision-making compared to running a single experiment at a time, and introduces other challenges in ensuring efficient use of the allocated budget, e.g. how to choose the batch in each iteration for maximum return.52,53 Many practical decisions need to be automated to reduce this material optimization process to practice, as we show later.
Second, the Experiment can be highly complex, sometimes broadly thought as a “black-box”, with no gradients to aid in optimization. This complexity may necessitate the use of an array of methods to compose f, including not only surrogate models, but also physical or empirical models, physical laws (e.g. thermodynamics), or heuristics garnered by the scientists. In addition, one may need to try many such “compositions” of f to find the most suitable one for the task. Hence, drawing inspiration from the basic intelligent agents of AI,54 we adopt a similar abstraction hereafter referred to as a research agent (or simply agent), which, in our case, is a computational entity that manages the decision-making phase of the scientific-method; i.e. chooses candidate materials as hypotheses to test next, making use of any past or recent results and any available model, data, intuition, heuristics, logic or uncertainty as well as exploration–exploitation strategies. The agent is capable of performing this duty recursively, each time with added knowledge of recent experiments. Agents can communicate with the outside-world, namely, with external experimental facilities, by means of an experiment-specific application programming interface (API).
An outline of this framework for computational autonomy for materials discovery (CAMD) is illustrated in Fig. 1, where the main process in a campaign is a back and forth between an agent and an experiment. Since the API translates the requests of the agent to the experimental facility and delivers the results back to the system when experiments finish, the agent is disentangled from where or how the experiments are run. This abstraction yields a modularity that allows seamless transition between experimental resources or between agents. For instance, as we show in Section 2.2, the framework can simulate the performance of a series of agents using existing data along with an “after-the-fact” experiment API that emulates running experiments but simply returns results from a lookup table, and can later swap in the relevant experiment API to run the actual experiments with an agent of choice.
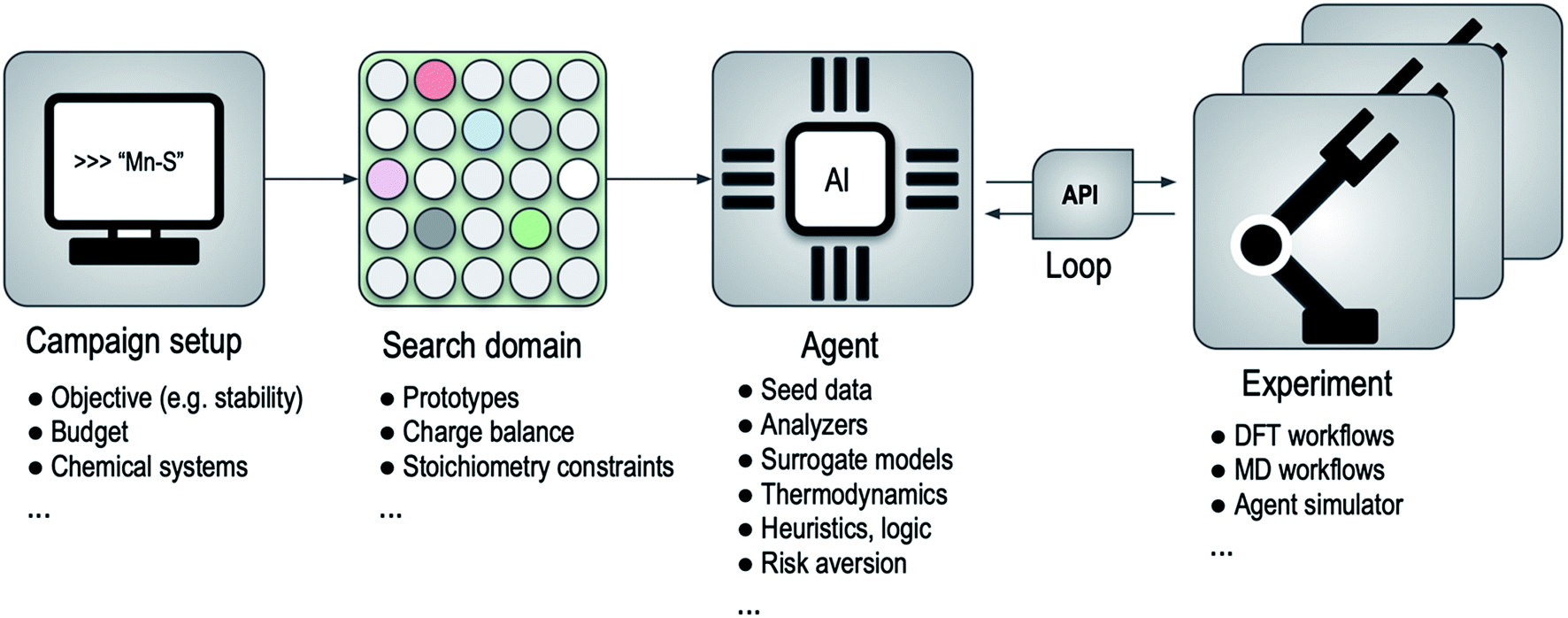 | ||
| Fig. 1 High-level flow-diagram for the agent-based sequential framework for computational autonomy for materials discovery (CAMD). Bullet points below each major component list examples or parts of the respective component. API, DFT and MD denote application programming interface, density functional theory and molecular dynamics, respectively. | ||
In designing agents for materials discovery, a practical challenge stems from the composition–structure space of materials being infinite, and the difficulty in formulating it while ensuring distinct materials to have distinct representation vectors. Despite recent promising efforts,55,56 the difficulty in inversion of such vectors to materials is a compounding factor in the overall problem. A means of circumventing these issues is creating a finite search space; a domain of candidates, such as from decoration of prototype crystal structures in our current effort for finding materials of relative stability. While not restricted to it, we isolate the domain creation process from the agents (Fig. 1), as an independent stage, again to benefit from the modularity. We note also that the nature of given materials domain, particularly in the distinction between categorical and continuous variables, may strongly influence the effectiveness of a given strategy for exploring it.57 In our primary example, domain generation blends heuristics, such as an integral grid-based stoichiometric formula generation and chemically-selective enforcement of nominal charge-balance constraints over formulas, and prototype extraction from structure databases (see the Methods section for details).
We consider several strategies in our agent-design for stable material discovery. As a common design element, agents are allowed to take existing results from DFT calculations that were already run, train formation energy regression models, modify predictions on the candidate space having the objective of stability in mind (e.g. by tagging on an uncertainty derived component or other exploration–exploitation strategies), construct a thermodynamic energy-composition convex-hull and use distance to this estimated hull as part of their decisions. The use of convex-hull within agents is a concrete example of a physical construct that is leveraged in the form of relational-information between materials, that is otherwise not easy to incorporate as part of standard optimization frameworks. Agents can also use logic; e.g., current agents enforce a floor for how low predicted energies can go, so they do not inadvertently skew the results in early iterations where energy predictions might be poor. Or they can use heuristics, e.g. to minimize the risk of acquiring similar points, or terminate if their performance is not satisfactory. Further details of agents are provided in the Methods section.
There are a large array of approaches for how acquisitions can be decided for finding materials that meet the objective of stability, often taking into account the variance in model predictions, such as the probability of improvement, expected improvement, lower (or upper) confidence bound or greedy (including ε-greedy) approaches.25,61,62 We frequently use the latter two, as they more easily generalize to optimization in the data-regimes we navigate. Some related algorithms the current set of agents adopt include query-by-committee (QBC)63,64 which can decorate a learner with variance in predictions, ensemble learners where prediction variances can be approximated in a similar way, or Bayesian approaches, including Gaussian process (GP) and its variants which circumvent its unfavorable computational complexity.65 Voronoi-based descriptors by Ward et al.66 are incorporated to represent materials in models, and for stability agents that require a regressor, we use a simple two hidden-layer (84 × 50) fully-connected neural network (NN) (see Methods). We found that for the present application, these choices for the formation energy models provide a good compromise in terms of accuracy and the speed of model training and prediction.
The value delivered by the discovery system should be measured carefully against baselines. A randomly choosing (RC) agent is therefore always simulated for comparison. For the task of finding new materials, however, this agent is not too difficult to outperform, unlike an active-learning scenario, where the goal is to minimize the variance over the prediction. In that case, an RC is a strong baseline. Thus, for discovery tasks, we simulate additional “one-shot” baselines, to test whether the sequential learning paradigm (i.e. incremental improvement of agent) is helping the discovery or not. These, unlike an RC agent, are difficult to beat, as they harness models trained on the entire past data, and can use acquisition techniques, but in contrast to sequential agents with access to accumulating data, are simply not given the opportunity to learn from each prior iteration. They make their choices in a chemical system, in one-shot, at the same overall budget as other simulated agents.
2.2 Campaign simulations with agents for materials discovery
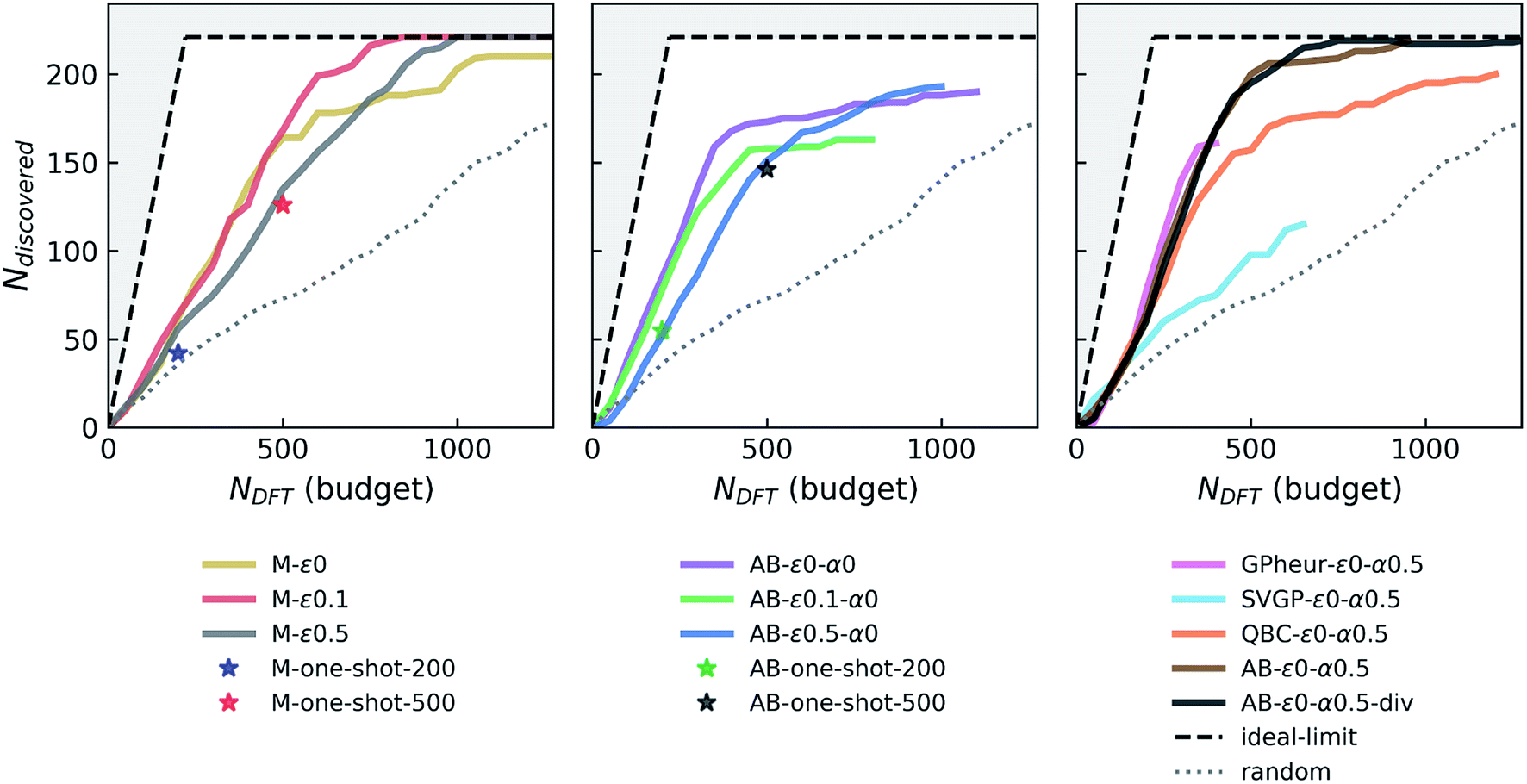 | ||
| Fig. 2 Simulated performance of various agents in discovering stable Fe–X binary compounds. Ndiscovered and NDFT correspond to the total number of materials that are within 0.1 eV per atom of the instantaneous convex-hull, and the DFT budget (in terms of total number of DFT calculations agent is allowed to request), respectively. Agents are labeled as follows: M, AB, GPheur, SVGP and QBC which respectively correspond to “greedy” agent with a surrogate model, AdaBoost based agent, heuristic GP agent, stochastic variational GP agent, and query-by-committee based agent. The parameters ε (fraction of random exploration) and α (mixing parameter for uncertainty) are followed by their respective values in labels. The “one-shot” points refer to direct acquisition of N = 200 or 500 candidates, with the noted agent (M refers to the NN regressor, and AB refers to the same regressor boosted with AdaBoost), with no sequential (batch-mode) acquisitions. Presence of “div” in the label means the agent uses diversification. Ideal-limit shows the maximum achievable performance, i.e. if every agent-designated material was deemed stable and therefore “discovered”, outside of which is not accessible by any agent and hence grayed out. | ||
000 ICSD-derived OQMD entries and newly “acquired” examples in each campaign), similar to the random forest model trained on ∼30000 examples in the original study that introduced the Voronoi-based descriptors we use.66 We did not see a notable drift from this performance with growing data set size or switching between models with enough complexity (e.g. random forest vs. NN). However, we find that adaptive boosting (AdaBoost, or simply AB) of the original regressor notably increases the model accuracy, reducing the CV-MAE with the same data to about 77 meV per atom. This boosting yields an improvement in the discovery rate (Fig. 2, second panel) for the greedy agent (AB-ε0-α0), but a pronounced saturation happens at about the same level as the non-boosted greedy agent. Interestingly, switching to an ε-greedy strategy for the boosted agent (e.g. AB-ε0.1-α0) does not help avoid early saturation (which will be studied in detail in a later section). Since results of the DFT or convex-hull computations do not have any notable uncertainty element, the accuracy of the formation energy model remains as the main bottleneck in the overall performance of the agents. Considerations of prediction uncertainties in decisions could therefore be useful for improving the agent performance.
While a GP is the “go-to” Bayesian approach for this purpose, its computational complexity prohibits its straightforward use in the data regimes (>30000 examples with 273 dimensional representations) we operate in. We first try a simple workaround: a GP is trained on a seed that combines ICSD-derived OQMD compounds in Fe–X chemistries, with 5000 additional samples randomly drawn from the rest of the ICSD-OQMD seed. We find that an agent employing this heuristic GP strategy (GPheur-ε0-α0.5) is on par with the boosted greedy agents, despite its lower MAE of ∼110 meV per atom, but saturates early like the fully-greedy agents. Despite its ability to handle big data in a GP setting, stochastic variational inference GP65 yielded relatively large MAEs (∼130 to 140 meV per atom) with the surrogate matrix sizes we could afford, and using the same LCB strategy, the agent SVGP-ε0-α0.5 displayed a relatively poor performance (third panel in Fig. 2).
Given the above difficulties, we turn to decorating predictions of non-Bayesian regression models with uncertainty estimates. QBC is an ensemble method analogous to bagging and estimates prediction variance from the disagreement of the committee members (each trained on a subset of data), and can be used for this purpose.64 A QBC agent that uses LCB (α = 0.5) with our standard NN regressor did not produce results better than a fully-greedy agent. The random acquisitions, as in ε-greedy agent, seem to play a more significant role than uncertainty estimates from QBC, to overcome the observed saturation. Given that boosting forms an ensemble of models, we can decorate predictions of AB with proxies for uncertainties derived from the variance among that of each regressor in the ensemble. An AB agent that uses such uncertainties with LCB and α = 0.5 (AB-ε0-α0.5 in Fig. 2) has the same discovery rate as its boost-only parents, but outperforms them by saturating much later, at a point where about only 20 stable materials left in the candidate pool. This is so far the best performing agent.
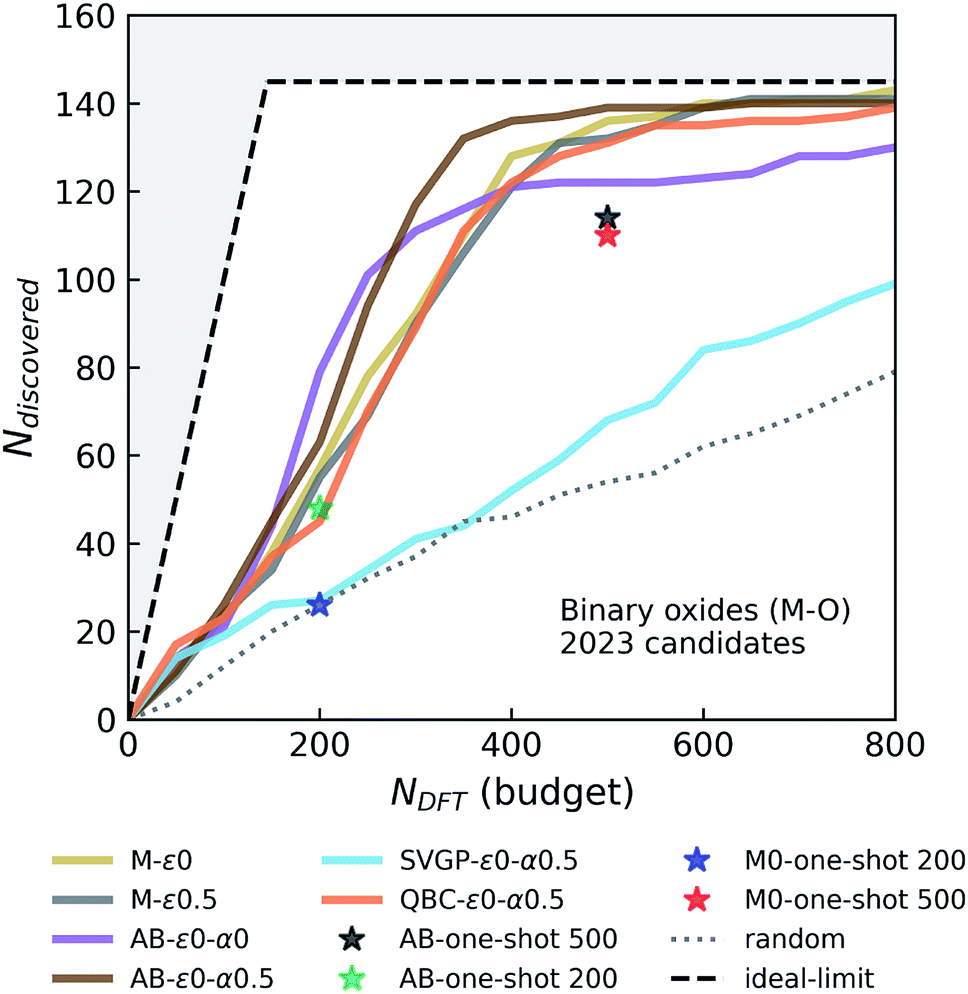 | ||
| Fig. 3 Simulated performance of various agents in discovering stable M–O binary compounds. Ndiscovered and NDFT correspond to the total number of materials that are within 0.1 eV per atom of the instantaneous convex-hull, and the DFT budget (in terms of total number of DFT calculations agent is allowed to request), respectively. See the caption of Fig. 2 for the descriptions of the layout and agent labels. | ||
In these variable-seed campaigns, we still focus on the Fe–X chemistry due to its chemical diversity and candidate set size, as well as for consistency with the previous campaigns, but this time we remove all Fe–X phases from the seed regardless of its origin, and reserve all of the 1933 Fe–X phases as candidates. We start these campaigns by randomly choosing and adding 50 candidates from this Fe–X candidate space to the above-mentioned seed. While we keep the chemistry and other settings (e.g. number of queries in each iteration) consistent as much as possible with Fig. 2, these new campaigns by design have a slightly larger candidate pool, and hence more stable materials (473 within 0.1 eV per atom of the convex-hull) available compared to prior deterministic campaigns. For each agent, we repeatedly run these campaign simulations with varying initial seeds at least eight times for the primary agents of interests that we selected from Fig. 2. The results of these variable-seed campaigns are presented in Fig. 4, where ±2σ confidence bounds are shown around the mean performance at each iteration of the campaign. Although there are minor performance differences compared to Fig. 2 due above-mentioned slight differences in campaign structure (for example M-ε0 and M-ε0.1 are closer now, but variation in performance with ε is still present as seen for M-ε0.5), most notable aspects, such as the improvements in performance with AB agents, their early saturation in greedy or ε-greedy strategies, and further improvement with LCB in AB, are all observed in this setting as well. Fig. 4 clearly shows that the confidence intervals are relatively tight, especially in the early stages of the campaigns, and tend to broaden only slightly as campaigns progress, the largest σ remaining within ∼13 compounds in AB agents. These results indicate that the incomplete phase information in partially explored chemical spaces do not result in significant variations in agent performance.
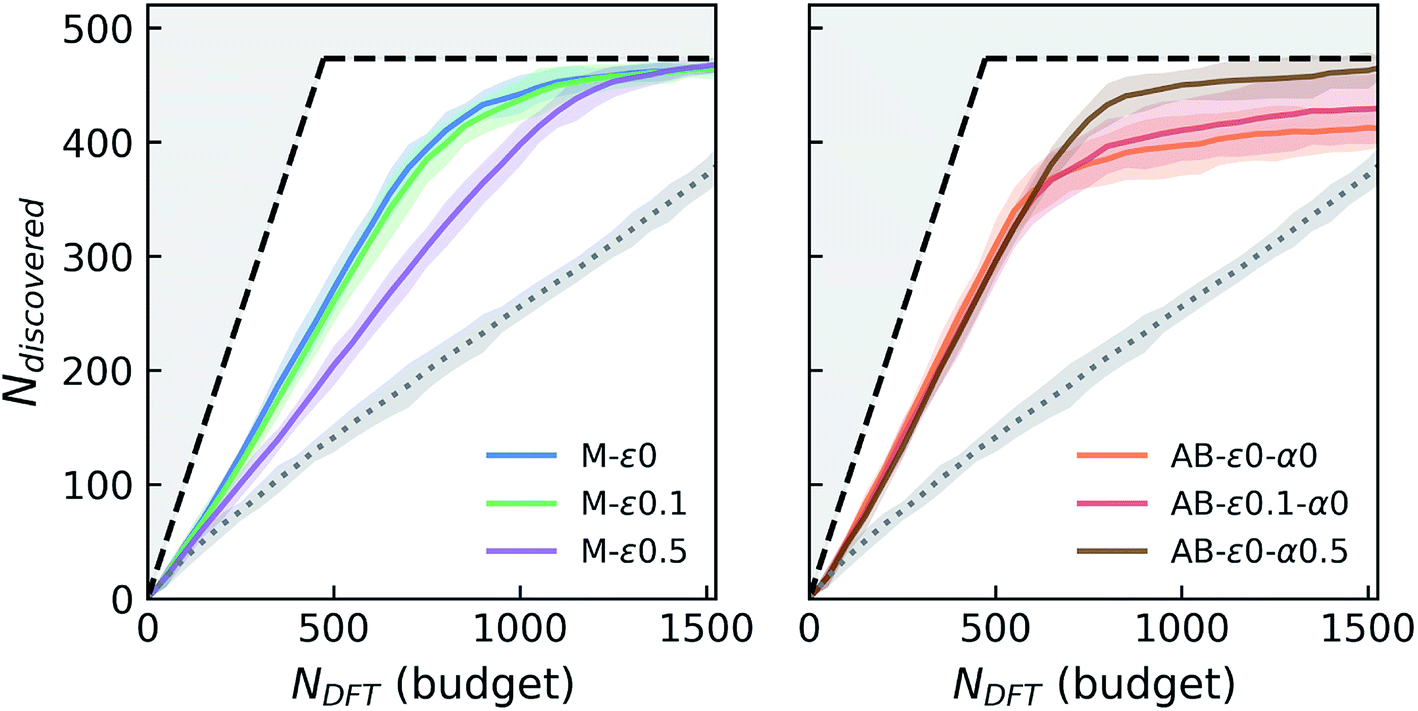 | ||
| Fig. 4 Simulated performance of selected agents under variable-seed conditions. For each agent, the solid line and the shaded area around it show the average number of discoveries and the ±2σ (95%) confidence interval around these averages, from at least eight random campaign initializations. See the caption of Fig. 2 for the descriptions of the layout and agent labels. | ||
 | ||
| Fig. 5 Analysis of the errors in predicted energies of candidates by various energy-models underlying several agents at early (iteration = 3) and later (iteration = 25) stages of respective campaigns. Top panels show parity plots comparing predicted formation energies to DFT values for candidates at the selected early and late stages of each campaign, where dashed-lines are a guide for the eye depicting ideal fit (i.e. x = y). The bottom panels show corresponding histograms of the difference between predicted and DFT values of formation energies for candidates in the same stages of the campaign as the top panels. Each same-colored set of vertical dashed-lines correspond from left to right to μ − σ, μ and μ + σ where μ and σ are the mean and standard deviation of their corresponding (same colored) distributions at two separate stages shown. Corresponding μ and σ (in units of meV per atom for clarity) are also shown as insets along with MAE (as “m”), color-coded the same way. The agent labels are shown above the parity plots. | ||
2.3 Active deployment of agents: DFT-based campaigns of materials discovery
For the domain creation, we use an approach that decorates crystal structure prototypes derived from databases of existing materials, combined with chemical, stoichiometric and charge-balance heuristics (see the Methods section). Briefly, the inputs to a stable materials discovery campaign are a list of elements which define the chemical system in which materials are to be discovered. With these elements, reasonable chemical formulas are generated algorithmically using heuristics from charge balance and permissible integral stoichiometric coefficients of elements in formulas. Candidate structures at these compositions are generated from the large array of anonymized (prototype) crystal structures. This process of domain generation renders a dataframe of structures keyed by element-specific stoichiometry and structure prototype and provides it to the campaign as the “candidate data”. The “seed data” is provided as the ICSD-derived entries in the OQMD (as explained before), because they reasonably capture the current stability landscape from experimentally known materials, and hence form a strong baseline against which to measure the stability of new findings. In effect, the energy-composition convex-hull construction for determination of the stability of new discoveries explicitly includes experimental data to the extent possible in the present framework, ensuring the predictions are valid in comparison to real, known materials. We note that these are merely the implementation defaults, and framework allows using other choices for all these components.
Defining experiments typically requires more effort than defining the campaign domain, as ensuring compatibility of the experimental data generated and the existing seed data may require knowledge of that seed data's provenance. For our initial implementation, we employ a DFT workflow that mirrors calculations done by the OQMD (see the Methods section), which ensures compatibility with DFT data that already exists therein. We employ a strict time limit for DFT runs to prioritize rapid turnover, and therefore agents may often receive a fraction of their requests back (further explained in the Methods section). Upon completion of a given experiment, a stability analyzer computes the convex hull corresponding to data from both new experimental results and prior seed data. We note that stability is an aggregate property of the dataset and thus requires a post-processing step independent of the experiment that includes data from the same seed data used as part of the agent. This batch-mode sequential workflow of the agent and the experiment iterating back and forth continues until the budget is exhausted or the campaign is terminated for one of the reasons outlined in Methods. A post-campaign structure analysis is performed, as prototype derived structures can often relax into a similar structure after the DFT relaxation, and hence become duplicates. The settings and other practical details of active campaigns reported here are listed in the Methods section.
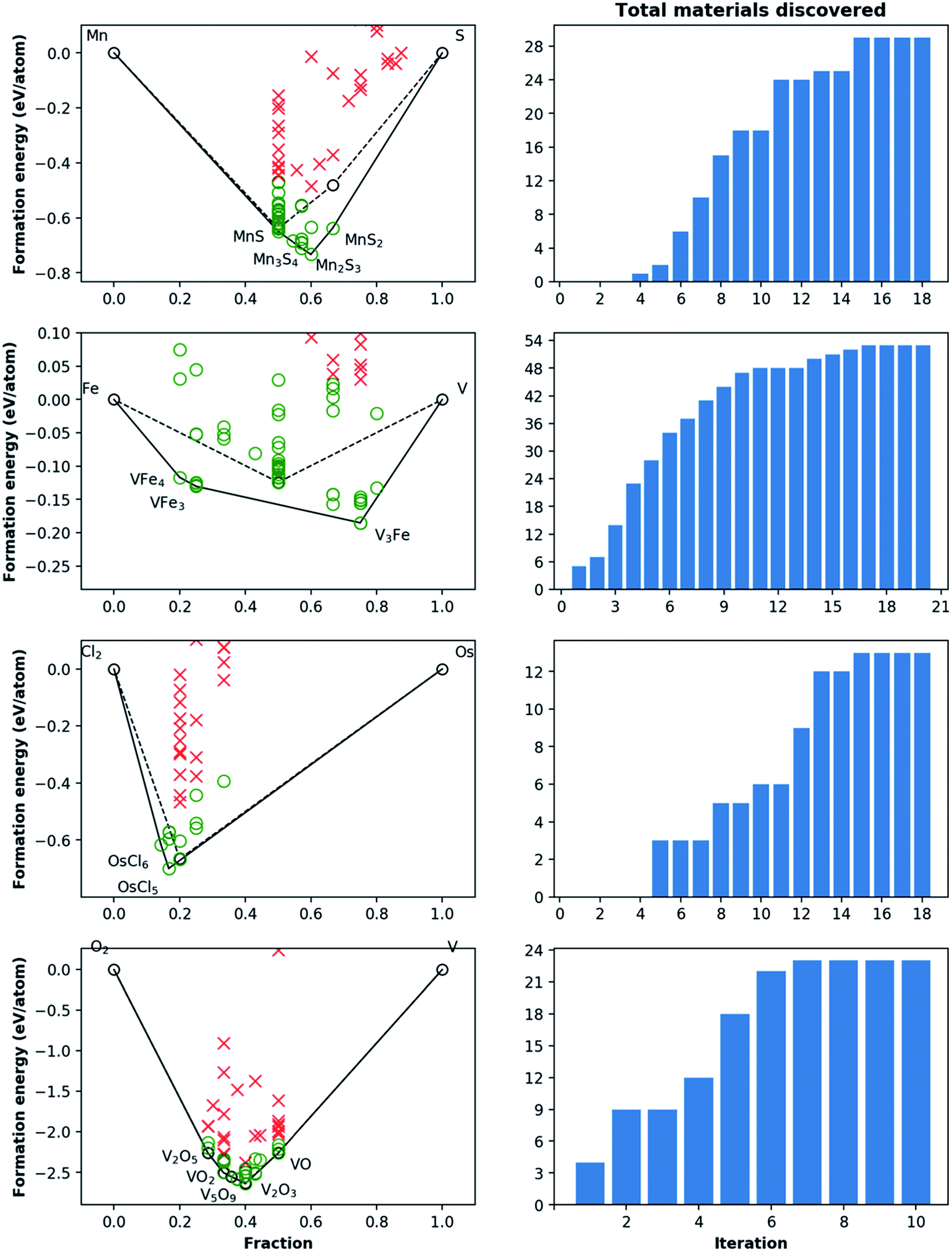 | ||
| Fig. 6 Phase diagram and cumulative discovery histograms for CAMD campaigns in Mn–S, Fe–V, Os–Cl, and V–O chemical systems. The 2-D phase diagrams are shown on the left with both pre-campaign convex hull (dashed line) and post-campaign convex hull (solid line). Material discoveries, i.e. computed structures within 0.2 eV per atom of the convex hull, are shown as green circles, while materials higher than this threshold are shown as red crosses. The number of total materials discovered for each chemical system as the campaign iterations progress are shown in the right-hand bar charts. For materials from pre-campaign convex-hulls (i.e. existing materials), only the most stable ones are shown for simplicity. | ||
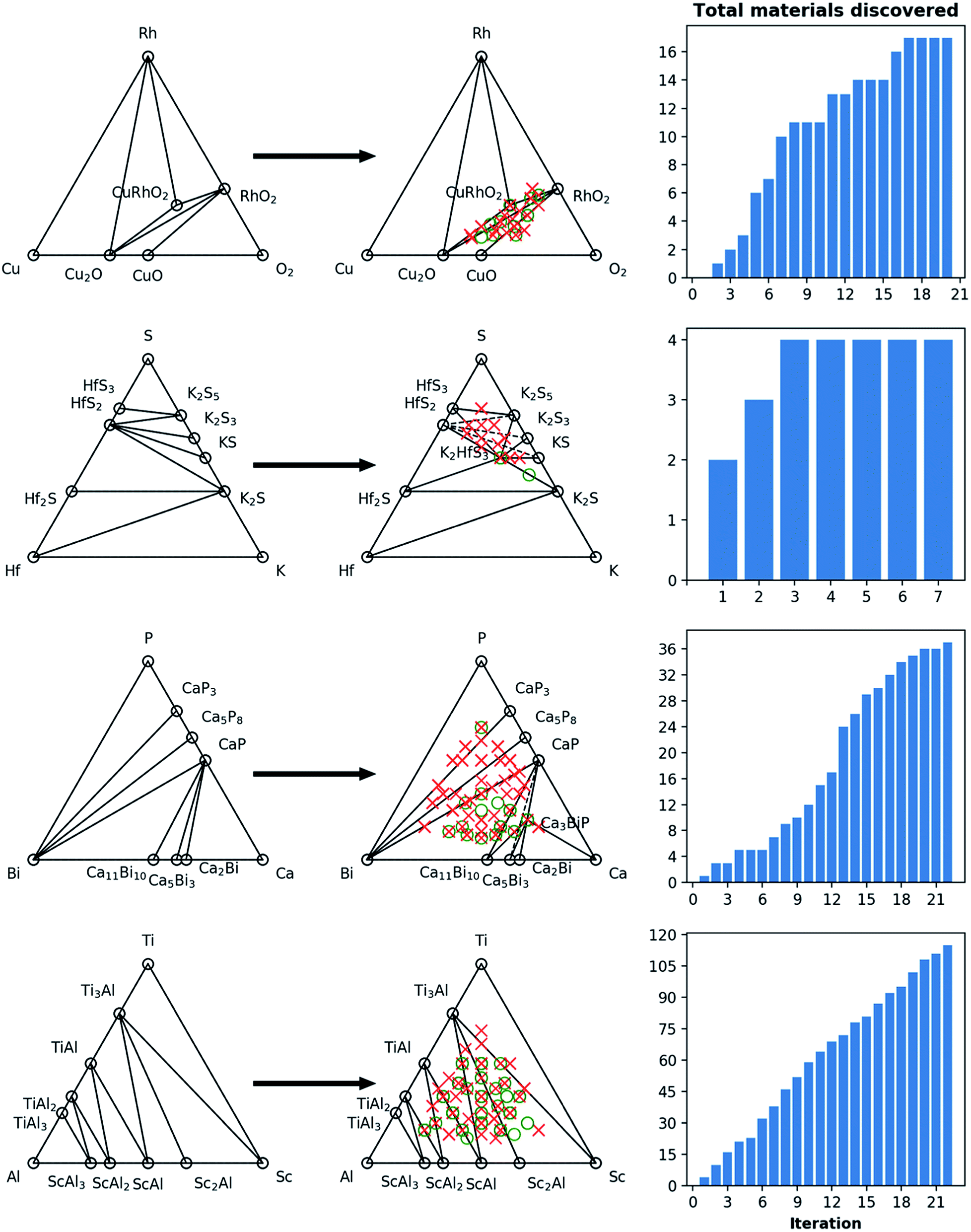 | ||
| Fig. 7 Phase diagram and cumulative discovery histograms for CAMD campaigns in Cu–Rh–O, Hf–K–S, Ca–Bi–P, and Al–Ti–Sc chemical systems. The ternary phase diagrams are shown on the left with both pre-campaign convex hull (left column) and post-campaign convex hull (center column). Material discoveries, i.e. computed structures within 0.2 eV per atom of the convex hull, are shown as green circles, while materials higher than this threshold are shown as red circles. The number of total materials discovered for each chemical system as the campaign iterations progress are shown in the right-hand bar charts. Note that, for Hf–K–S and Ca–Bi–P, discoveries K2HfS3 and Ca3BiP are on the convex-hull, i.e. current ground-states at those compositions. | ||
| System | Agent requests | DFT received | Gross discovery | Duplicate discovery | Experimental | Unique discovery |
|---|---|---|---|---|---|---|
| B–Fe | 152 | 146 | 32 | 28 | 3 | 19 |
| C–Fe | 91 | 81 | 37 | 14 | 2 | 26 |
| Cl–Os | 101 | 70 | 13 | 1 | 1 | 11 |
| Fe–N | 66 | 47 | 17 | 10 | 1 | 10 |
| Fe–S | 158 | 133 | 22 | 23 | 6 | 14 |
| Fe–V | 147 | 130 | 53 | 30 | 1 | 32 |
| Mn–S | 99 | 73 | 29 | 5 | 2 | 22 |
| Ni–S | 220 | 191 | 94 | 30 | 6 | 66 |
| O–V | 66 | 52 | 23 | 5 | 3 | 17 |
| S–V | 174 | 129 | 31 | 22 | 3 | 20 |
| Al–Sc–Ti | 216 | 199 | 115 | 32 | 0 | 90 |
| Bi–Ca–P | 220 | 157 | 37 | 10 | 0 | 27 |
| Ca–Mo–P | 73 | 53 | 6 | 2 | 0 | 4 |
| Cu–O–Rh | 123 | 72 | 17 | 4 | 1 | 14 |
| Hf–K–P | 89 | 63 | 8 | 4 | 0 | 7 |
| Hf–K–S | 38 | 23 | 4 | 0 | 0 | 4 |
For the 16 chemically-diverse campaigns presented in this manuscript, we report 383 autonomously-discovered distinct structures that could not be matched to an experimental entry in the OQMD and are within 0.2 eV per atom of the convex hull. The discovery rate for unique structures within this threshold range from around 10% to 45% across different campaigns (Table 1), averaging close to 22%. Out of these structures, 13 are on the convex-hull, and 36 are within 0.025 eV per atom, 67 are within 0.05 eV per atom and 153 are within 0.1 eV per atom of the convex-hull. Of the structures discovered as unique, only 9 are found in the MP database, suggesting that 374 of the discovered structures have not been previously reported there either. In addition, we note that the structures generated by CAMD have a natural structural diversity that is limited only by the structural diversity of the seed from which the prototype set is generated. From the 383 discovered structures, 7 crystal systems and 87 distinct space groups are represented. Furthermore, it can be observed in Fig. 6 and 7 that the breadth of compositions CAMD samples varies notably across different chemistries. Particularly in spaces where a charge-balance can be enforced (e.g. Cu–Rh–O), agents naturally focus mostly on the respective parts of the phase diagram enclosed by existing charge-balanced compounds, avoiding potentially unphysical compositions (see Methods for details). While this choice comes at the expense of potentially missing a few plausible candidates (e.g. peroxides or superoxides), we find it to be essential to ensure efficient use of computational resources in the current “exploratory” campaigns. In cases when charge-balance is not as plausible and hence is not enforced, the composition space broadens (e.g. Ca–Bi–P and Al–Sc–Ti). Some of the stable or nearly stable structures identified by CAMD are displayed in Fig. 8.
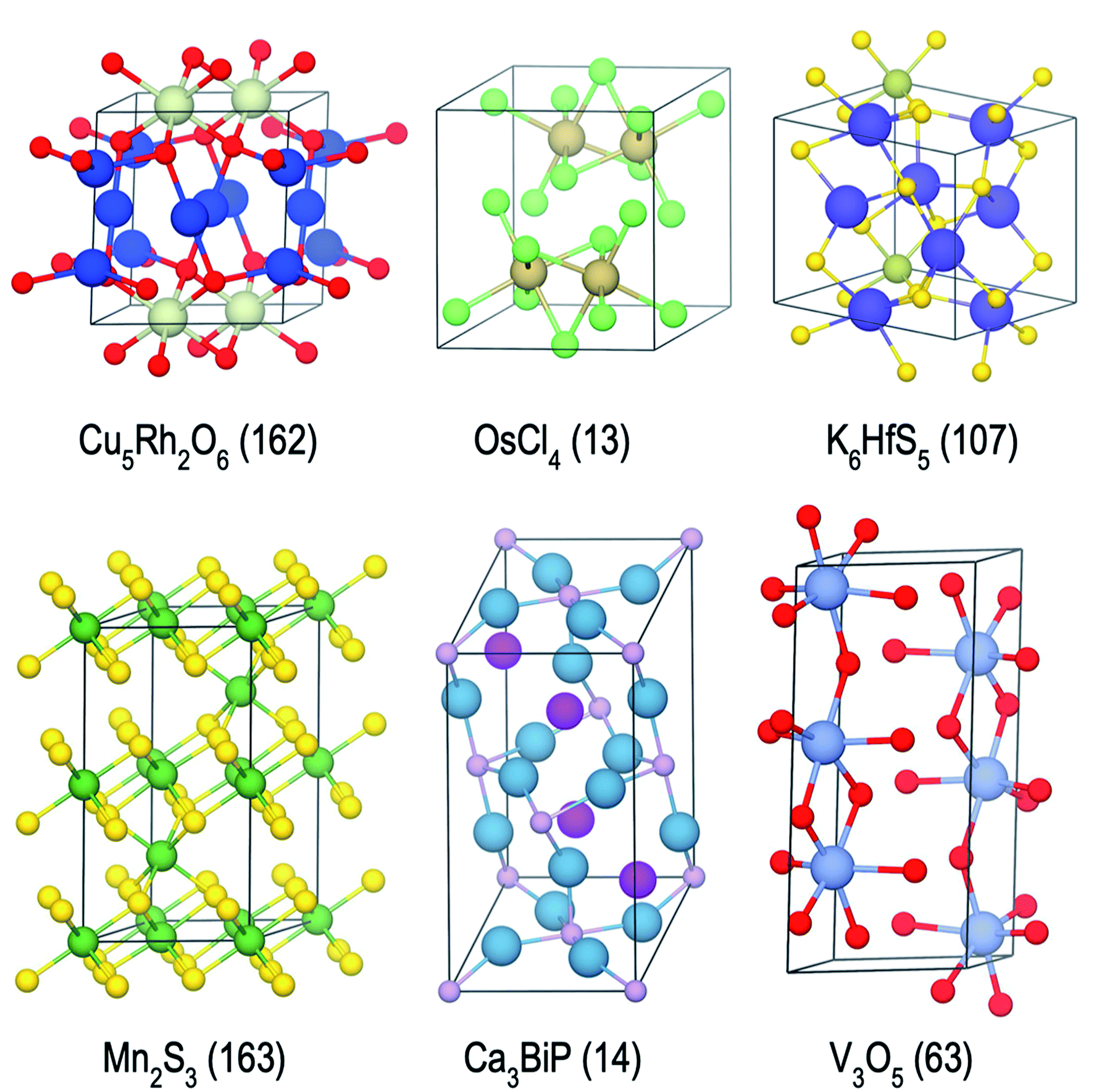 | ||
| Fig. 8 Examples of stable or nearly stable crystal structures found in the present campaigns. Space group numbers are given in parentheses. | ||
We find it encouraging that the CAMD framework identified many new, unique stable (or nearly stable) phases in this diverse set of binary and ternary chemical systems, even in systems that one expects to have been thoroughly-explored, such as Mn–S or Fe–V. Nevertheless, a quick survey for these two examples reveals that for the Mn–S system, Okamoto68 mentions that the S-rich side is still speculative due to insufficient experimental data, and for the Fe–V system, Bloch et al.69 performed an ab initio high-throughput search and found new ordered Fe3V and FeV3 phases, and further provide experimental evidence that kinetic barriers to formation of such previously unobserved phases in this system can be overcome by hydrogen absorption. These examples hint at the need to revisit even such well-known systems for new materials, and also show CAMD can rapidly uncover such gaps in our knowledge of materials without any intervention. While searches in MP13 and OQMD15 show that both databases naturally identified several hypothetical structures in the current chemical systems (and many others) as part of high-throughput prototyping studies run over the course of years, we showed that by streamlining the discovery process, CAMD far exceeds prior work in terms of the number of viable unique material structures and the breadth of new compositions such structures are found at (Table 1). Thus, our current, and future results as they are being produced by the present framework are expected to be valuable additions to such open material databases.12–14
3. Outlook and open questions
The CAMD framework is suitable for sequential campaigns in materials space in general, but we expect it to be more effective in optimization efforts for identifying new inorganic materials as showcased in this study, where the search space is large and representations of entries are high-dimensional (i.e. thousands of candidates and complex vector representations), with no obvious gradients to trace, and where acquisition decisions require blending of multiple scientific paradigms (such as machine-learning, phase-diagram construction, risk-aversion algorithms, etc.) under strict budget constraints. The design of the framework, and in particular the agents, as well as many of the hyperparameters or practical implementation details of the system are geared towards our goal of achieving rapid searching of entire chemical systems at minimal computational cost. While there are no restrictions to investigating individual compositions, as their core advantage, the agents in this work process entire systems and are capable of taking into account the relative stability of materials not only against their polymorphs but against decomposition into other materials in their entire chemical systems.The framework currently assumes a priori creation of a search domain, and hence is not generative in the sense that it would not consider an entirely new crystal structure that does not exist in our list of chemically anonymized prototypes derived from databases of known materials. But constraining the search space this way enables speed, in other words, we can scan entire chemical spaces (such as binaries or ternaries) for new stable materials at relatively small computational costs. With the current settings, a budget of maximum 220 DFT calculations yields many new stable (or nearly stable) materials in entire chemical systems (Table 1), often spending a lot less than the allocated budget. For these reasons, we expect that the present use case of finding new stable materials will be complementary to the existing approaches to crystal structure prediction.70–72 We should also emphasize there is no obstacle against the implementation of a generative approach in the presented framework. Such generative-agents, for example, can hypothesize the same way and suggest new structures while they are active or a generative approach can deliver a large set of candidate structures upfront to form the search domain.
We expect that the future work can extend the framework to achieve improved performance in the present application, and to add new target applications and functionality. For example, more intelligent agents can be developed for tasks including and beyond finding stable materials, such as studies that consider other material properties and specific material applications. For this purpose, both traditional73 and newly advanced a priori74 multi-objective approaches may serve as useful components of the CAMD discovery agents. Those agents can adapt new representations, surrogate models, rules, empirical relations, physical or chemical laws etc. In our initial implementation to find stable inorganic materials, no entropic effects are considered in the determination of whether a material is to be marked as a discovery. We note that these may serve an important role and are an objective for future implementations of our experimental and analysis functionality, particularly in alloy and higher-order systems.75,76 Since thermodynamically accessible energy ranges of making polymorphs vary across material classes,58,59 agents that utilize a chemically-informed hull-distance threshold are likely to deliver more performant, and less-biased campaigns across chemistries. A priori identification of underexplored chemical spaces with higher likelihood of discoveries can improve performance in exploratory searches.43,77 Incorporation of cost estimation of acquisitions, dynamic budgeting, and better error handling (e.g. whether to attempt to heal a failed request) are planned as additions to the framework. Agents can adopt strategies that alter their hyperparameters (e.g. ε and α discussed above) in response to observed performance metrics. Besides, it is foreseeable that, as if forming a team of scientists, a team of agents would be able to attack a scientific problem from different angles, or agents can demonstrate self-improvement skills in campaigns, beyond minimizing the variance of surrogate models and would be more risk averse. Intersection with, and potential benefits from other black-box optimization approaches and reinforcement learning are expected.78
4. Conclusion
In this work, we presented a sequential, agent-based optimization framework for complex scientific objectives, designed explicitly with the constraints and utility of materials discovery in mind. The framework is designed to be modular such that it allows simulations of research agents before their active deployment in real discovery campaigns, enabling their efficient, a priori design by the human researchers. We demonstrated agent simulations in a case study of Fe–X systems and showed how complexity can be built into agents, from baseline one-shot models to uncertainty-driven decision-making. We further demonstrated active DFT-based deployment of agents we designed in 16 sample chemistries. The framework found hundreds of new, structurally-unique materials in these chemistries with no human intervention, validating, and also demonstrating the utility of having an end-to-end framework for discovery of new materials autonomously. The supporting code is open-sourced for community use.5. Methods
5.1 Code, models and data availability
We have developed an open-source python-package, named computational autonomy for materials discovery (CAMD) to carry out the work presented in this publication, available at https://github.com/TRI-AMDD/CAMD. The CAMD software comes with instructions and examples that are complementary to the descriptions presented in this work. Main components of CAMD build on open-source materials science and machine-learning software such as pymatgen,79 qmpy,15 matminer,20 scikit-learn,80 gpflow81 and tensorflow.82 Stability calculations are carried out using a modified (parallelized) implementation of the linear-programming approach in qmpy,15 available in the CAMD library. Structure visuals in Fig. 8 are generated using VESTA.83 Datasets used or generated in this work are accessible at http://data.matr.io/3. The NN models use a 84 × 50 layer configuration and the default arguments of MLPRegressor class in scikit-learn, with a rectified linear unit activation function, L2 regularization parameter of 0.0001, a learning rate of 0.001 and the adam optimizer. The mean absolute error of the models are computed by 3-fold cross validation during each campaign iteration over the seed data to keep track of satisfactory model training and accuracy.5.2 Active campaign settings
In active DFT-based campaigns, agents were allowed to request up to 10 DFT calculations in each iteration. Each campaign is allowed to run for at least 5 iterations regardless of its performance, after which, a campaign would be automatically terminated (i) if the agent could not identify new materials meeting the stability goal within any of the three most recent iterations or (ii) if the campaign reaches 25% consumption of its candidate space or (iii) if the campaign reaches 22 iterations. If an agent cannot suggest at least one structure to acquire with DFT, it terminates the campaign regardless of the campaign step count. Hence, every campaign was allowed effectively a maximum budget of 220 DFT calculations. The stability goal was set as maximum 0.2 eV per atom above the evolving convex-hull. Agents were allowed to choose structures that have up to 20 atoms, and each DFT calculation was allowed a wall-time of 8 hours on 16 CPUs on an AWS EC2 instance. Calculations lingering beyond that point are terminated and agents move to the next stage. While this step is not optimal, and more intelligent stopping criteria for the experiments are planned as future work, the current setting is geared towards the ability to search rapidly and preservation of resources, as relaxations that linger longer often correspond to prototype-derived structures that might be much harder to optimize. Post-campaign unique structure matching tests to determine uniqueness and phase diagram generations are carried out using pymatgen.79 Certain agents are allowed to attempt to diversify their requests to minimize the regret of acquiring too many similar candidates using a computationally cheap risk-aversion algorithm. This algorithm measures similarity as Euclidean-distance in a standardized feature space, and finds a diverse subset of the stability-ordered list of candidates meeting the hull-distance threshold. The algorithm eliminates points listed higher on the list (i.e. further away from the hull) that are too similar to those lower on this list (i.e. closer to the hull), based on a similarity distance threshold that self-adjusts by attempting to iteratively find the smallest such subset larger than the allowed acquisition batch size. The algorithm has no hyperparameters and reduces to the ranked stability list if there are not enough candidates. In a test campaign with a synthetic scenario where we perturbed structure features of materials in the Si–O system to overpopulate the pool with similar candidates, the agent incorporating this algorithm acquired more unique structures.5.3 Generation of search domains for stable material discovery
During formula generation, charge balance was enforced based on the allowed valence state of elements available in pymatgen, if one or more of the elements O, Cl, F, S, N, Br, or I are present in the target chemical system. Formula generation follows a certain set of rules for stoichiometric compound formation. For example, for a system A–B, coefficients x and y in formulas AxBy are allowed to take integer values from the set {1, 2, …, gmax}. For systems where no charge balance was enforced, the maximum integer, gmax is set as 4 (inclusive) for both binaries and ternaries. For charge balanced systems, larger values of gmax are allowed, and gmax is determined explicitly by incrementing the default maximum above until at least the 20 candidates are generated, upto a hard limit of 7 (inclusive). It should be noted all these parameters can be adjusted by the user, and these are merely the settings we picked for the purposes of the present study.Structure candidates at each composition are generated with a crystallographic prototype enumeration scheme, as implemented in protosearch84,85 using the space group and Wyckoff positions to identify symmetrically unique structures. We have limited the search to experimentally observed crystal structure prototypes, by parsing the ICSD67 entries present in the OQMD database,14,15 which has a large structural diversity. This consists of approximately 32000 structures which can be classified into 8050 unique crystal prototypes based on our scheme, including 131 unary (N = 1), 1070 binary (N = 2), 3196 ternary (N = 3), 1970 quaternary (N = 4), 1013 quinary (N = 5), 542 sexinary (N = 6), 104 septenary (N = 7), and a few higher-order structures (where N is the number of elemental components). For each selected prototype, a structure (or several structures depending on the symmetry of available sites) with the desired composition is constructed by elemental substitution. Subsequently, lattice constants are approximated by rescaling the unit cell, while avoiding atomic overlap, assuming a hard-sphere radius for atoms as 90% of the elements' covalent radii. Anisotropic scaling is applied to relevant crystal systems, while internal coordinates and angles are kept fixed.
Featurization of all crystal structures (including the OQMD entries used in agent simulations and as seed data, and structures generated for active DFT campaigns as described above) throughout this work was performed using the composition and structure derived (Voronoi-based) material descriptors introduced by Ward et al.66 producing a vector of 273 features for each material as implemented in Matminer.20
5.4 Density functional theory calculations
The DFT workflow consists of a structure optimization followed by a static calculation of the final, optimized structure using the Vienna ab initio simulation package (VASP).86,87 The DFT parameters are generated using OQMD's qmpy interface, which renders OQMD-compatible computational data from the seed data derived from the OQMD.15 Note that the actual DFT calculation is done within a docker container on AWS batch, and the Experiment API within CAMD submits, monitors, and fetches the resultant data from the batch job in order to provide formation energy–structure pairs back to the seed dataset.Author contributions
JM and MA wrote the python package for agent based computational discovery. KW, RF and TB developed the structure generation methods. JM, JH and MA conceived the project, with input from TB. MA and JM wrote the manuscript, with input from all authors.Conflicts of interest
M. A. has a related U.S. patent application. The remaining authors declare no conflict of interest.References
- G. E. P. Box, J. Am. Stat. Assoc., 1976, 71, 791–799 CrossRef.
- P. Langley, H. A. Simon, G. L. Bradshaw, J. M. Zytkow and University of Chicago Staff, Scientific Discovery: Computational Explorations of the Creative Processes, MIT Press, 1987 Search PubMed.
- D. Kulkarni and H. A. Simon, Cognit. Sci., 1988, 12, 139–175 CrossRef.
- J. M. Zytkow and H. A. Simon, Synthese, 1988, 74, 65–90 CrossRef.
- J. M. Żytkow, Mach. Learn., 1993, 12, 7–16 Search PubMed.
- R. K. Lindsay, B. G. Buchanan, E. A. Feigenbaum and J. Lederberg, Artif. Intell., 1993, 61, 209–261 CrossRef.
- R. E. Valdés-Pérez, Artif. Intell., 1995, 74, 191–201 CrossRef.
- J. M. Żytkow, in Advances in Artificial Intelligence, Springer Berlin Heidelberg, 2000, pp. 443–448 Search PubMed.
- D. P. Tabor, L. M. Roch, S. K. Saikin, C. Kreisbeck, D. Sheberla, J. H. Montoya, S. Dwaraknath, M. Aykol, C. Ortiz, H. Tribukait, C. Amador-Bedolla, C. J. Brabec, B. Maruyama, K. A. Persson and A. Aspuru-Guzik, Nat. Rev. Mater., 2018, 3, 5–20 CrossRef CAS.
- L. Ward, M. Aykol, B. Blaiszik, I. Foster, B. Meredig, J. Saal and S. Suram, MRS Bull., 2018, 43, 683–689 CrossRef.
- M. Aykol, J. S. Hummelshøj, A. Anapolsky, K. Aoyagi, M. Z. Bazant, T. Bligaard, R. D. Braatz, S. Broderick, D. Cogswell, J. Dagdelen, W. Drisdell, E. Garcia, K. Garikipati, V. Gavini, W. E. Gent, L. Giordano, C. P. Gomes, R. Gomez-Bombarelli, C. Balaji Gopal, J. M. Gregoire, J. C. Grossman, P. Herring, L. Hung, T. F. Jaramillo, L. King, H.-K. Kwon, R. Maekawa, A. M. Minor, J. H. Montoya, T. Mueller, C. Ophus, K. Rajan, R. Ramprasad, B. Rohr, D. Schweigert, Y. Shao-Horn, Y. Suga, S. K. Suram, V. Viswanathan, J. F. Whitacre, A. P. Willard, O. Wodo, C. Wolverton and B. D. Storey, Matter, 2019, 1, 1433–1438 CrossRef.
- S. Curtarolo, W. Setyawan, G. L. W. Hart, M. Jahnatek, R. V. Chepulskii, R. H. Taylor, S. Wang, J. Xue, K. Yang, O. Levy, M. J. Mehl, H. T. Stokes, D. O. Demchenko and D. Morgan, Comput. Mater. Sci., 2012, 58, 218–226 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, J. Met., 2013, 65, 1501–1509 CAS.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 15010 CrossRef CAS.
- J. R. Hattrick-Simpers, J. M. Gregoire and A. G. Kusne, APL Mater., 2016, 4, 053211 CrossRef.
- K. Choudhary, I. Kalish, R. Beams and F. Tavazza, Sci. Rep., 2017, 7, 5179 CrossRef.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, J. Chem. Phys., 2018, 148, 241722 CrossRef.
- Q. Zhou, P. Tang, S. Liu, J. Pan, Q. Yan and S.-C. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6411–E6417 CrossRef CAS.
- L. Ward, A. Dunn, A. Faghaninia, N. E. R. Zimmermann, S. Bajaj, Q. Wang, J. Montoya, J. Chen, K. Bystrom, M. Dylla, K. Chard, M. Asta, K. A. Persson, G. J. Snyder, I. Foster and A. Jain, Comput. Mater. Sci., 2018, 152, 60–69 CrossRef.
- D. Davies, K. T. Butler and A. Walsh, Chem. Mater., 2019, 31, 7221–7230 CrossRef CAS.
- C. A. Sutton, M. Boley, L. M. Ghiringhelli, M. Rupp, J. Vreeken and M. Scheffler, Identifying Domains of Applicability of Machine Learning Models for Materials Science, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9778670.
- S. Sun, N. T. P. Hartono, Z. D. Ren, F. Oviedo, A. M. Buscemi, M. Layurova, D. X. Chen, T. Ogunfunmi, J. Thapa, S. Ramasamy, C. Settens, B. L. DeCost, A. G. Kusne, Z. Liu, S. I. P. Tian, I. M. Peters, J.-P. Correa-Baena and T. Buonassisi, Joule, 2019, 3, 1437–1451 CrossRef CAS.
- F. Ren, L. Ward, T. Williams, K. J. Laws, C. Wolverton, J. Hattrick-Simpers and A. Mehta, Sci. Adv., 2018, 4, eaaq1566 CrossRef PubMed.
- T. Lookman, P. V. Balachandran, D. Xue and R. Yuan, npj Comput. Mater., 2019, 5, 21 CrossRef.
- L. Bassman, P. Rajak, R. K. Kalia, A. Nakano, F. Sha, J. Sun, D. J. Singh, M. Aykol, P. Huck, K. Persson and P. Vashishta, npj Comput. Mater., 2018, 4, 74 CrossRef.
- P. Nikolaev, D. Hooper, F. Webber, R. Rao, K. Decker, M. Krein, J. Poleski, R. Barto and B. Maruyama, npj Comput. Mater., 2016, 2, 16031 CrossRef.
- A. Mannodi-Kanakkithodi, G. Pilania, T. D. Huan, T. Lookman and R. Ramprasad, Sci. Rep., 2016, 6, 20952 CrossRef PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- J. Ling, M. Hutchinson, E. Antono, S. Paradiso and B. Meredig, Integrating Materials and Manufacturing Innovation, 2017, 6, 207–217 CrossRef.
- F. T. Bolle, N. Rask Mathiesen, A. Juul Nielsen, T. Vegge, J. M. Garcia Lastra and I. E. Castelli, Batteries Supercaps, 2020, 3, 488–498 CrossRef.
- B. Rohr, H. S. Stein, D. Guevarra, Y. Wang, J. A. Haber, M. Aykol, S. K. Suram and J. M. Gregoire, Chem. Sci., 2020, 11, 2696–2706 RSC.
- V. Dragone, V. Sans, A. B. Henson, J. M. Granda and L. Cronin, Nat. Commun., 2017, 8, 15733 CrossRef PubMed.
- P. J. Kitson, G. Marie, J.-P. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS PubMed.
- D. Xue, P. V. Balachandran, J. Hogden, J. Theiler, D. Xue and T. Lookman, Nat. Commun., 2016, 7, 11241 CrossRef CAS PubMed.
- M. Krenn, M. Malik, R. Fickler, R. Lapkiewicz and A. Zeilinger, Phys. Rev. Lett., 2016, 116, 090405 CrossRef PubMed.
- P. B. Wigley, P. J. Everitt, A. van den Hengel, J. W. Bastian, M. A. Sooriyabandara, G. D. McDonald, K. S. Hardman, C. D. Quinlivan, P. Manju, C. C. N. Kuhn, I. R. Petersen, A. N. Luiten, J. J. Hope, N. P. Robins and M. R. Hush, Sci. Rep., 2016, 6, 25890 CrossRef CAS PubMed.
- S. Langner, F. Häse, J. D. Perea, T. Stubhan, J. Hauch, L. M. Roch, T. Heumueller, A. Aspuru-Guzik and C. J. Brabec, Adv. Mater., 2020, 32, 1907801 CrossRef CAS PubMed.
- L. M. Roch, F. Häse, C. Kreisbeck, T. Tamayo-Mendoza, L. P. E. Yunker, J. E. Hein and A. Aspuru-Guzik, Science Robotics, 2018, 3, eaat5559 CrossRef.
- L. M. Roch, F. Häse, C. Kreisbeck, T. Tamayo-Mendoza, L. P. E. Yunker, J. E. Hein and A. Aspuru-Guzik, PLoS One, 2020, 15, e0229862 CrossRef CAS PubMed.
- I. M. Pendleton, G. Cattabriga, Z. Li, M. A. Najeeb, S. A. Friedler, A. J. Norquist, E. M. Chan and J. Schrier, MRS Commun., 2019, 9, 846–859 CrossRef CAS.
- C. C. Fischer, K. J. Tibbetts, D. Morgan and G. Ceder, Nat. Mater., 2006, 5, 641–646 CrossRef CAS PubMed.
- G. Hautier, C. C. Fischer, A. Jain, T. Mueller and G. Ceder, Chem. Mater., 2010, 22, 3762–3767 CrossRef CAS.
- R. Gautier, X. Zhang, L. Hu, L. Yu, Y. Lin, T. O. L. Sunde, D. Chon, K. R. Poeppelmeier and A. Zunger, Nat. Chem., 2015, 7, 308–316 CrossRef CAS PubMed.
- A. Seko, H. Hayashi, H. Kashima and I. Tanaka, Phys. Rev. Mater., 2018, 2, 013805 CrossRef CAS.
- T. Ueno, T. D. Rhone, Z. Hou, T. Mizoguchi and K. Tsuda, Mater. Discovery, 2016, 4, 18–21 CrossRef.
- Y. Okamoto, J. Phys. Chem. A, 2017, 121, 3299–3304 CrossRef CAS PubMed.
- A. Dunn, J. Brenneck and A. Jain, Journal of Physics: Materials, 2019, 2, 034002 Search PubMed.
- R. A. Fisher, The design of experiments, Hafner Publishing Company, New York, NY, 1971 Search PubMed.
- F. Pukelsheim, Optimal design of experiments, SIAM, 2006 Search PubMed.
- R. Leardi, Anal. Chim. Acta, 2009, 652, 161–172 CrossRef CAS PubMed.
- T. Desautels, A. Krause and J. W. Burdick, J. Mach. Learn. Res., 2014, 15, 3873–3923 Search PubMed.
- J. Azimi, A. Fern and X. Z. Fern, in Advances in Neural Information Processing Systems 23, ed. J. D. Lafferty, C. K. I. Williams, J. Shawe-Taylor, R. S. Zemel and A. Culotta, Curran Associates, Inc., 2010, pp. 109–117 Search PubMed.
- S. Russel, P. Norvig, et al., Artificial intelligence: a modern approach, Pearson Education Limited, 2013 Search PubMed.
- J. Noh, J. Kim, H. S. Stein, B. Sanchez-Lengeling, J. M. Gregoire, A. Aspuru-Guzik and Y. Jung, Matter, 2019, 1, 1370–1384 CrossRef.
- J. Hoffmann, L. Maestrati, Y. Sawada, J. Tang, J. M. Sellier and Y. Bengio, 2019, arXiv:1909.00949v1.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, 2020, arXiv:2003.12127v1.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- M. Aykol, S. S. Dwaraknath, W. Sun and K. A. Persson, Sci. Adv., 2018, 4, eaaq0148 CrossRef PubMed.
- M. Aykol, V. I. Hegde, L. Hung, S. Suram, P. Herring, C. Wolverton and J. S. Hummelshøj, Nat. Commun., 2019, 10, 2018 CrossRef PubMed.
- E. Brochu, V. M. Cora and N. de Freitas, 2010, arXiv:1012.2599v1.
- K. K. Vu, C. D'Ambrosio, Y. Hamadi and L. Liberti, Int. Trans. Oper. Res., 2017, 24, 393–424 CrossRef.
- H. S. Seung, M. Opper and H. Sompolinsky, in Proceedings of the Fifth Annual Workshop on Computational Learning Theory, ACM, New York, NY, USA, 1992, pp. 287–294 Search PubMed.
- R. Burbidge, J. J. Rowland and R. D. King, in Intelligent Data Engineering and Automated Learning - IDEAL 2007, Springer Berlin Heidelberg, 2007, pp. 209–218 Search PubMed.
- J. Hensman, N. Fusi and N. D. Lawrence, 2013, arXiv:1309.6835.
- L. Ward, R. Liu, A. Krishna, V. I. Hegde, A. Agrawal, A. Choudhary and C. Wolverton, Phys. Rev. B, 2017, 96, 024104 CrossRef.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef PubMed.
- H. Okamoto, J. Phase Equilib. Diffus., 2011, 32, 78 CrossRef CAS.
- J. Bloch, O. Levy, B. Pejova, J. Jacob, S. Curtarolo and B. Hjörvarsson, Phys. Rev. Lett., 2012, 108, 215503 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- K. Sindhya, A. Sinha, K. Deb and K. Miettinen, in 2009 IEEE Congress on Evolutionary Computation, 2009, pp. 2919–2926 Search PubMed.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Chem. Sci., 2018, 9, 7642–7655 RSC.
- V. I. Hegde, M. Aykol, S. Kirklin and C. Wolverton, Sci. Adv., 2020, 6, eaay5606 CrossRef.
- C. Toher, C. Oses, D. Hicks and S. Curtarolo, npj Comput. Mater., 2019, 5, 69 CrossRef.
- S. Lotfi, Z. Zhang, G. Viswanathan, K. Fortenberry, A. M. Tehrani and J. Brgoch, Matter, 3, 261–272 CrossRef.
- K. M. Choromanski, A. Pacchiano, J. Parker-Holder, Y. Tang and V. Sindhwani, in Advances in Neural Information Processing Systems 32, ed. H. Wallach, H. Larochelle, A. Beygelzimer, F. d'Alché-Buc, E. Fox and R. Garnett, Curran Associates, Inc., 2019, pp. 10299–10309 Search PubMed.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- A. G. D. G. Matthews, M. Van Der Wilk, T. Nickson, K. Fujii, A. Boukouvalas, P. León-Villagrá, Z. Ghahramani and J. Hensman, J. Mach. Learn. Res., 2017, 18, 1299–1304 Search PubMed.
- M. Abadi, P. Barham, J. Chen, Z. Chen, A. Davis, J. Dean, M. Devin, S. Ghemawat, G. Irving, M. Isard, et al., in 12th USENIX Symposium on Operating Systems Design and Implementation, 2016, pp. 265–283 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Protosearch, https://github.com/SUNCAT-Center/protosearch Search PubMed.
- A. Jain and T. Bligaard, Phys. Rev. B, 2018, 98, 214112 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |