
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVitrimers: directing chemical reactivity to control material properties
Marc
Guerre†
 *a,
Christian
Taplan†
b,
Johan M.
Winne
*c and
Filip E.
Du Prez
*b
*a,
Christian
Taplan†
b,
Johan M.
Winne
*c and
Filip E.
Du Prez
*b
aLaboratoire des IMRCP, Université de Toulouse, CNRS UMR5623, Université Paul Sabatier, 118 route de Narbonne, 31062 Toulouse Cedex 9, France. E-mail: guerre@chimie.ups-tlse.fr
bDepartment of Organic and Macromolecular Chemistry, Polymer Chemistry Research Group, Center of Macromolecular Chemistry (CMaC), Faculty of Sciences, Ghent University, Krijgslaan 281 (S4-bis), 9000 Ghent, Belgium. E-mail: filip.duprez@ugent.be
cDepartment of Organic and Macromolecular Chemistry, Laboratory of Organic Synthesis, Faculty of Sciences, Ghent University, Krijgslaan 281 (S4-bis), 9000 Ghent, Belgium. E-mail: johan.winne@ugent.be
First published on 16th April 2020
Abstract
The development of more sustainable materials with a prolonged useful lifetime is a key requirement for a transition towards a more circular economy. However, polymer materials that are long-lasting and highly durable also tend to have a limited application potential for re-use. This is because such materials derive their durable properties from a high degree of chemical connectivity, resulting in rigid meshes or networks of polymer chains with a high intrinsic resistance to deformation. Once such polymers are fully synthesised, thermal (re)processing becomes hard (or impossible) to achieve without damaging the degree of chemical connectivity, and most recycling options quickly lead to a drop or even loss of material properties. In this context, both academic and industrial researchers have taken a keen interest in materials design that combines high degrees of chemical connectivity with an improved thermal (re)processability, mediated through a dynamic exchange reaction of covalent bonds. In particular vitrimer materials offer a promising concept because they completely maintain their degree of chemical connectivity at all times, yet can show a clear thermally driven plasticity and liquid behavior, enabled through rapid bond rearrangement reactions within the network. In the past decade, many suitable dynamic covalent chemistries were developed to create vitrimer materials, and are now applicable to a wide range of polymer matrices. The material properties of vitrimers, however, do not solely rely on the chemical structure of the polymer matrix, but also on the chemical reactivity of the dynamic bonds. Thus, chemical reactivity considerations become an integral part of material design, which has to take into account for example catalytic and cross-reactivity effects. This mini-review will aim to provide an overview of recent efforts aimed at understanding and controlling dynamic cross-linking reactions within vitrimers, and how directing this chemical reactivity can be used as a handle to steer material properties. Hence, it is shown how a focus on a fundamental chemical understanding can pave the way towards new sustainable materials and applications.
 Marc Guerre | Marc Guerre completed his Ph.D degree in polymer chemistry in 2017 from the Chemistry School of Montpellier (ENSCM, France) where he studied and developed RAFT polymerization techniques applied to fluoropolymers. Then, he joined Prof. Filip Du Prez's laboratory at Ghent University to work on the development of fluorinated vitrimers. Since October 2019, he is an independent CNRS researcher at the IMRCP laboratory (Toulouse, France) in the team Precision Polymers by Radical Process (P3R). His research interest includes the synthesis of well-defined polymeric systems directed towards self-assembled structures for the design of sophisticated dynamic covalent materials. |
 Christian Taplan | Christian Taplan obtained his master's degree in chemistry in 2017 from Dresden University of Technology (TU Dresden, Germany). In October 2017, he joined the PCR group at Ghent University to work on the next generation of polymeric materials such as fluorinated and tailor-made vitrimers, which is supported by a PhD Fellowship from the Research Foundation Flanders (FWO) to further extend his interest in advanced, smart and sustainable polymer materials. |
 Johan M. Winne | Johan Winne was appointed as an assistant-professor at Ghent University in 2015. There, he leads a research group of organic synthetic chemists that share his fascination for the chemistry of natural products, heterocycles and cycloadditions. Apart from tackling fundamental problems in organic reactivity and explorative reaction design for the synthesis of complex molecules, he has picked up a keen interest in applications of organic reactivity within polymer chemistry, through joint projects with Filip Du Prez and his research group in the design and exploration of novel materials based on dynamic covalent chemistries. |
 Filip E. Du Prez | Filip E. Du Prez is since 1999 heading the Polymer Chemistry Research Group (http://www.PCR.UGent.be) at Ghent University in which about 25 researchers are dealing with the design of dynamic/renewable polymer materials, the development of new polymer structures such as sequence-defined polymers and the exploration of powerful functionalization methods in polymer chemistry. In 2008, he had a Visiting Professor position at the CAMD Research Center (UNSW, Sydney). He is author of about 300 peer-reviewed publications, 14 patents, 10 book chapters and (co-)chairman of more than 10 (inter)national conferences on polymer chemistry related topics. Since 2018, he is associate editor of Polymer Chemistry. |
Introduction
We now live in the ‘plastic age’, at a time where societal efforts and focus are strongly driven by an increase in environmental awareness. If the aim is to preserve our world for future generations, society and industry need to start the transition from a linear towards a circular economy.1,2 Chemistry will have a crucial part in providing tools and materials enabling this disruptive transition. Plastics or polymers, the materials that define our epoch, will be allies and not enemies in this transition. Their low cost, light weight, high performance, easy production, and overall durability and adaptability render them ideal materials for modern applications with a low ecological footprint.While the majority of plastics is in principle recyclable as a raw material, yet plastic recycling is limited by the intrinsic incompatibility of many polymers and the presence of fillers and impurities on end-product properties, thus requiring a high degree of sophisticated sorting, separating and cleaning. In addition to incompatibility issues, an important caveat in the success story of plastics is the fact that many plastics are in practice not recycled. Next to technical challenges that need to be overcome, there are also clear fundamental scientific challenges.
Polymer materials derive their bulk properties from their covalent bonds, mostly based on carbon atoms within macromolecules. In contrast to low molecular weight organic substances that are only held together by weak intermolecular forces and easily lose structural integrity, polymer materials possess viscoelastic properties arising from their chain entanglements, providing strength and durability to objects. The benefit of this class of materials is that, in contrast to others e.g. glass or metal, moderate heating can often be enough to make polymer chains flow and rearrange their position, allowing for typical plastic processing and, in principle, easy recycling (thermoplastic behaviour). The clear disadvantage lies in the decrease of the polymer chain-length upon recycling, especially regarding mechanical performance, creep and chemical resistance. This intrinsic behaviour is one of the reasons why recycled plastics are often only usable as ‘downgraded’ materials for low end applications. One attractive way to increase robustness and resistance against mechanical stress or chemicals, is to drive up the degree of connectivity to a situation where all chains are integrated into a covalent network (thermoset). Hence, thermosets should be ideally recyclable plastics, with long useful lifetimes, yet they are in fact unrecyclable today, and even a major concern for plastic recycling and treatment in general. This is because their permanent covalent network also makes them rather rigid, and covalent cross-links prevent the relative mobility of chains. Once synthesized, a thermoset will keep its initial shape and becomes intractable as a raw material.
In recent years, attention in the polymer science community has been increasingly turned towards the unrecyclable thermoset class of polymeric materials, not only as a problem, but also as an opportunity. The clear scientific challenge now consists in developing robust polymer networks, with a high degree of cross-linking, providing enduring material properties, which can also overcome the inherent non-recyclability due to the total lack of relative chain mobility. The most straightforward chemical strategy to transform a cross-linked polymer network into a more recyclable material is by introducing dynamic covalent bonds into it. If enough of such chemically reactive bonds are introduced, one arrives at the concept of a covalent adaptable network (CAN), introduced into the polymer field by Bowman and others.3–10 Such shape-shifting networks can rearrange their connectivity in response to an external stimulus, with individual dynamic and reversible chemical bond exchange reactions mediating a molecular network rearrangement process.
As a result of the above-mentioned developments, the classical distinctions between thermoplastic and thermoset polymer materials became blurred. Inspired by the ideal situation of ‘constant connectivity’ as it is found in inorganic glasses (ionic networks), Leibler and co-workers took the ideas found in classical reversible polymer networks (such as supramolecular systems),11–14 and in covalent adaptable networks, one step further.15 They designed polymer networks with a permanent degree of connectivity, yet capable of undergoing a macromolecular network rearrangement.15 To achieve this, it was necessary to introduce a specific type of dynamic covalent bonds, which would undergo an exchange, without going through an intermediate state of disconnection. Mechanistically put, the bond exchange should happen by first forming a new (additional) network bond, and only then releasing the original network bond (associative bond exchange). By implementing this simple, yet elegant idea, they created polymer networks that seemed to defy the classical distinctions within polymer materials. Whereas reversible (or supramolecular) polymer networks can be thought of as ‘switchable’ polymers that behave as networks under certain conditions, but can also be triggered to behave as thermoplastics (via de-crosslinking), Leibler's networks do not show thermoplastic behaviour, and do not lose chemical connectivity. Yet, they are intrinsically malleable, just like inorganic silica. However, macroscopic flow is not mediated through the rearrangement of an ionic network, but through rearrangement of a molecular network. This gives a thermal malleability, but quite unlike regular thermoplastics, there is no sudden loss of overall integrity. In order to make the unique features found in this new class of polymers clear, distinct from the original two polymer classes, Leibler and co-workers coined the name ‘vitrimers’. The first covalent adaptable ‘permanent’ networks that can undergo a reversible exchange (not reversible debonding) were introduced by Cook and co-workers.16 These could undergo stress relaxation via a triggered free-radical mediated thiol exchange reaction. While the concept of vitrimers, and the accompanying first thermally triggered associative polymer networks, were introduced to the field by Leibler and co-workers in 2011.15 In 2016 we summarized this emerging area from a chemical perspective in a first mini-review.17 Since their conceptual introduction, the main research goals in the chemical development of vitrimers was to explore and develop new chemical platforms for so-called associative exchange chemistries into various backbones. A selection of chemistries is provided in our original mini-review and other related overviews and perspectives that recently appeared.18–21 Very recently, Winne, Leibler and Du Prez also published a mechanistic perspective on the chemistry underlying vitrimers and dynamic covalent networks in general, with the attempt to clarify the sense and nonsense of distinguishing different types of bond exchange mechanisms, and their relation to mechanical properties.22
Having established a broad toolbox of building blocks and chemistries that can be used to design vitrimers, researchers are now facing the challenge of gaining control over the reactivity profile, in order to rationally design materials with desired properties in terms of processability and recyclability. The upcoming challenges are thus to gain full control over the dynamic exchanges in vitrimers, as they are directly related to their material properties. Complementary to previous (mini-) reviews and perspectives on vitrimers, this mini-review will target specifically at providing comprehensive, yet condensed information of what efforts have been taken to address and ultimately deliberately manipulate specific exchange mechanisms, in order to adjust the viscoelastic material behaviour and simultaneously the resulting material properties. It is therefore subdivided in four different sections detailing the contribution of several chemical parameters towards the control of the viscoelastic behaviour of vitrimers (catalytic activity and chemical mechanism, supramolecular interactions, matrix effect and reinforced vitrimers), and closing with a discussion of the topology freezing temperature (Tv) of vitrimers. This will show the potential, the challenges and also the pitfalls that need to be considered in this exciting and highly interdisciplinary field of research.
Vitrimer properties: chemistry all the way down
In the area of reversible polymer networks and CANs, chemistry and in particular chemical reactivity aspects are very important. The rate at which bonds are broken and formed at a given temperature are important factors, while assessing the equilibrium between bonded and de-bonded states is also crucial in material design. However, the architecture and dynamic properties of the underlying de-cross-linked states also ‘build on’ to the rheological profile. Specifically, for vitrimers, the situation is more clear-cut as virtually all (segmental) polymer chain dynamics are conditional upon the rate of a chemical reaction. From a fundamental point of view, vitrimers are thus an excellent case study to investigate the link between chemical reactivity and the dynamic properties of polymer materials.External catalysis
One straightforward approach to introduce the desired dynamic exchanges is to use catalytically active moieties or compounds that can manipulate the exchange rate or mechanism. If those compounds are not covalently bound in the matrix, they are herein referred to as external catalysts. The purpose of this additive is to provide a more viable exchange pathway, or to lower the barrier for the rate-determining reaction step in the uncatalysed pathway, and thus to alter the kinetics of an exchange reaction. This catalytic control provides a strong handle to adjust the vitrimer's viscous-flow behaviour on demand, and was indeed implemented in the very first vitrimers.15 With regard to the desired performance of vitrimers, picking the right catalyst often involves making a compromise between promoting a swift exchange, while keeping a high enough energy barrier to achieve a strong temperature dependency of the exchange, providing high exchange rates at processing temperature but dramatically decreasing or inhibiting them in the application temperature range.17 To achieve this degree of control, several classical catalysts, more commonly used to promote organic solution chemistry, have all been embedded within vitrimer materials. Thus, various strong Brønsted/Lewis acids and bases have found their way as useful additives into vitrimers (Fig. 1). In their seminal studies, Leibler and co-workers demonstrated elegantly that polyester-based vitrimers were drastically influenced by different catalyst types. For example, such as amidine bases (1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)), P(Ph)3 and Zn(OAc)2 were respectively compared in the same polymer matrices, and shown to result in different activation energies (Fig. 1a2).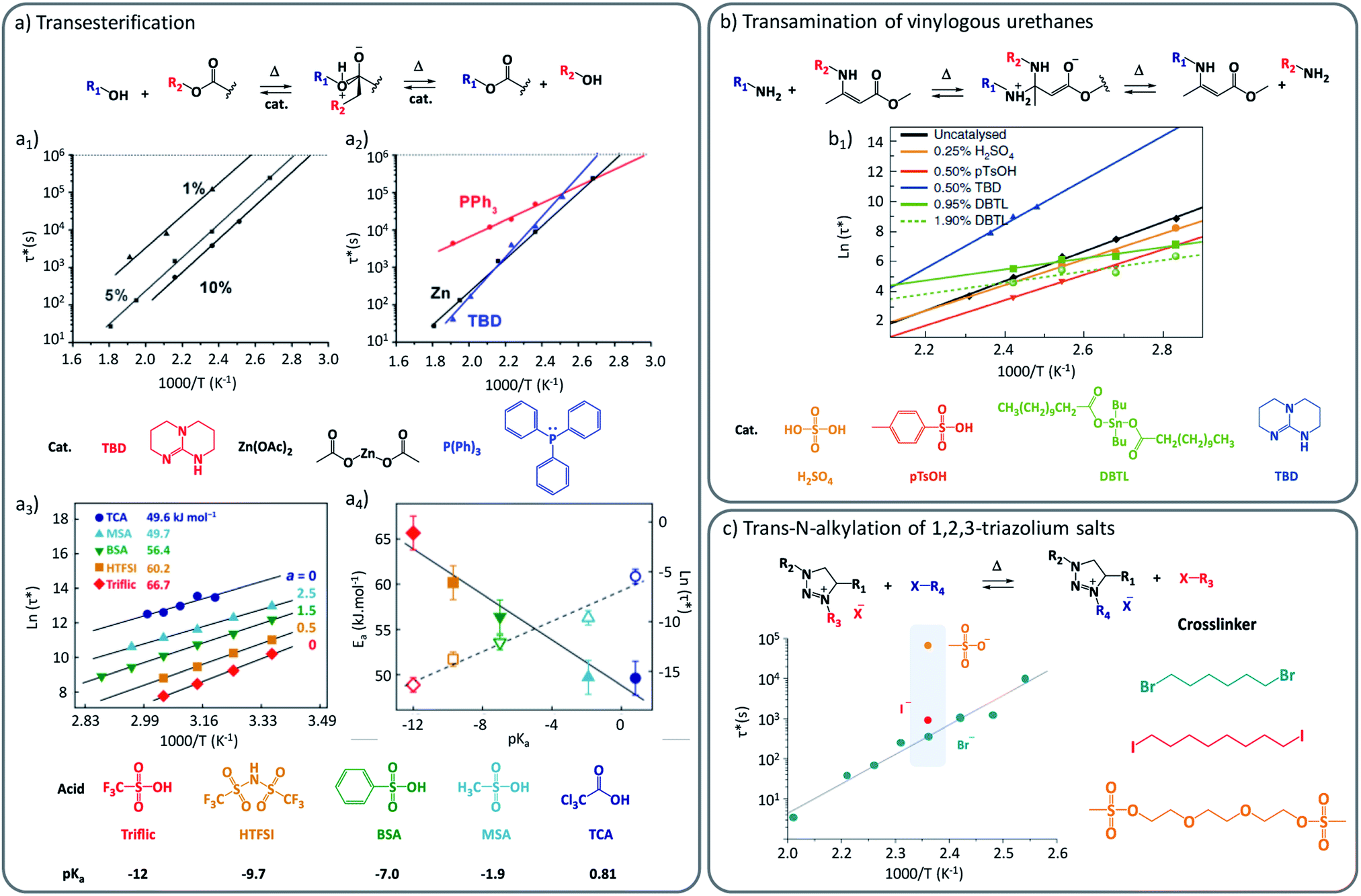 | ||
| Fig. 1 Influence of external catalysis on viscoelastic properties of vitrimers. (a) Catalysed transesterification between alcohol and ester. (a1) Influence of catalyst concentration (1%, 5% and 10% of Zn2+) over Arrhenius plot, showing an accelerating effect with the increasing concentration. (a2) Modification of the Arrhenius plot and resulting activation energy with the use of different external catalysts (TBD, Zn(OAc)2 and P(Ph)3). Adapted with permission from ref. 23. Copyright 2012 American Chemical Society. (a3) Characteristic relaxation time as a function of the inverse temperature for several Brønsted acids having different pKa values. (a4) Linear relation between activation energy (Ea) and respective Brønsted acidity. Adapted with permission from ref. 24. Copyright 2018 American Chemical Society. (b) Exchange mechanism of the transamination of vinylogous urethanes. (b1) Arrhenius plot of several vinylogous urethane vitrimers loaded with different external catalysts (H2SO4, pTsOH, DBTL, TBD). Adapted from ref. 25 with permission from Springer Nature. (c) Exchange mechanism of the trans-N-alkylation of 1,2,3-triazolium salts. (c1) Characteristic relaxation time of poly(ionic liquids) made of different cross-linkers and alkylating agents (Br, I and mesyl-based). Reconstructed with permission from ref. 26. Copyright 2015 American Chemical Society. Notice to reader: further permissions related to the material excerpted of DOI: 10.1021/jacs.5b02653 should be directed to the ACS. | ||
This difference in viscoelastic behaviour can thus be readily rationalized by the expected different catalytic efficiency of P(Ph)3 compared to its stronger Lewis base counterpart. In addition to this comparative study, the authors showed that an increase of the concentration of catalytically active Zn2+ led to a linear increase of the exchange rate, while maintaining a similar activation energy (Fig. 1a1). Similar observations of a concentration dependency, provided by Tournilhac and co-workers, demonstrated in small-molecular studies that even small amounts of merely 0.05 mol% were enough to significantly enhance the transesterification rate.27 Further, they proposed that more complex catalytic effects can be related to the mobility of a catalytic species with a vitrimer network. For Zn(II)-catalysts, their findings suggest that catalytic function is mediated through coordination of the metal ion to either the carbonyl or the β-hydroxyl group in proximity to the ester functionality, fixating the catalyst to the reactive moieties.
In a more recent study on the activation of polyester vitrimers, Bates and co-workers explored various simple Brønsted acid additives in their polyester networks and observed a clear relationship between pKa values of protic mono-functional acids and the stress relaxation times of the materials (Fig. 1a3–4).24 Next to a large accelerating effect on the overall rheology in these ‘Fischer transesterification’ vitrimers, the stronger acids also gave a slight increase of the mechanical activation energy, even though the exchange itself was much faster. These Brønsted acids are highly effective catalysts and gave stress relaxation times in the order of hours already at room temperature. Bates and co-workers also observed significant differences between Brønsted and Lewis acids, with the latter clearly being less effective catalysts in their system. However, Schmidt together with Reynaud and co-workers28 reported a study where no significant differences in rate and activation energy were observed. At this point, however, it is important to point out that these comparisons to Lewis acid additives did not take into consideration the similarity of polymer matrices, cross-linking density, or measurement conditions. Also, Bates and co-workers performed their experiments between 25 °C and 75 °C whereas Schmidt, Reynaud and co-workers performed theirs at much higher temperatures being 240 °C to 270 °C. Therefore, it can be expected that the differences in relaxation time might arise from a variety of different parameters that should be considered with caution. Indeed, even within the same polymer matrix and temperature window, different exchange mechanisms may manifest themselves (vide infra). Similar to the studies of transesterification vitrimers, Du Prez, Winne and co-workers demonstrated that the normally ‘catalyst-free’ dynamic covalent vitrimer chemistry of vinylogous urethanes can be further modified by simply adding small amounts of TBD, pTsOH, or dibutyltinlaurate (DBTL), which gives remarkable effects on the resulting viscoelastic properties.25 With these catalytic additives, the viscoelastic profile could be adjusted over several orders of magnitude (Fig. 1b). Remarkably, each catalyst was also shown to result in a quite different activation energy. In the absence of catalyst, or when a Brønsted acid was added, the amine exchange proceeds via a “protic pathway” involving an activated iminium intermediate with a measured enthalpic barrier (Ea) of about 75 kJ mol−1. In contrast, when strong bases were used as additives, this protic pathway was not only inhibited – as can be expected due to the lack of acidic protons – at the same time a new exchange mechanism manifests itself, showing a much higher energy barrier in the range of 100 to 140 kJ mol−1 (Fig. 1b).
This was rationalized to correspond to a much slower Michael-type direct amine addition pathway, not involving pre-existing ammonium species, but going through higher energy neutral zwitterion intermediate adducts. Thus, an interesting level of control arises in vinylogous urethane vitrimers, where small amounts of additives cannot only speed up but also slow down dynamic bond exchange reactions.
A similar and interesting effect was also reported in materials based on the 1,2,3-triazolium trans-N-alkylation mechanisms studied by Drockenmuller and Montarnal. Here, the bond exchange pathway is mediated and effectively catalysed through the counterion of the triazolium cross-links.26 Although, stricto sensu, counter ions are not fully external catalysis (due to ionic bonding), their rate enhancing effects in these vitrimer-like networks is worth closer consideration. In a work reported by the same research group dealing with poly(ionic liquid)s (PILs) as vitrimer materials, the chemical nature of the counter-ions was changed between Br−, I− and mesylate−, and a profound impact on the exchange rate and thus the visco-elastic properties was observed (Fig. 1c).26,29 While PILs composed of Br− and I− counter ions lead to relaxation times in the order of 300 s and 900 s, respectively, mesylate-based PILs impacted significantly the exchange speed with a relaxation time in the order of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. As this is a case where the catalytic role is not performed by different (Lewis) acid or bases, but rather by different nucleophilic catalysts, these studies reveal the power and diversity of external catalysis when targeting specific viscoelastic profiles. We believe that current research has only scratched at the surface of catalytic fine-tuning of vitrimer properties.
000 s. As this is a case where the catalytic role is not performed by different (Lewis) acid or bases, but rather by different nucleophilic catalysts, these studies reveal the power and diversity of external catalysis when targeting specific viscoelastic profiles. We believe that current research has only scratched at the surface of catalytic fine-tuning of vitrimer properties.
However, a word of caution should be issued to those interested in studying catalysis-vitrimer property relationships. For different polymer systems, observed properties such as relaxation time, or activation energy for the viscous flow, should not be directly compared. As it is shown in the following sections, even within highly similar systems, not only external additives will influence the rheological properties but also its network topology, cross-linking-density, Tg and other material parameters need to be considered.
Internal catalysis and neighbouring group participation (NGP)
Although the use of additives is quite common in polymer materials, external catalysts often have a low compatibility and chemical stability for common polymer applications. Thus, it can be advantageous to provide the polymers themselves with a catalytically active moiety, preventing catalyst leaching and aggregation. In an elegant article by Altuna and co-workers, a tertiary amine catalyst was covalently introduced in epoxy vitrimers using a primary or secondary amine that reacted with the matrix constituents.30 Due to the ability of tertiary amines to catalyse transesterification reactions through general base catalysis, this material design enabled exchange reactions without the addition of external catalysts, solving partially some related drawbacks thereof. A similar but more elaborate strategy was introduced by Guo, Zhang and co-workers who introduced very hydroxyl-rich monomers, like glycerol or hyperbranched epoxy precursors to accelerate the rate of the non-catalysed transesterification.31 The rate-enhancing effect in these vitrimers is likely caused by the high concentration or clustering of hydroxyl functions that can provide a more polar/less hydrophobic reaction environment in which exchanges with ionic intermediates are promoted. This supposed proximity effect is far away from what is usually understood as the function of an ‘immobilized’ catalyst, and this proximity-driven rate enhancement starts to border on the classical organic chemistry concept of “neighbouring group participation” (NGP). Similarly, Altuna and co-workers already reported in 2013 a catalyst-free dynamic polymer network, capable of some degree of stress relaxation, relying on the proximity of an additional carboxylic acid on the cross-linker (in casu citric acid used to cross-link epoxidized soy bean oil, Fig. 2a).32 The resulting epoxy materials surprisingly showed a dynamic behaviour upon heating without added catalyst. Although they assigned this effect to hydroxyl group concentration, the presence of unreacted free carboxyl acid in close proximity to ester bonds was probably an early example of internal catalysis or perhaps a NGP in dynamic materials. In fact, the very first reported vitrimers, Leibler's epoxy-ester networks, may also incorporate a degree of internal catalysis. Compared to normal ester cross-links, the β-hydroxy ester links are particularly well suited for transesterifications, as the proximity of the hydroxyl will facilitate the exchange reaction.15,23,33 | ||
| Fig. 2 Examples of “neighbouring group participation (NGP) effect” in vitrimers enabling a fine tuning of exchange reactions and thus viscoelastic properties. Neighbouring groups are displayed in green and exchanging moieties in blue. (a) Citric acid-based esters with unreacted free carboxyl acid in close proximity to ester bonds.32 (b) Boronic ester exchanges and amino-NGP influence.34 (c) Silyl ether exchanges and amino-NGP influence.35 (d) Phthalate monoesters with free carboxyl or sulfonic acid groups in close proximity.36,37 | ||
The difference between the concepts of internal catalysis and NGP is a subtle one but should normally reflect clear mechanistic differences. Neighbouring group participation is reserved for rate-enhancing (transition state stabilizing) substituent effects wherein the substituent becomes covalently bonded to the reaction centre, implicating either an internal group transfer (e.g. protonation) or cyclisation reaction. All other proximity-driven rate-enhancing effects (electrostatic) should be termed as internal or intramolecular catalysis. The internal catalysis concept was further developed and explicitly studied by Guan and co-workers who demonstrated the strong rate enhancement on transesterification of di(o-aminophenylboronic) ester formed cross-links compared to non-substituted diphenylboronic esters (Fig. 2b).34 As an explanation, they suggested that the o-amino substituent would facilitate the exchange based on its function as a proximal base for faster proton-transfer. As these amines also seem suitably placed to form a five-membered ring intermediate, a classical NGP effect could indeed also be at play here.
More recently, the same group introduced amino groups in gamma position of a silyl-ether-exchange based dynamic network, which resulted in a dramatic acceleration of the exchange rate of three orders of magnitude (Fig. 2c).35 Further studies, aimed at comparing the effect to externally added amines as catalyst have not been reported, thus it is hard to judge if a five-membered ring intermediate would be involved in the catalysed exchange pathway, but this certainly seems feasible. With the explicit objective to implement a NGP effect in a transesterification-based vitrimer, we recently developed a new approach to achieve catalyst-free (or internally catalysed) transesterifications (Fig. 2d).36 The carboxylic acid substituent was found to effect a NGP, not through internal proton transfer, but rather through a cyclisation reaction to an anhydride intermediate. The resulting materials exhibited a linear viscosity profile over a range from 100 °C until 180 °C, based on the internal activation of the ester bonds through the ortho-acid substituent. While the objectives where successfully achieved, this NGP installs a clearly dissociative bond exchange mechanism, which should not result in vitrimer properties. In fact, the same is likely true for many vitrimers, as debonded states can always arise as alternative mechanisms.19 For a closer discussion of the relationship between exchange mechanisms and material properties, we refer to our recent perspective on the topic.22 A similar concept was very recently reported by Heuts, Sijbesma and co-workers who demonstrated internal activation but this time using a benzenesulfonic ortho-acid substituent (Fig. 2d).37 This sulfonic acid based NGP resulted in significantly faster stress relaxation compared to carboxylic acid counterparts through a similar reversible mechanism based on a cyclised sulfobenzoic anhydride intermediate. Furthermore, the resulting materials seem to be less sensitive towards an undesired ester formation in comparison to a purely carboxylic acid-based system.
We believe that further research efforts should be undertaken to fully extend the concepts of catalytic control over vitrimers. However, the field needs to take care that these effects are not confused and over-simplified, as exchange mechanisms can respond in unpredictable ways to even minor changes in the reaction set-up.17 Trying to enhance one pathway, may inadvertently promote another one. An illustrative example of that can be found in the next subsection.
Competition of different exchange mechanisms
In principle, material characteristics can be tuned through using different exchange reactions within a polymer network (vide infra). A more direct and more attractive approach lies in the possibility to have different reaction mechanisms for one and the same bond exchange system. This is because implementing two different synergetic and orthogonal dynamic covalent chemistries within the same matrix is quite tricky and can be marred by cross-reactivity.Our group recently demonstrated the concept in perfluorinated as well as in non-fluorinated polyether based VU vitrimers. These systems show a marked transition from a low activation energy-dependence of around 60 to 70 kJ mol−1 at lower temperatures, to a higher energy dependence of around 130 to 170 kJ mol−1 at higher temperature (Fig. 3), showing an unusual rheology profile.38 We surmised that this mechanical transition was related to a chemical transition between two different previously proposed underlying reaction mechanisms. These different pathways, i.e. a protic (cationic) pathway, and an aprotic (zwitterionic) pathway, lead to exactly the same reaction products but involved different intermediates and reactive species.
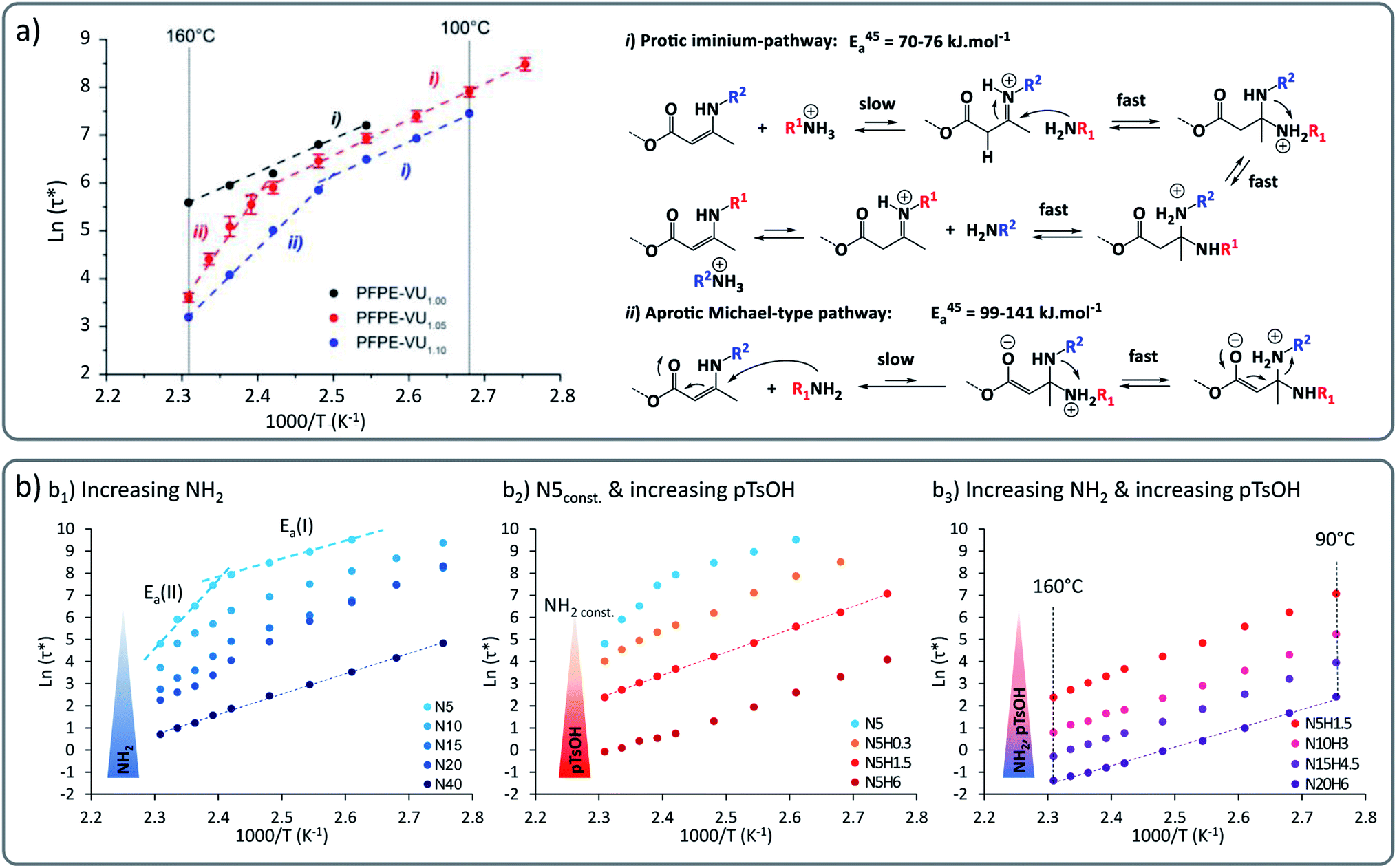 | ||
| Fig. 3 (a) Vinylogous urethane vitrimers exhibiting a dual temperature response based on two concomitant exchange mechanisms: iminium-type mechanism in lower temperature range with Ea of 60–70 kJ mol−1 and Michael-type addition at higher temperature with higher energy barrier of 130–170 kJ mol−1. Reproduced with permission from ref. 38. Copyright 2018 American Chemical Society. (b) Arrhenius plot of poly(propylene glycol) and vinylogous urethane based vitrimers showing a control of the bond exchange rate by playing with: (b1) the concentration of active nucleophilic moieties (primary amine groups), (b2) the concentration of acid catalyst (pTsOH), (b3) the concentration of both active nucleophilic moieties and acid catalyst. Adapted from ref. 39 with permission from the Royal Society of Chemistry. | ||
It could be further shown that this unusual behaviour occurs within a wide range of matrices, pointing towards a chemical rather than a material effect. In more recent work, we could demonstrate that this phenomenon could be applied to strategically enhance the exchange rate to achieve ever faster stress relaxation.39 By altering the stoichiometry and catalysts, the systems can be pushed into a rheology profile with a single activation energy (Fig. 3b1). Furthermore, addition of the protic acid para-toluene sulfonic acid (pTsOH) resulted in dramatic accelerations (Fig. 3b2). This constitutes an example of how a good control of the viscoelastic properties of vitrimers can be achieved, varying relaxation times over the range of four orders of magnitude, down to ultrafast relaxation times below one second (Fig. 3b3).
The co-existence of two mechanisms within the same network has also been reported in polyhydroxyurethane vitrimers based on the opening of 5-membered cyclic carbonates with amines.40 In this case, the referred additional mechanism was dissociative and based on reversible cyclic carbonate aminolysis that co-existed with associative transcarbamoylation bond exchanges. However, there have been no studies directed at gaining control over either or both mechanisms by changing stoichiometries and catalytic systems. Such type of research seems to hold a particular promise, both for gaining insight into chemical reactivity within vitrimers as well as for purposely designing new vitrimers with ‘on demand’ dynamic properties.
Vitrimer hybrids: combination of dynamic bond exchanges and supramolecular interactions
In idealised vitrimer materials, chain mobility is exclusively governed or at least dominated by chemical reaction rates. In real life vitrimers, not only unexpected additional reactions, but also many other non-covalent effects can add on to the rheological behaviour. Polymer networks can also be purposely designed by a combination of transient supramolecular interactions and more robust permanent, yet dynamic covalent cross-links, leading to improved properties.For example, hydrogen bonding has been shown to be beneficial in combination with dynamic disulfide cross-links.41 These intermolecular forces can effectively reduce the dynamic exchange based on (very fast) disulfide exchange at lower temperatures. Here, the fast covalent exchange is only revealed upon heating to processing temperature as the hydrogen bonding interactions are broken and enable rapid exchange based on disulfide metathesis in poly(urea-urethane) networks (Fig. 4a).42–44 Other ‘additional’ interactions or polymer ‘stickers’ such as metal coordination have been combined with covalent exchange reactions to tailor the viscoelastic properties of vitrimers. For instance, epoxidized natural rubber (ENR) was modified with a boronic ester containing cross-linker and additional ZnCl2 was introduced in the created networks (Fig. 4b).45
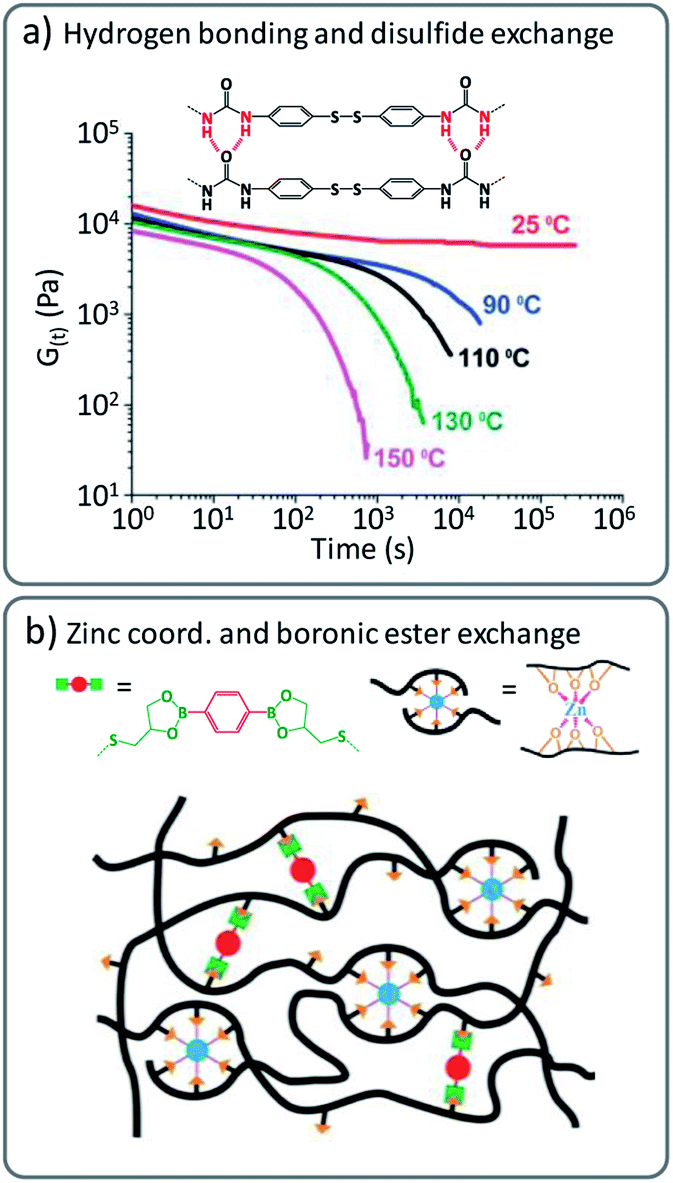 | ||
| Fig. 4 (a) Combination of disulfide exchange and hydrogen bonds, leading to vitrimer materials with strong viscosity-temperature dependence. Adapted from ref. 42 with permission from the Royal Society of Chemistry. (b) Schematic representation of vitrimer networks with dual cross-links composed of dynamic boronic ester bonds and non-covalent sacrificial Zn2+–O coordination bonds. Adapted with permission from ref. 45. Copyright 2019 American Chemical Society. | ||
The latter vitrimers were proposed to show a dual cross-linking, based on dynamic boronic ester exchange and by coordinated Zn2+ ions (via available alcohol moieties from the opened epoxy) as sacrificial bonds upon mechanical stress. It must be noted that similar effects could already be at play in Leibler's original Zn(II)-doped polyhydroxyl transesterification vitrimers. For the boronic ester/ZnCl2 ENR vitrimers, Guo and co-workers also explored ‘sacrificial’ supramolecular bonds based on hydrogen bond formation of dangling amides, in combination with a transesterification catalysed via TBD.46 Later, they also introduced a benzimidazole group in styrene–butadiene rubber (SBR) to form Zn2+–imidazole complexes that induced micro-phase separation and led to significant enhancement of the mechanical properties.47 Likewise, Zhang, Sun and co-workers reported a strategy where 2-ureido-4[1H]-pyrimidinone (Upy) dimers were combined with boronic ester cross-linkers, to enhance mechanical properties of supramolecular networks by addition of physical cross-links.9
However, not only supramolecular interactions can be combined with dynamic covalent cross-linking. Also combining different covalent exchange chemistries can be a viable, yet possibly tricky, strategy to gain better control of vitrimer properties, especially in the area targeting more responsive materials, by making sure that each exchange chemistry is responsive to different triggers (for example light and heat). As an illustrative example, Wolf and co-workers demonstrated that linear polymers formed of aliphatic 1,12-dodecanebisacetoacetate, chain-extended with 1,12-dodecanediamine and 1,5-diaminoanthracene displayed both thermal and light responsiveness.48 Herein, the combination of UV-active anthracene with thermally triggered vinylogous urethane exchange reactions enabled an increase in Tg and a marked change in rheological properties upon UV irradiation via dimerization and partial network (or vitrimer) formation, starting from otherwise linear polymers. Interestingly, the authors described that linear Arrhenius-plots could be obtained in a purely linear polymer condensate, indicating that the chemical VU exchange is likely to be the dominant factor in the stress relaxation of the non-cross-linked system. This observation again supports the fact that the existence of a linear Arrhenius-plot as such is far from a sufficient criterium for a crosslinked polymer material to be considered as a vitrimer. Also, this feature is certainly also not unique for vitrimers. In another vitrimer chemistry combination study, synergetic effects were demonstrated by Chen and co-workers by using disulfide metathesis in combination with transesterification, where both exchange chemistries appear to allow faster reprocessing when built-in together in one network (Fig. 5a).49
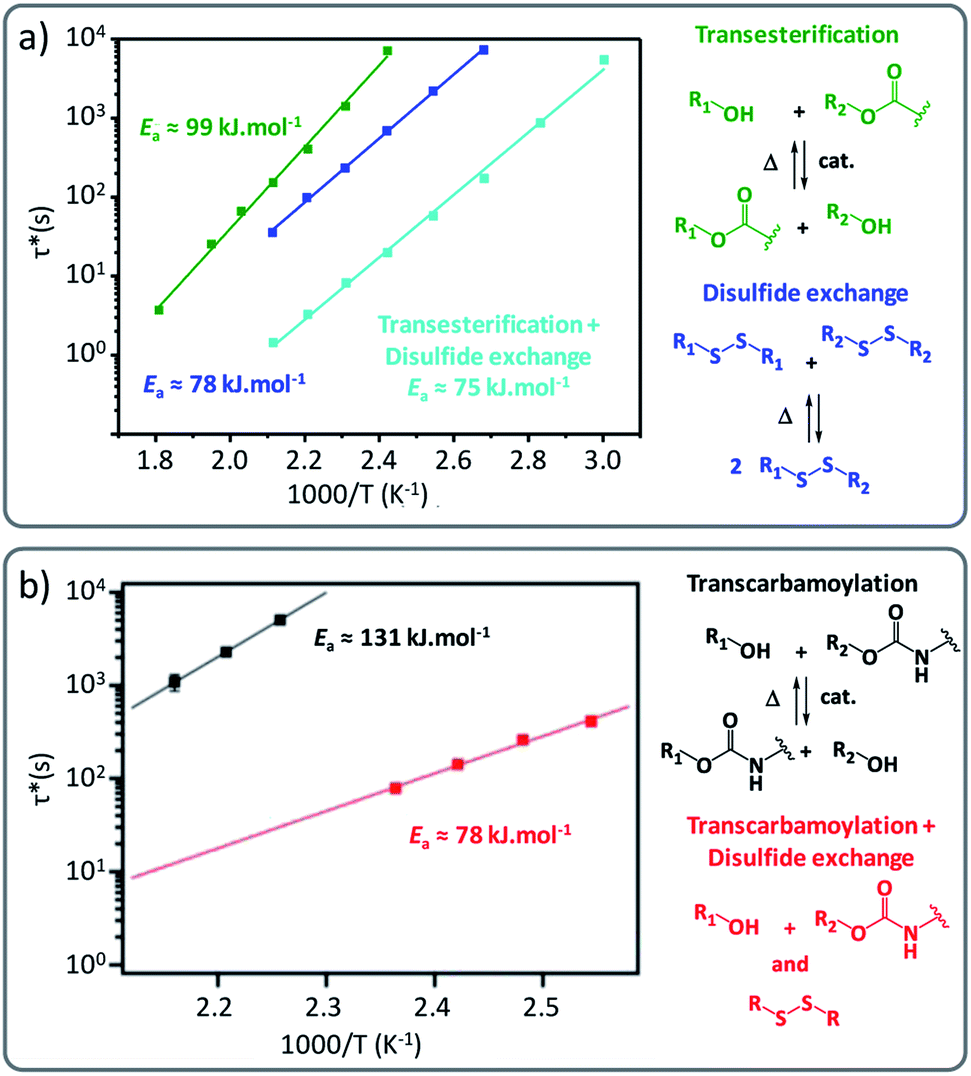 | ||
| Fig. 5 (a) Comparison of Arrhenius plots of epoxy vitrimers constituted of a single exchange pathway (green for transesterification reactions/blue for disulfide exchange) and dual exchange pathway (cyan for transesterification and disulfide exchange). Reconstructed with permission from ref. 49. Copyright 2019 American Chemical Society. (b) Arrhenius plots of polyhydroxyurethane vitrimers comparing single exchange (black: transcarbamoylation exchange reaction) and dual exchange-based vitrimers (red: transcarbamoylation and disulfide exchange). Reproduced with permission from ref. 50. Copyright 2018 American Chemical Society. | ||
Also, Dichtel and co-workers observed that applying the disulfide exchange in polyhydroxyurethane networks resulted in more rapid reprocessing of PHU networks, i.e. within 30 min at about 150 °C (Fig. 5b).50 Their study also suggested that the incorporation of disulfide containing diamine linkers not only changed the exchange rate but also the underlying temperature dependency (activation energy) from 131 ± 8 kJ mol−1 to 78 ± 4 kJ mol−1.
Thus, although the materials design requires more caution from a chemical perspective, the combination of covalent exchanges seems a strategy with a high potential to obtain a tailor-made control of the viscoelastic behaviour of vitrimers. We are convinced that further combinations should be investigated in the future, which allows to achieve additional control of vitrimer properties and opens new application areas.
Vitrimers: matrix effects
Even if vitrimers are not explicitly combined with other dynamic links, and in the absence of obvious catalytic effects, their reactivity (and thus dynamics) are still likely to be influenced by the exact composition and architecture of their polymer matrix. Indeed, when small molecule reactions are performed in different stoichiometries and different solvents, dramatic effects on the reactivity can occur. Similar effects should thus be expected in vitrimers.Network composition or vitrimer ‘solvent’ effects
Many vitrimer chemistries have been implemented in different polymer matrices, and indeed they invariably show small to even quite pronounced matrix dependencies. A clear example are the vinylogous urethane vitrimers, where stress relaxation times can depend heavily on the polymer matrix, regardless of catalyst levels.38 For this reason, we recently reported a systematic comparison of the viscoelastic behaviour of vinylogous urethane-based vitrimers, resulting from the same exchange reaction embedded in different polymer backbones (PDMS, PPG, PTHF, etc.).51 The results obtained from stress relaxation experiments evidenced that for the same reversible chemical platform, the calculated activation energy (Ea) can be significantly affected by the nature of the matrix, with values ranging from 68 kJ mol−1 for aliphatic-based vitrimers to 117 kJ mol−1 for PDMS-based vitrimers.51This significant difference of Ea as a function of the matrix was also reported in imine-based vitrimers where diphenylmethane, dicyclohexylmethane and hexamethylene-derived building blocks resulted into Ea-values of 81, 74 and 49 kJ mol−1, respectively (Fig. 6a).52 Dichtel, Hillmyer and co-workers also reported a comparable effect in hydroxyurethane vitrimers, in which an ether based-vitrimer displayed an Ea of about 100 kJ mol−1 while a corresponding aliphatic-based material generated a higher energy barrier of 136 kJ mol−1.53
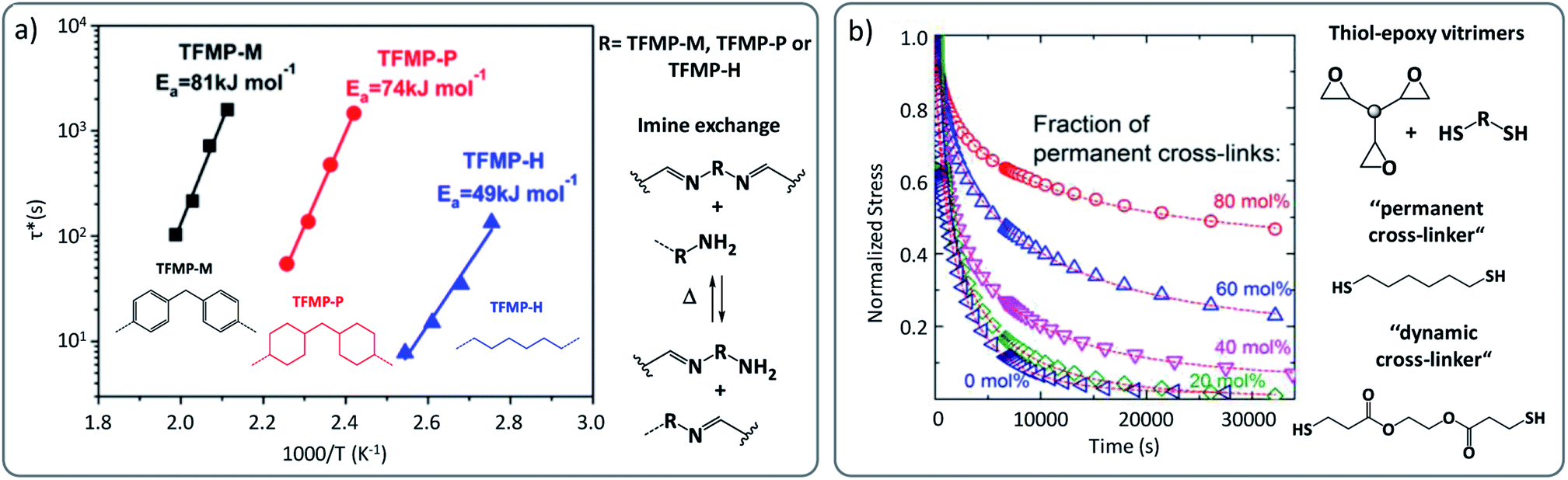 | ||
| Fig. 6 (a) Arrhenius plots of imine-based vitrimers constituted of diphenylmethane (TFMP-M), dicyclohexylmethane (TFMP-P) and hexamethylene (TFMP-H)-derived building blocks, showing significant change in relaxation times and related activation energy. Adapted with permission from ref. 52. Copyright 2018 American Chemical Society. (b) Stress relaxation recorded for epoxy-based vitrimers possessing different ratio of permanent cross-links. Incomplete relaxation is observed in composition, having more than 20% permanent linkers. Adapted with permission from ref. 54. Copyright 2018 American Chemical Society. | ||
A similar comparison was done by Torkelson and co-workers in hydroxyurethane vitrimers.55 Three different vitrimers, constituted of polybutadiene (PB), poly(tetramethylene oxide) (PTMO), and polydimethylsiloxane (PDMS), have been evaluated and compared with regard to their recycling efficiency. In this case, the incompatibility of the backbone and dynamic chemistry induced the formation of phase-segregated nanostructures that have been proven to affect substantially the good property recovery of the referred vitrimer materials.
It must be noted that most of these striking variabilities have not been systematically studied so far and that a lot more work along these lines will need to be performed before we can draw clear rationales for these behaviours. Hydrophobicity or polarity seems like an obvious factor to address in the case of vitrimers that rely on an ionic exchange mechanism. However, catalyst mobility is also a factor that should not be underestimated in the overall dynamics of vitrimers and may be greatly influenced by the matrix properties.
Phase separation within vitrimers
Probably due to an initially intense focus on chemical factors, the influence of phase separation on viscoelastic properties, as an obvious and well-known physical phenomenon in polymers, was so far barely investigated in the context of vitrimers. However, it is currently attracting growing attention.55 Recently, Ricarte, Leibler and co-workers reported a poly(ethylene) vitrimer in which phase separation causes an uneven distribution or partial confinement of the dynamic cross-links. This explains the relatively low grafting densities, as well as the high soluble fractions (between 52–78 wt% soluble fraction).56 This study emphasizes the importance to consider phase separation and related effects in the design of vitrimer materials. When observing the notable influence of phase separation on the final mechanical properties, this effect was quickly and purposely exploited to enhance the vitrimer mechanical properties. Very recently, Sumerlin, Epps and co-workers implemented phase segregated domains in acrylic based vinylogous urethane vitrimers.57 In this study, incompatible block copolymer architectures were prepared by RAFT polymerization and the resulting block-derived materials displayed different nanostructural and rheological properties relative to their statistically-derived counterparts.Although the direct repercussion of the nanostructuration on the overall mechanical properties observed in vitrimers is not as marked as other effects that have been observed so far, it is clearly a factor that should not be ignored or forgotten, and more significant effects may still be observed in the future.
Partial irreversible cross-linking of the vitrimer matrix
A current major challenge for vitrimers is to design elastomers that show good processability at high temperature (for reprocessing) but show limited reactivity at lower temperature to avoid creep and plastic deformations. Factors that will improve one aspect, typically negatively affect the other. Creep behaviour is an important drawback that could prevent certain types of vitrimers from being developed for a wide variety of applications. By judiciously designing the polymer matrix, creep can be affected and minimised. In an elegant approach proposed by Torkelson and co-workers, irreversible permanent cross-links were introduced into an otherwise reversible permanent covalent network, i.e. an epoxy-based vitrimer network (Fig. 6b).54 It was found that creep behaviour can be reduced in the order of 71% relative to the reference vitrimer without non-dynamic cross-links. However, the strategy was not equally effective for the entire range of compositions, and a minimum of 60% reversible cross-links was necessary to obtain good recyclability and good recovery of properties (Fig. 6b), creating an upper limit to this strategy. A similar concept was in fact simultaneously reported by Sumerlin and co-workers in the context of self-healing boronic ester networks.58 Recently, the idea was further taken up by Nicolaÿ and co-workers in polybutadiene vitrimers59 and by our group in vinylogous urethane epoxy-based vitrimers.60 While this concept represents an effective ‘generic’ strategy to suppress creep, the reprocessability also suffers qualitatively and quantitatively, and it seems hard to overcome this intrinsic limitation. Nevertheless, targeted irreversible cross-linking is a straightforward way to improve some of the adverse properties of a vitrimer.Stoichiometry of reactive functions within vitrimer matrices
Achieving low creep and highly processable vitrimer elastomers will likely require a very good understanding of all the factors that control covalent exchange. A relatively obvious, yet often overlooked factor in vitrimer design is the availability of the reactive centres or cross-links with respect to each other. In vitrimers, the associative mechanism requires association of two polymer chain segments before a cross-link can be exchanged. In this way, stoichiometry can thus have a considerable impact on vitrimer properties. This influence has been very-well exemplified by Dušek and co-workers61 in an old study dealing with epoxy thermosets, lately resituated in the context of vitrimers by Williams and co-workers.62 These two studies are highly relevant for the understanding of epoxy ester vitrimers since they take into account many known side reactions that could potentially occur in the case of (off)-stoichiometric formulations. These in-depth studies especially highlight the impact that the complete reactivity profile can have on material properties, with a special focus on the network formation itself (under off-stoichiometry conditions), and how this influences reprocessing at high temperature over longer reaction times.Networks that are formed under exact stoichiometric conditions will show less complex reactivity profiles, but for most vitrimers off-stoichiometry conditions are actually prerequisites to achieve fast exchanges, especially when an exclusive associative-type mechanism is involved.39,63 As a rule, the introduction of free reactive groups (or the proximity of reactive cross-links) is an absolute necessity to enable exchange reactions to happen. Driving up the concentration or availability of reactive groups is not without risk, as these are typically not the most inert functions within a polymer material, and will thus give important changes to the overall polymer properties if they start to be more abundant throughout the network. For example, a higher concentration of reactive hydroxyl groups in a transesterification vitrimer will create a higher chance for exchange, which will in principle lead to faster stress relaxation. However, regardless of the kinetics, such off-stoichiometric hydroxyl-rich materials (beneficial in case of transesterification based materials) will also result in higher swelling ratios and lower dimensional stability, and will also become more hydrophilic and thus more sensitive to hydrolysis. Therefore, vitrimer design through tuning the stoichiometry not only involves in-depth knowledge of the chemical reactivity parameters, but must also take into account the specific material applications that are targeted, balancing bulk material properties and ‘dynamic covalent’ processability.
Cross-link density of vitrimer matrices
A key architectural feature, apart from stoichiometry and the introduction of partial irreversible cross-linking in vitrimer networks, is its actual cross-linking density. This will also have a strong effect on the viscoelastic behaviour. Indeed, by adjusting the cross-linking density, and thus the relative distance between network points, different parameters that govern the macroscopic flow will be affected. Obviously, in line with the arguments given for stoichiometric considerations, the cross-link density will also influence the diffusion, relative mobility and availability of the reactive groups within a network. Dynamic bonds may be part of cross-links between chains and can also be embedded into linear chains. If the dynamic bonds are limited to the cross-linking points, increasing the average distance of the cross-link points, a decrease in exchange kinetics is the consequence of a simple dilution effect. Also when reversibly exchanging bonds are embedded in the main chain, the mobility and availability might be limited by increasing the cross-link density between chains.In an illustrative study by Nicolaÿ and co-workers, a polybutadiene (PB)-based vitrimer, wherein PB chains are cross-linked by dioxaborolanes, a clear increase of stress relaxation times was observed as a function of the increasing cross-link density (Fig. 7a) and thus decreased chain mobility.59 Nonetheless, in a similar case study dealing with polyesters, COOH side groups were crosslinked by di-epoxy compound and the opposite phenomenon was reported.64 Indeed, Hayashi and co-workers observed the fastest relaxation rate for the highest cross-link density. It is important to keep in mind that despite the use of comparable network architectures, different exchange mechanisms were involved (boronic ester metathesis vs. transesterification). We can speculate in the latter case that the close proximity of the hydroxyl groups in the β-hydroxy ester links facilitates the exchange reactions. Nonetheless, a similar tendency to the one reported by Hayashi and co-workers was observed by Drockenmuller and co-workers in ionic vitrimers based on 1,2,3-triazolium cross-links.29 They showed that an increase in the cross-link density resulted in a decrease of relaxation time. It is however important to note that the latter system remains very peculiar since higher molar fraction of cross-linker (i.e. bis-alkylating agent in this case), does not necessarily imply higher cross-link density due to the presence of different chemical species (triazole, triazolium, unreacted alkylating agent, …). Indeed, beyond a certain molar fraction of cross-linker, further addition induces a decrease of cross-link density as well as strong modification in the composition of chemical species. It is interesting to compare these results with those obtained by us, where a range of vitrimer formulations were made based on a polytetrahydrofuran (PTHF) backbone.51 Here, the crosslink density was varied within the same network architecture, while the overall network topology was not affected (A3 + B2 structure). In this study, increasing the distance between cross-links and thus decreasing the resulting cross-link density clearly results in a slower stress relaxation (Fig. 7b),51 as also observed by Hayashi and co-workers.64 However, in all these studies the change in cross-link density was accompanied by a modification of the concentration of reactive/exchangeable groups.
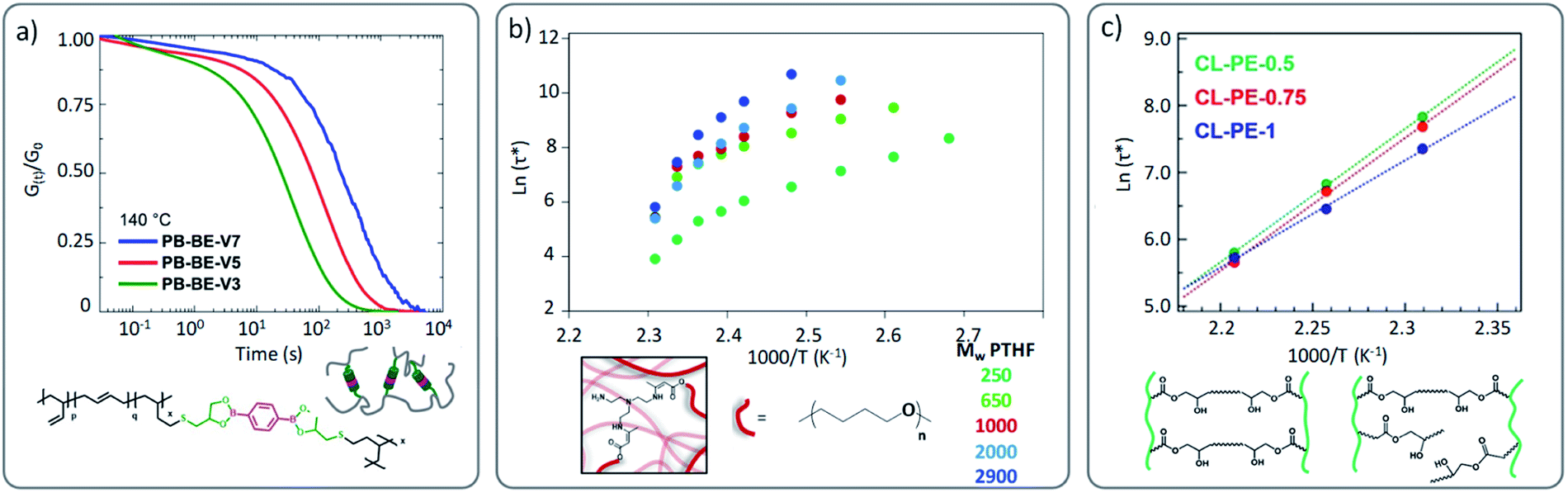 | ||
| Fig. 7 (a) Stress relaxation of poly(butadiene)-based vitrimers cross-linked with different ratio of dioxaborolane cross-linker (V3, V5 and V7 in PB-BE-VX is standing for the number of cross-linkers per chain). Adapted with permission from ref. 59. Copyright 2019 American Chemical Society. (b) Arrhenius plots of PTHF-based vitrimers composed of different molecular weights with high (PTH250) to lower crosslinking density (PTHF2900). Adapted from ref. 51 with permission from the Royal Society of Chemistry. (c) Arrhenius plots of polyester vitrimers with different cross-linking density but same OH concentration (low crosslink density for CL-PE-0.5 to high cross-linking density for CL-PE-1, with X standing in CL-PE-X to the mole fraction of the epoxy groups derived from the di-epoxy ratio to the total epoxy groups: for example, 1 = all di-epoxy). Adapted with permission from ref. 65. Copyright 2020 American Chemical Society. | ||
Therefore, the establishment of a clear correlation between cross-link density and vitrimer properties, without considering other network parameters, is not possible. Very recently, Hayashi and co-workers examined this aspect based on an already reported polyester vitrimer material.64 By using mono- and di-functional epoxy groups, the authors were able to prepare polyester based vitrimers with varying cross-link density while keeping the concentration of reactive hydroxyl groups constant.65 Similar trends were observed in comparison to their first study, although to a much lesser extent (Fig. 7c). Fine-tuning of the cross-link density and the distribution of cross-link junctures in vitrimers thus also sits on a delicate balance.
A good example of ‘compromise materials’ that can be achieved in this regard is the design of “reinforced vitrimer thermoplastics” as introduced by Leibler and Nicolaÿ in 2017.66 In this interesting system, the inclusion of some additional dynamic ‘vitrimer-type’ cross-links in otherwise fully thermoplastic materials significantly increased solvent resistance compared to non-modified materials, whereas the cross-links do not in fact excessively slow down the processability as a result of their dynamic nature, as would be the case for irreversibly cross-linked materials. Because of this combination of vitrimer and thermoplastic processing, the high molecular weight thermoplastic materials were able to be reprocessed several times by means of extrusion or compression- or injection-moulding, similar to commercial thermoplastics, while retaining their enhanced properties. Compared to real vitrimers, such weakly cross-linked thermoplasts show very limited resistance to dissolution by solvents.66–68 In spite of such drawbacks, these ‘vitrimerised thermoplasts’ have attracted considerable attention,66–74 considering the fact that they can be processed using usual thermoplastic processing techniques, while their properties can be improved in comparison to the bulk thermoplastic counterparts.
Interpenetrating vitrimer networks
In contrast to typical network parameters that were partially investigated and discussed in this minireview, it seems that the combination of different interpenetrated vitrimer networks is still subject of research. Indeed, this concept was exclusively reported by Rong, Zhang and co-workers who combined boronic esters and disulfide exchanges in two different yet interpenetrated networks.75 Thanks to this network interlocking, the phase separation of the two incompatible networks was stabilized generating a single intermediate glass transition. Furthermore, this interlocking also resulted in a nonlinear improvement in static and dynamic mechanical properties. However, the associative nature of the exchanges as well as the dynamic characterisation were not investigated. The dynamic exchange was only exploited for reprocessing through the dissociative mechanism (selective boronic ester hydrolysis) which led to selective network unlocking and material recovery. We are convinced that intensive research in this direction still needs to be done, yet, it can be a promising road to take for creating unprecedented, because possibly otherwise incompatible, properties.Hence, as can be seen from the subsections above, it should be clear that what we perhaps naively call ‘vitrimer matrix effects’ in fact encompasses an extremely wide scale of factors that need to be taken into consideration in the design and understanding of vitrimers.
Reinforced vitrimer materials
As materials, part of the excitement for the developments in the area of vitrimers is also driven by the fact that they also hold promise as thermoset substitutes for the development of weldable, recyclable and reshapable composites.76 Therefore, the development of vitrimer (nano)composites have attracted considerable interest in the past few years, as demonstrated by the numerous varieties of already implemented reinforcing agents, such as glass and carbon fibers,77–81 silica and alumina-based fillers,82–89 carbon dots and nanotubes,90–95 graphene,96–99 cellulose,100 nanocrystals,101etc.102 The most frequently reported systems used in this context are based on the original transesterification vitrimer chemistry, because of the widespread commercially available hydroxy-functionalized fillers.82,84,86 Yet, other chemistries such as imine,96 siloxane88 or transcarbamoylation86 have also been reported.However, most of these reports have understandably focused on the mechanical properties and only few studies have evaluated the impact that these reinforcing agents may have on the actual viscoelastic properties of the vitrimer. Indeed, the introduction of inorganic materials such as for instance glass fibres are not inert with regard to the dynamic exchanges, which can affect the resulting viscoelastic properties. In a well-conducted study by Soulié-Ziakovic and co-workers, the influence of covalently and non-covalently bound silica nanoparticles was evaluated within an epoxy based vitrimer.82 Their results showed that even though the nanoparticles slow down the stress relaxation in comparison to an unfilled vitrimer analogue, a full relaxation of the applied stress in association to a higher modulus can be achieved, which is a combination that is hard to achieve otherwise (Fig. 8a). However, it is not clear what the impact of these ‘covalently linked’ fillers would be on recyclability and property recovery. This aspect has been studied more in detail by Torkelson and co-workers in hydroxyurethane nanocomposites.86 They reported a similar accelerating effect in vitrimers composed of covalently bonded nanofillers compared to non-reactive nanofillers. However, while non-functionalized modified vitrimers showed full recovery of their material properties, covalently reinforced vitrimers resulted in a loss of mechanical properties associated to a decrease in cross-link density. The decrease in recovery of the desired properties was reported to be likely caused by side reactions between silica functionalities and the underlying vitrimer chemistry.
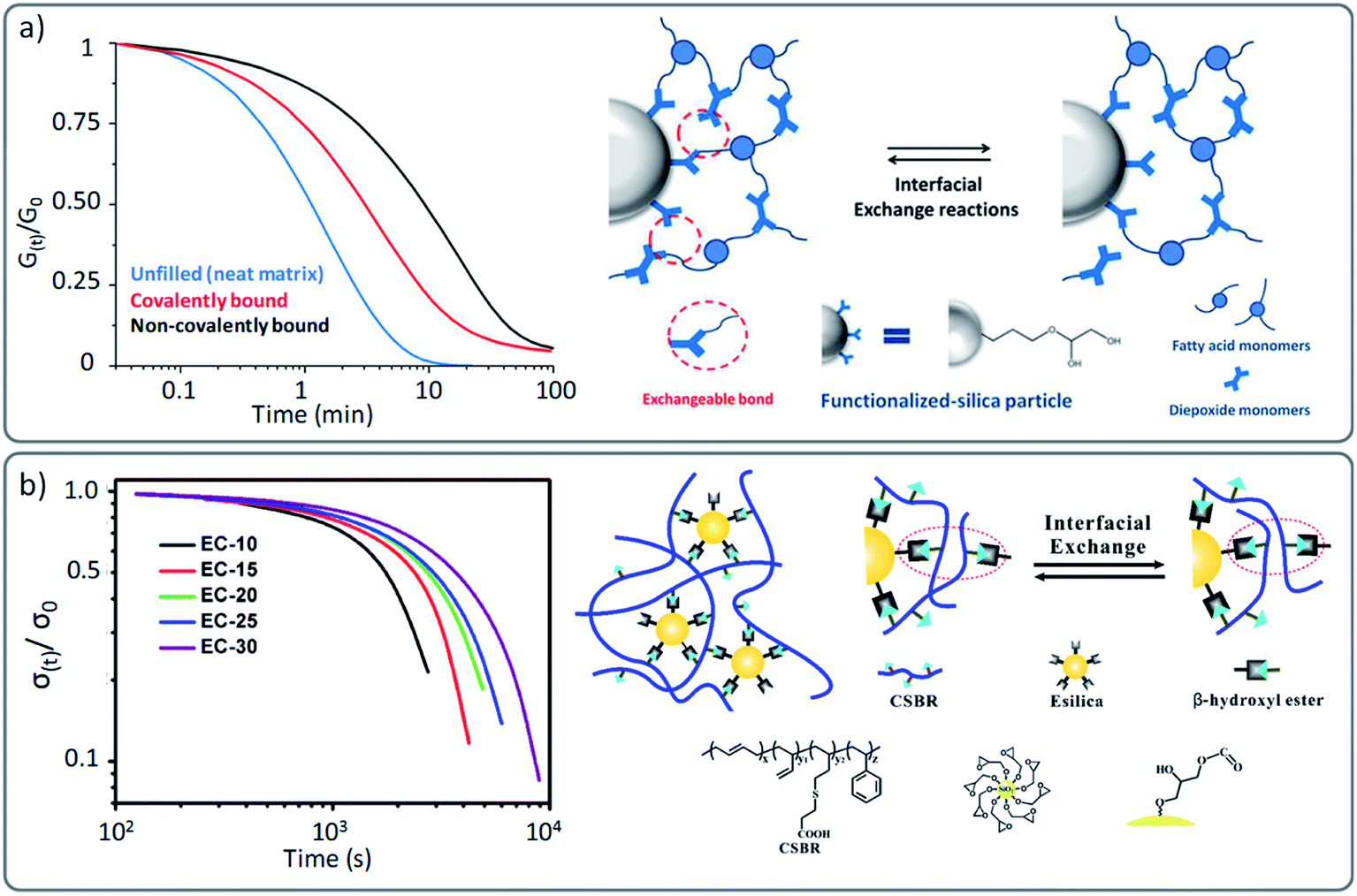 | ||
| Fig. 8 (a) Stress relaxation of vitrimers and vitrimer nanocomposites: neat matrix (blue), filled with epoxide-functionalised fillers (covalently bond) and non-functionalised silica particles (non-covalently bond). Schematic representation of interfacial exchange reactions of covalently bound fillers. Adapted with permission from ref. 82. Copyright 2016 American Chemical Society. (b) Stress relaxation of filled vitrimer nanocomposites with a different ratio of covalently bound fillers, resulting in different cross-link density and relaxation behaviour (from the least (EC10) to the highest cross-linked network (EC30)). Adapted with permission from ref. 84. Copyright 2018 American Chemical Society. | ||
It is also important to point out that a “cross-link density effect” has been reported in nanocomposites in analogy with non-reinforced networks (see previous section). Guo and co-workers reported in different studies a gradual decrease of stress relaxation as a function of the amount of added covalently bound fillers (Fig. 8b).84,91 This effect can be interpreted as localized multiple cross-linking points that hinder the overall macroscopic flow and thus result in a denser network with slower stress relaxation. Next to the expected effects on mechanical properties, the introduction of fillers can also be a good way to tune the viscoelastic properties (and thus reactivity) of vitrimers. We recently demonstrated this by using vinylogous urethane PDMS-based vitrimers.87 In this work, the nature of the filler was also found to effect the dynamics. Although neither the relaxation time nor the creep resistance of the material were barely varied, these results are a promising and elegant starting point for further research which takes advantage of functional fillers as both reinforcing agents and catalysts. In other studies, also the temperature dependence was found to be affected by fillers in vitrimer (nano)composites. For instance, the apparent activation energy of the earlier described hydroxyurethane vitrimers developed by Torkelson and co-workers was affected by the introduction of silica fillers (135 kJ mol−1 for a neat vitrimer compared to 142–155 kJ mol−1 for a filled vitrimer).86
In conclusion, the reinforcement of vitrimer materials seems to be highly challenging yet interesting, and cross-influences of mechanical reinforcement and good reprocessability need to be considered carefully.
Vitrimers and their topology freezing temperature (Tv)
Since the introduction of the concept of vitrimer materials, the polymer community has struggled with the contemporaneous introduction of the topology freezing temperature (Tv) of vitrimers. This was somewhat arbitrarily introduced as a temperature below which chemical exchanges would become negligible, taking the value of 1012 Pa s as an ‘upper limit’ for the viscosity. Quite unlike physics, chemistry tends not to show phase transitions, and any temperature relating to a chemical phenomenon is necessarily arbitrary and context-dependent. Very highly viscous may not be quite viscous enough for some circumstances. However, the conceptual grounds for the definition of a Tv are quite sound: above this temperature, dynamic covalent exchanges become relevant for the experimentally observed time frames, and below this temperature, they are not relevant. This is clearly not a binary situation, but a spectrum, and is also highly dependent on the time frame that is relevant for your experiment. Treating this temperature as an absolute threshold, or even as an experimentally verifiable physical constant, can be highly misleading. By convention, Tv is determined as a hypothetical value, which is obtained by extrapolating viscosity trends into regions where viscosity almost by definition becomes unmeasurable within reasonable experimental time frames. This extrapolation sometimes spans more than 100 K (see Fig. 9), making a reasonable comparison of Tv-values between different systems problematic and perhaps unwarranted.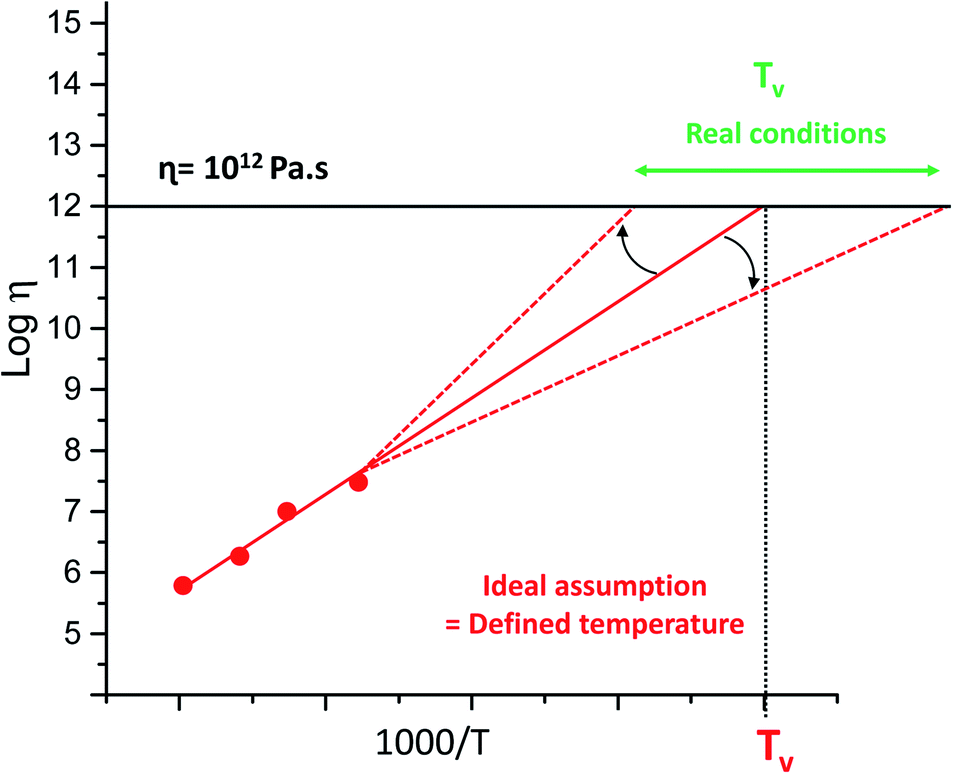 | ||
| Fig. 9 Example of imperfect evaluation of the topology freezing temperature (Tv) caused by measurement errors, based on standard deviations and small temperature window or unconsidered low temperature interactions. | ||
This extrapolation also hinges on the fundamental assumption that the viscosity will continue to show the same temperature dependency, whereas both physical and chemical factors have now been shown to affect the actual viscosity profile, giving off-linear deviations in both possible directions. However, this extrapolation is theoretically interesting, as it has something to say about the temperature at which a given exchange mechanism will cease to be relevant for stress relaxations, but Tv will often be unobtainable from empirical data. Thus, this temperature should not be treated as a temperature belonging to a material, but rather as a temperature belonging to the specific dynamic exchange mechanism present within the particular material, which may or may no longer be relevant for the material properties at a given temperature. Indeed, we have found instances where the mechanism is ‘overtaken’ by a different exchange pathway that is not relevant at higher temperatures, but gains importance at lower ones due to a lower temperature dependence related to a lower activation energy (Tv right shift in Fig. 9).38
Another well-known limitation of Tv, which was already addressed by Leibler and in our original mini-review,17 is the fact that as the polymer viscosity increases upon cooling, the observed macroscopic flow is inhibited because of the decrease in mobility of chains and functional groups, and chemistry no longer is the rate-limiting step in overall polymer dynamics (Tv left-shift Fig. 9). However, when one material matrix is used, with a similar cross-linking-density and exchange chemistry, a value for Tv is indeed a simple and intuitive measure for comparing the vitrimer-type bond exchanges, giving a single value qualitative measure of the exchange dynamics at lower temperatures. In our opinion, a better definition of Tv than that originally introduced cannot be given, and should not be given, as long as it is clear what this conceptual value means. For example, it is interesting to compare its value to Tg of a material, in order to quickly determine what will be the more important phenomenon governing polymer chain dynamics: exchange chemistry or intermolecular forces.
Hence, the Tv was mainly introduced in order to give an estimation of the relative importance of a given dynamic covalent exchange mechanism, especially relevant for the consideration of low Tg materials.
Indeed, the Tv in high Tg materials is of little interest since the exchange moieties are frozen by the slow segmental motion associated to the glass transition.17 When considering Tv in low Tg materials, we recommend the researcher not to focus too much on these numbers, but to rather perform stringent creep experiments at low temperature instead of relying on extrapolations from high temperature, high frequency stress-relaxation experiments.38
Conclusions
While our previous mini-review highlighted for the first time the early chemical developments surrounding the intriguing concept of vitrimers, and described how different reversible bond exchange reactions can be used to achieve different vitrimer properties and characteristics, this review aimed to provide a complementary summary of different governing parameters. These context-dependent chemical parameters are often overlooked or at least not fully appreciated, and can nevertheless offer excellent handles to control and direct material properties towards useful applications. In order to arrive at applications that will help us transition from a world where plastic waste is treated as a nuisance and a hazard to a situation where they are treated for the valuable raw materials that they truly are. Polymer chemists, physicists and engineers will need to team up to tackle the fundamental scientific challenges, and figure out how to understand, and ultimately control and predict this promising new class of polymers.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
C. T. acknowledges the Research Foundation-Flanders (FWO) for the PhD fellowship. The authors would also like to thank Dr Nezha Badi for valuable input. F. D. P. thanks BOF-UGent (GOA-funding). We thank Prof. Ludwik Leibler for many helpful discussions over the last years.References
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- C. Zhang, Z. Yang, N. T. Duong, X. Li, Y. Nishiyama, Q. Wu, R. Zhang and P. Sun, Macromolecules, 2019, 52, 5014–5025 CrossRef CAS.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- C. M. Moorhoff, W. D. Cook, F. Chen, D. Nghiem, C. Braybrook, S. H. Thang, J. Sun, T. F. Scott and C. N. Bowman, Aust. J. Chem., 2011, 64, 1083–1093 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 1906876 CrossRef PubMed.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 101233 CrossRef.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- A. Demongeot, S. J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Polym. Chem., 2016, 7, 4486–4493 RSC.
- W. Liu, D. F. Schmidt and E. Reynaud, Ind. Eng. Chem. Res., 2017, 56, 2667–2672 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- H. Zhang, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 272–277 CrossRef CAS.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- M. Liu, J. Zhong, Z. Li, J. Rong, K. Yang, J. Zhou, L. Shen, F. Gao, X. Huang and H. He, Eur. Polym. J., 2020, 124, 109475 CrossRef CAS.
- R. Martin, A. Rekondo, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, J. Mater. Chem. A, 2014, 2, 5710–5715 RSC.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193–199 CrossRef CAS.
- Y. Liu, Z. Tang, D. Wang, S. Wu and B. Guo, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- T. Wright, T. Tomkovic, S. G. Hatzikiriakos and M. O. Wolf, Macromolecules, 2019, 52, 36–42 CrossRef CAS.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 255–260 CrossRef CAS.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, submitted.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- X. Chen, L. Li and J. M. Torkelson, Polymer, 2019, 178, 121604 CrossRef CAS.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Macromolecules, 2019, 52, 432–443 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 RSC.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- Y. Spiesschaert, M. Guerre, I. De Baere, W. Van Paepegem, J. M. Winne and F. E. Du Prez, Macromolecules, 2020, 53(7), 2485–2495 CrossRef.
- L. Matějka, S. Pokomý and K. Dušek, Polym. Bull., 1982, 7, 123–128 Search PubMed.
- I. F. Altuna, E. C. Hoppe and J. R. Williams, Polymers, 2018, 10, 43 CrossRef PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047–2056 RSC.
- M. Hayashi and R. Yano, Macromolecules, 2020, 53, 182–189 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- F. Caffy and R. Nicolaÿ, Polym. Chem., 2019, 10, 3107–3115 RSC.
- J. Tellers, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polym. Chem., 2019, 10, 5534–5542 RSC.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- Y. Zhou, J. G. P. Goossens, S. van den Bergen, R. P. Sijbesma and J. P. A. Heuts, Macromol. Rapid Commun., 2018, 39, 1800356 CrossRef PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- Y. Yang, F.-S. Du and Z.-C. Li, Polym. Chem., 2020, 11, 1860–1870 RSC.
- F. Lossada, D. Jiao, X. Yao and A. Walther, ACS Macro Lett., 2020, 9, 70–76 CrossRef CAS.
- Z. Wang, Y. Gu, M. Ma and M. Chen, Macromolecules, 2020, 53, 956–964 CrossRef CAS.
- W. L. Peng, Y. You, P. Xie, M. Z. Rong and M. Q. Zhang, Macromolecules, 2020, 53, 584–593 CrossRef CAS.
- D. A. Kissounko, P. Taynton and C. Kaffer, Reinf. Plast., 2018, 62, 162–166 CrossRef.
- T. Philip, N. Huagang, Z. Chengpu, Y. Kai, L. Samuel, J. Yinghua, Q. H. Jerry and Z. Wei, Adv. Mater., 2016, 28, 2904–2909 CrossRef PubMed.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabanero, J. Rodriguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, W. Yuan, S. Zhou, S. You and J. Zhu, Green Chem., 2019, 21, 1484–1497 RSC.
- L. Yu, C. Zhu, X. Sun, J. Salter, H. Wu, Y. Jin, W. Zhang and R. Long, ACS Appl. Polym. Mater., 2019, 1, 2535–2542 CrossRef CAS.
- A. Legrand and C. Soulié-Ziakovic, Macromolecules, 2016, 49, 5893–5902 CrossRef CAS.
- Z. Huang, Y. Wang, J. Zhu, J. Yu and Z. Hu, Compos. Sci. Technol., 2018, 154, 18–27 CrossRef CAS.
- Y. Liu, Z. Tang, Y. Chen, C. Zhang and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 2992–3001 CrossRef CAS PubMed.
- C. Xu, R. Cui, L. Fu and B. Lin, Compos. Sci. Technol., 2018, 167, 421–430 CrossRef CAS.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- Y. Spiesschaert, M. Guerre, L. Imbernon, J. M. Winne and F. Du Prez, Polymer, 2019, 172, 239–246 CrossRef CAS.
- S. Wu, Z. Yang, S. Fang, Z. Tang, F. Liu and B. Guo, J. Mater. Chem. A, 2019, 7, 1459–1467 RSC.
- A. L. Dobson, N. J. Bongiardina and C. N. Bowman, ACS Appl. Polym. Mater., 2020, 2(3), 1053–1060 CrossRef CAS.
- Y. Yang, Z. Pei, X. Zhang, L. Tao, Y. Wei and Y. Ji, Chem. Sci., 2014, 5, 3486–3492 RSC.
- Z. Tang, Y. Liu, B. Guo and L. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS.
- F. Zhou, Z. Guo, W. Wang, X. Lei, B. Zhang, H. Zhang and Q. Zhang, Compos. Sci. Technol., 2018, 167, 79–85 CrossRef CAS.
- M. Qiu, S. Wu, S. Fang, Z. Tang and B. Guo, J. Mater. Chem. A, 2018, 6, 13607–13612 RSC.
- G. Zhang, X. Zhou, K. Liang, B. Guo, X. Li, Z. Wang and L. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 11712–11720 CrossRef CAS.
- Z. Tang, Q. Huang, Y. Liu, Y. Chen, B. Guo and L. Zhang, ACS Macro Lett., 2019, 8, 1575–1581 CrossRef CAS.
- Y. Liu, Z. Tang, Y. Chen, S. Wu and B. Guo, Compos. Sci. Technol., 2018, 168, 214–223 CrossRef CAS.
- G. Chen, K. Wu, Q. Zhang, Y. Shi and M. Lu, Macromol. Res., 2019, 27, 526–533 CrossRef CAS.
- J. Chen, H. Huang, J. Fan, Y. Wang, J. Yu, J. Zhu and Z. Hu, Front. Chem., 2019, 7, 632 CrossRef CAS PubMed.
- B. Krishnakumar, R. V. S. Prasanna Sanka, W. H. Binder, C. Park, J. Jung, V. Parthasarthy, S. Rana and G. J. Yun, Composites, Part B, 2020, 184, 107647 CrossRef.
- F. Lossada, J. Guo, D. Jiao, S. Groeer, E. Bourgeat-Lami, D. Montarnal and A. Walther, Biomacromolecules, 2019, 20, 1045–1055 CrossRef CAS PubMed.
- C. Xu, Z. Zheng, W. Wu, L. Fu and B. Lin, Compos. Sci. Technol., 2019, 181, 107677 CrossRef CAS.
- D. Zhou, Y. Wang, J. Zhu, J. Yu and Z. Hu, Polymer, 2019, 167, 202–208 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |