
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of theranostic nanocapsules with lanthanide doped nanoparticles†
Miao
Wang
 ae,
Yu
Zhang
b,
Michael
Ng
c,
Artiom
Skripka
a,
Ting
Cheng
a,
Xu
Li
abf,
Kishore Kumar
Bhakoo
c,
Alex Y.
Chang
de,
Federico
Rosei
a and
Fiorenzo
Vetrone
*a
ae,
Yu
Zhang
b,
Michael
Ng
c,
Artiom
Skripka
a,
Ting
Cheng
a,
Xu
Li
abf,
Kishore Kumar
Bhakoo
c,
Alex Y.
Chang
de,
Federico
Rosei
a and
Fiorenzo
Vetrone
*a
aInstitut National de la Recherche Scientifique (INRS), Centre Énergie, Matériaux et Télécommunications, Université du Québec, Varennes (QC), Canada. E-mail: vetrone@emt.inrs.ca
bInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), Singapore
cSingapore Bioimaging Consortium (SBIC), Agency for Science, Technology and Research (A*STAR), Singapore
dDepartment of Oncology, Johns Hopkins University, Baltimore, Maryland, USA
eJohns Hopkins Singapore, Singapore
fDepartment of Chemistry, National University of Singapore, Singapore
First published on 1st June 2020
Abstract
We report a one-pot synthesis strategy for a new theranostic nanoplatform by simultaneously encapsulating Er3+, Yb3+ doped NaGdF4 upconverting nanoparticles (UCNPs) and photosensitizer zinc phthalocyanine (ZnPc) into polymeric micelle/silica nanocapsules. This approach consists of interfacial templating condensation, using triblock copolymers, namely (ethylene oxide)106(propylene oxide)70(ethylene oxide)106 (PEO-PPO-PEO) as the templating and protecting agent. The encapsulation follows a straightforward microemulsion mechanism in an aqueous environment at a near-neutral pH. To prevent the interaction between the hydrophobic oleic acid (OA) ligands of UCNPs and the silanol groups of hydrated tetramethoxysilane (TMOS), we adjusted the addition sequence of TMOS. It allowed us first to encapsulate UCNPs in PEO-PPO-PEO micelles, and then grow the silica shell within the micellar PPO core and PEO corona interface. The silica shell is incorporated for its chemical and mechanical stability, while the PEO corona gives additional steric balance to the nanocapsule. Using this strategy we successfully co-encapsulated UCNPs and ZnPc in one-pot, and minimized the distance between the two payloads to facilitate the energy transfer from UCNPs to ZnPc. The integrated nanocapsule has an average hydrodynamic size of 85 nm with a low polydispersity index of 0.1, and demonstrates excellent colloidal stability, biocompatibility, enhanced negative contrast for T2-weighted imaging and photodynamic therapy. The latter is obtained through indirect excitation of co-encapsulated ZnPc by UCNPs, resulting in singlet oxygen generation and in vitro eradication of BT474 breast cancer cells. Overall, the presented one-pot approach shines light on the co-encapsulation of OA-capped inorganic UCNPs with hydrophobic photosensitizers, constituting an important step forward in the surface engineering of UCNPs, as well as upconversion based photodynamic therapy systems.
1 Introduction
Photodynamic therapy (PDT) has been widely studied for cancer treatment including skin, prostate, head and neck, pancreatic, breast, and lung cancer1–4 and utilizes photosensitizers (PS) as light-sensitive drugs to locally treat the target tissue upon irradiation with light of appropriate wavelengths. The excited PS interacts with surrounding oxygen molecules to generate reactive oxygen species (ROS), including singlet oxygen (1O2), which causes oxidative damage to biological substrates and ultimately cell death.5 In addition to having less systemic side effects and cumulative cytotoxicity, PDT is able to overcome the multi-drug resistance caused by chemotherapy.6 Moreover, unlike chemotherapeutic drugs and radiotherapy that tend to be immunosuppressive, PDT is capable of triggering an antitumor immune response by activation of the innate and/or adaptive immune system, subsequently prolonging survival of patients.6,7 However, the efficacy of PS drugs is severely constrained by their inherent water-insolubility and poor pharmacokinetics.8,9 Moreover, the typical PS excitation wavelengths in the visible spectral range have limited tissue penetration depth. For this reason, PDT is limited to the treatment of tumours on or just under the skin, the lining of internal organs or cavities, thus being less effective in treating large and deep-seated tumours.10 In order to reach deep lying tumours, near-infrared (NIR) light with wavelengths in the optical transparency windows of biological tissues could be used.11,12 However, most efficient PS cannot be excited with NIR light directly (e.g. MC540, Ce6, ZnPc, etc.).13 To overcome this crucial challenge, the synergistic combination of photon upconverting materials and PDT drugs has been proposed and explored in recent years.14–20Lanthanide (Ln3+)-doped upconverting nanoparticles (UCNPs) have been widely studied for PDT because of their unique ability to convert NIR radiation to UV and/or visible wavelengths via the anti-Stokes, multiphoton process of upconversion.21,22 UCNPs decorated with a PS form the core of a theranostic (therapy + diagnostic) nanoplatform capable of both PDT (therapy) and imaging (diagnostic). In PDT, UCNPs play the role of energy transducers that facilitate indirect excitation of the attached PS under NIR stimulus. Not only do UCNPs enable PDT at greater tissue depth, but also provide the added benefit of disease visualization (e.g. optical and/or magnetic resonance imaging [MRI]).15,23–30 However, high quality UCNPs are usually synthesized in an organic environment with oleic acid (OA) and/or oleylamine (OM) as capping ligands, which prevents UCNPs from being readily dispersible in an aqueous milieu.31 The majority of efficient PS drugs are also hydrophobic in nature.9 To provide aqueous dispersity and stability under physiological conditions, surface engineering undoubtedly becomes a crucial step to enable the use of UCNPs in PDT (UC-PDT).
To date, several major strategies have been explored to combine PS and UCNPs, of which mesoporous silica coating is a common and well-studied method, which allows for high PS payloads.13,15,18,32,33 A typical process of mesoporous silica coating involves the self-assembly of cationic surfactants and silica precursors around the UCNPs in a basic solution, where the surfactants are used as a template for the formation of the silica shell. The mesopores are generated after subsequent removal of the surfactant micelles by either thermal decomposition or solvent extraction.34 The PS of interest is then loaded into the pores. However, leakage of the PS in the blood circulatory system is inevitable without capping the pores, which otherwise leads to potential whole-body phototoxicity and inadequate accumulation of the PS in the targeted tissue.13 Cetyltrimethylammonium bromide (CTAB), a surfactant, which is commonly employed to generate mesopores, is also highly toxic.22 Therefore, any incomplete removal of CTAB can impede the use of UCNPs in biomedicine. In addition, mesoporous silica coating requires a multi-step preparation as addressed above. Finally, additional surface modification of the silica shell is required to prevent silica coated UCNPs from aggregating.9,23
To enable a simpler one-pot synthesis of the UC-PDT system, another strategy known as polymer encapsulation has been proposed, in which amphiphilic polymers self-assemble into a micelle with both the UCNPs and PS based on their mutual hydrophobicity.26,35,36 While it is a versatile and cost-effective approach, using polymeric micelles does not address the problem of leakage. The polymeric micelles spontaneously dissociate when their concentrations fall below the critical micelle concentration (CMC), a scenario which such PS carriers encounter when diluted by several orders of magnitude upon intravenous delivery in vivo.37 As a result, the micelles could disintegrate and precipitate out their content.
To address the aforementioned issues, a silica–polymer hybrid system could be applied. The basic idea of this one-pot encapsulation is to confine the hydrolysis and condensation of the silica precursors to the core/corona interface of amphiphilic triblock copolymer [(ethylene oxide)106(propylene oxide)70(ethylene oxide)106 (PEO-PPO-PEO)] micelles. This forms a silica shell cross-linked micelle where the polymeric micelle is stabilised by silica cross-linking to prevent its disintegration. The free dangling PEO blocks can also prevent unwanted aggregation of the silica shells. In addition, the silica cross-linking provides a more effective diffusion channel for the movement of water and oxygen in and out of the silica shell without the need for generating pores. Importantly, in UC-PDT systems cytotoxic 1O2 is generated by PS which are activated via energy transfer (ET) from the NIR excited UCNPs. The ET can be either radiative14,18,26,35,36 or non-radiative,16,38–41 and its efficiency is affected by the distance between the UCNPs (energy donors) and PS (energy acceptors). To that end, co-encapsulation of UCNPs and PS in polymeric micelle/silica nanocapsules advantageously minimizes the distance between the two. The UCNP and the PS could be virtually in contact with each other, while the conventional approach of PS loading in the mesoporous silica coating results in greater separation between the two species.15,18,20,42
Here, we established an original one-pot synthesis strategy to co-encapsulate hydrophobic NaGdF4:Er3+ (2 mol%), Yb3+ (20 mol%) UCNPs and ZnPc (zinc phthalocyanine, hydrophobic PS) via the interfacial templating condensation approach. The resultant theranostic nanocapsules exhibited excellent colloidal stability, biocompatibility, and a PDT effect against BT474 chemo-resistant breast cancer cells. In addition, the encapsulated NaGdF4 UCNPs demonstrated promising negative contrast enhancement for T2 weighted MRI, to be used for imaging guided UC-PDT.
2 Results and discussion
2.1. Mechanism of polymeric micelle/silica co-encapsulation of UCNPs and PS by interfacial templating condensation
Scheme 1 illustrates the co-encapsulation of UCNPs and PS drugs via interfacial templating condensation. It is a single step approach for the synthesis of polymeric micelle/silica nanocapsules that can deliver UC-PDT and MRI at the same time. To achieve core/shell/corona nanocapsules, amphiphilic PEO-PPO-PEO triblock copolymers (F127), hydrophobic UCNPs and ZnPc are initially mixed in tetrahydrofuran (THF), a water-miscible organic solvent (Scheme 1a). The mixture is then injected into water where F127 will spontaneously self-assemble into micelles composed of a hydrophobic PPO core and a hydrophilic PEO corona. Simultaneously, UCNPs and ZnPc are localized in the PPO core due to their hydrophobic nature (Scheme 1b). Subsequently, silica precursors tetramethoxysilane (TMOS) are injected into the aqueous solution. Upon contact with water, TMOS starts to hydrolyse and condense at the interface of micellar core/corona, forming an ultrathin silica shell adjoining the micellar core (Scheme 1c). As the reaction proceeds, the unreacted silica precursors further hydrolyse and condense onto the outer surface of the silica shell, thickening it. Core/shell/corona nanostructures are eventually formed. The UCNPs and ZnPc are encapsulated in the micellar core, with the silica shell confined at the micellar core/corona interface, and free PEO chains that make up the corona protruding out into the water (Scheme 1d). This approach allowed for (1) integrating the cargo and carrier in one-pot; (2) high aqueous colloidal stability, antifouling and steric stabilization provided by the free PEO chains;43,44 (3) synthesis to be carried out in benign environments (in a near-neutral pH aqueous solution and at room temperature); (4) ZnPc to be brought closer to UCNPs, thus facilitating energy transfer from UCNPs to ZnPc; and (5) the control over the number of UCNPs encapsulated inside the nanocapsules. In addition, the silica shell surrounding the core of the polymeric micelles efficiently prevented the micelles from disintegrating even when the polymer concentration fell below its CMC,37,43 thus reducing the likelihood of ZnPc leaking.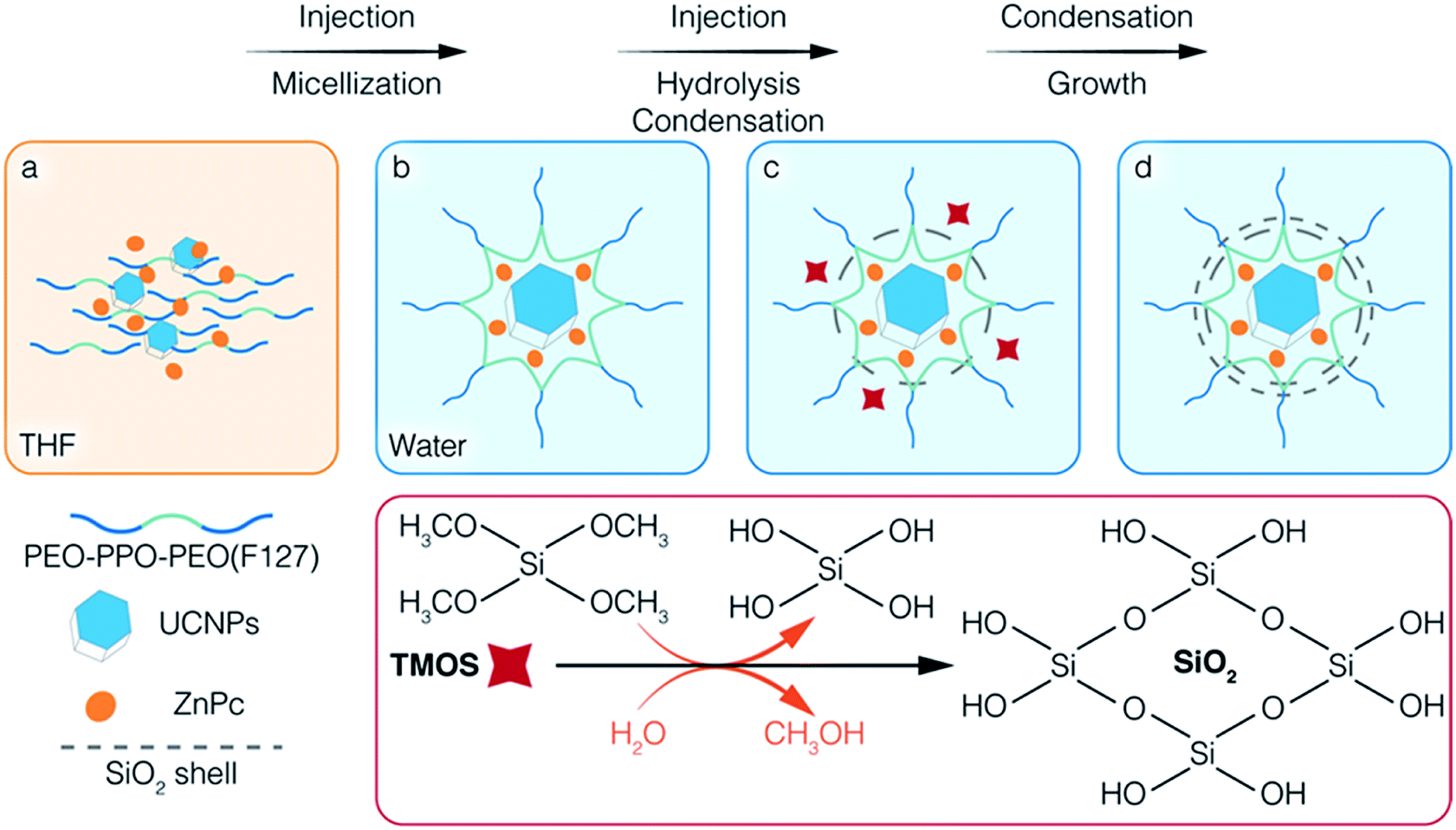 | ||
| Scheme 1 The formation of polymeric micelle/silica nanocapsules co-encapsulating UCNPs and organic PS drugs (ZnPc) via the interfacial templating condensation approach. The chemical equation illustrates the hydrolysis and self-condensation of silica precursor TMOS in water. | ||
2.2. Influence of TMOS on the encapsulation
According to the reported encapsulation protocol,43 F127, TMOS and payload mixture are injected simultaneously into water, where the payloads are encapsulated into the core of the F127 micelles and the silica precursors condense at the confined interface between the core and corona of the micelles. It was successful for the encapsulation of hydrophobic PS drugs, such as ZnPc; however, when we attempted to encapsulate UCNPs using the same protocol, the mixture became cloudy and precipitated. TEM images confirmed that highly aggregated UCNPs precipitated due to improper micellization (Fig. S1A–D†). Unlike organic molecules, the hydrophobicity of UCNPs originates from the OA capping ligand. The carboxylic end of OA is usually coordinated to the outermost Ln3+ ions by electrostatic interactions, which is also confirmed by the asymmetric stretching of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH from the FTIR results (Fig. 1C). When TMOS and OA-capped UCNPs were mixed with THF, TMOS hydrolysed to form silanol groups upon contact with the trace water in THF. The carboxylic groups (COOH) of OA are better anchoring points for the silanol groups via hydrogen bonding compared to the C–O groups of PEO.45 As a result, one or two Si–OH of silanol interacted with the COOH of OA-capped UCNPs, and others linked with each other to form a bridge between silanols (–O–Si–O–Si–), leading to the aggregation and precipitation of the UCNPs. On the other hand, the silanol groups might increase the hydrophilicity of surrounding UCNPs, resulting in the deficient absorption of UCNPs in the PPO core. Consequently, once such a mixture was injected into water, the preformed and newly formed silanol linking hampered the micellar encapsulation of the UCNPs. Based on this understanding, we modified the procedure by encapsulating UCNPs into the hydrophobic core prior to growing the silica shell.
C–OH from the FTIR results (Fig. 1C). When TMOS and OA-capped UCNPs were mixed with THF, TMOS hydrolysed to form silanol groups upon contact with the trace water in THF. The carboxylic groups (COOH) of OA are better anchoring points for the silanol groups via hydrogen bonding compared to the C–O groups of PEO.45 As a result, one or two Si–OH of silanol interacted with the COOH of OA-capped UCNPs, and others linked with each other to form a bridge between silanols (–O–Si–O–Si–), leading to the aggregation and precipitation of the UCNPs. On the other hand, the silanol groups might increase the hydrophilicity of surrounding UCNPs, resulting in the deficient absorption of UCNPs in the PPO core. Consequently, once such a mixture was injected into water, the preformed and newly formed silanol linking hampered the micellar encapsulation of the UCNPs. Based on this understanding, we modified the procedure by encapsulating UCNPs into the hydrophobic core prior to growing the silica shell.
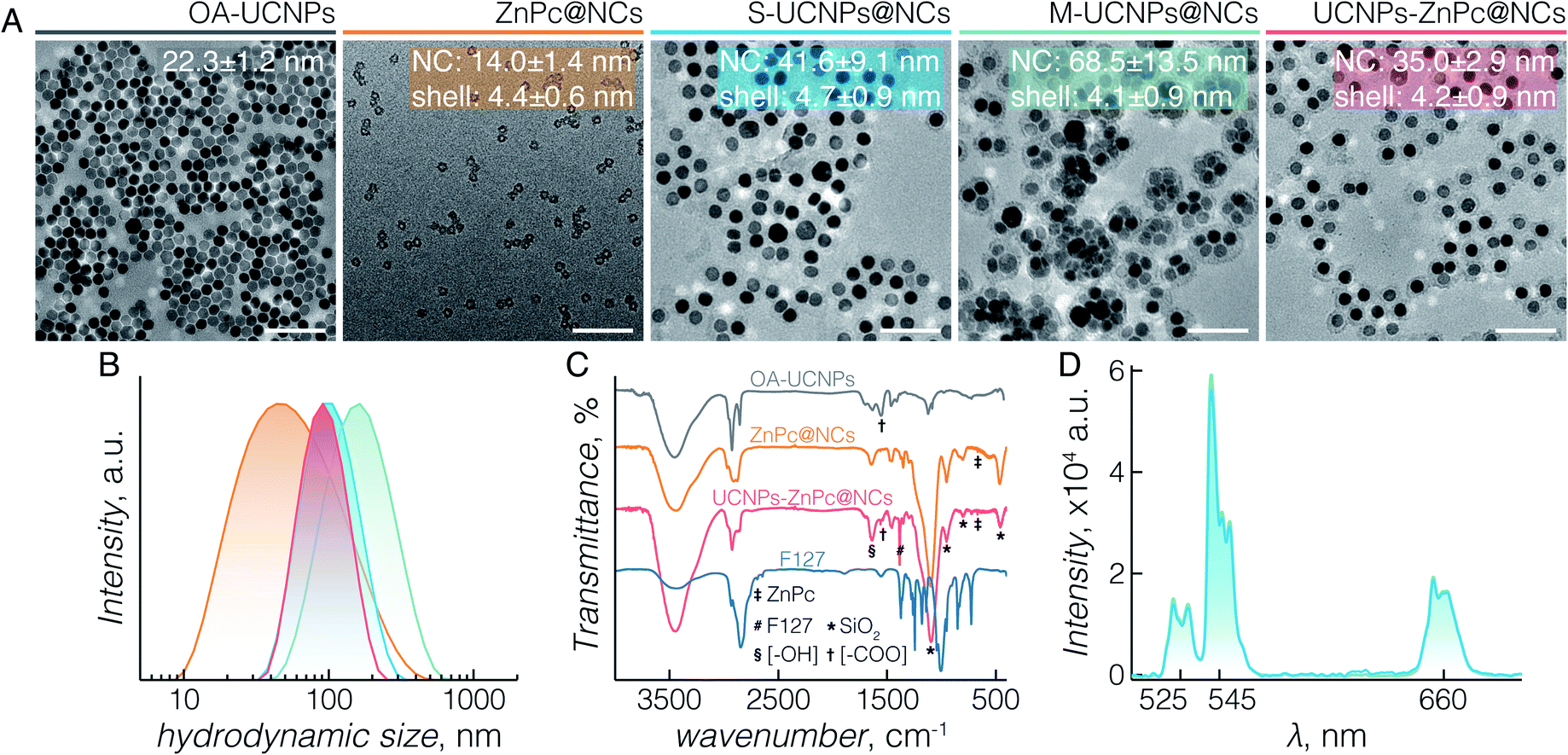 | ||
| Fig. 1 (A) TEM images demonstrating the various compounds studied. “NC” indicates the longest diameter of an individual nanocapsule; “shell” indicates the thickness of the silica shell. Estimated size is expressed as mean ± standard deviation (SD). Scale bars in all images correspond to 100 nm. (B) The hydrodynamic size of ZnPc@NCs, S-UCNPs@NCs, M-UCNPs@NCs, and UCNPs-ZnPc@NCs. (C) The organic/inorganic hybrid structure of the studied compounds characterized by FTIR analysis. (D) The upconversion luminescence spectra of S-UCNPs@NCs and M-UCNPs@NCs upon 980 nm laser irradiation. Note, colour coding of different compounds in (A) is maintained in (B), (C) and (D). | ||
First, only F127 and UCNPs were mixed in THF, injected into water, and left to stir for 2 days to encapsulate UCNPs into the PPO core through the micellization of F127. Then, TMOS dissolved in THF was injected into the micellar solution, in which the not fully hydrolysed TMOS diffused to the micelle and bonded to PEO chains through hydrogen bonding.46 Subsequently, the silanol groups condensed at the interface region to form a thin silica shell surrounding the core of the micelles. With the modified protocol we were successful in preventing the interaction between OA and silanol, and to achieve the formation of core/shell/corona nanocapsules loaded with OA-capped UCNPs. Moreover, we were able to control the multiplicity of UCNPs per nanocapsule by varying the concentration of the reactants. By adjusting the ratio of UCNPs/F127 added during the encapsulation, nanocapsules loaded with a different number (n) of UCNPs can be obtained, namely S-UCNPs@NCs and M-UCNPs@NCs synthesized with 1.7 × 1016 and 2.5 × 1016 UCNPs per mmol of F127, respectively. A clear stock solution of UCNPs, which is characteristic of stable and well dispersed UCNPs in the solvent, is crucial for successful encapsulation. Small hydrodynamic sizes and narrow size distributions are favourable in biomedical applications, thus we synthesised UCNPs-ZnPc@NCs where ZnPc was co-encapsulated with a single UCNP as follows. At fixed concentrations of F127, TMOS, and UCNPs, various amounts of ZnPc (0.3, 0.6, and 1.2 mg) were tested for co-encapsulation with UCNPs in the PPO core. From the TEM characterization, we observed that monodisperse nanocapsules can only be obtained by loading 0.3 mg ZnPc (Fig. 1A_UCNPs-ZnPc@NCs and Fig. S1E and F†). Increasing the loading amount to 0.6 and 1.2 mg caused ZnPc to be encapsulated alone without co-encapsulation with UCNPs (see the “empty” nanocapsules shown in Fig. S1G and H†). The loading efficiency for UCNPs-ZnPc@NCs is 1.2% (w/w), which is comparable with the previous literature values, such as encapsulation of UCNPs and ZnPc in lipid micelles [1.6% (w/w)],47 and loading of ZnPc in mesoporous silica decorated UCNPs [1.8% (w/w)].20
2.3. Characterization of hybrid nanocapsules
TEM images (Fig. 1A) demonstrate the morphologies of OA-capped UCNPs (grey), ZnPc@NCs (orange), S-UCNPs@NCs (blue), M-UCNPs@NCs (green), and UCNPs-ZnPc@NCs (red). OA-capped UCNPs are monodisperse in size and have a hexagon-shaped morphology with a size of 22 ± 1 nm. The XRD peaks (Fig. S2†) indicate a pure β (hexagonal)-phase (space group: P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) or P63/m), in excellent agreement with the reference data (JCPDS 27-699). All the nanocapsules formed are well dispersed with an ultrathin but complete silica shell of less than 5 nm thickness. The TEM size of nanocapsules is around 14 nm (ZnPc@NCs), 42 nm (S-UCNPs@NCs), 69 nm (M-UCNPs@NCs), and 35 nm (UCNPs-ZnPc@NCs). DLS analysis (Fig. 1B and Table S1†) reveals their hydrodynamic size (nm) and polydispersity indexes (PDI): ZnPc@NCs, 43/0.4; S-UCNPs@NCs, 93/0.2; M-UCNPs@NCs, 145/0.2; and UCNPs-ZnPc@NCs, 86/0.1. The PDI is an indication of the dispersity with respect to the size distribution. A value within 0.05–0.7 is considered highly monodisperse and acceptable for nanocarriers in biomedical applications.48 The contents of UCNPs and ZnPc in nanocapsules were analysed by ICP-AES (Table S2†). The amounts of nanocapsules used in this study were calculated based on the ICP data. TEM images (Fig. 1A_UCNPs-ZnPc@NCs, Fig. S1E and F†) clearly demonstrate that the UCNPs (dark colour) are centred in the core, and enclosed by the well-defined thin silica shell (∼4 nm thickness) (grey colour). UCNPs, compared with silica, have higher electron density and scatter electrons more strongly, thus appearing darker in colour. The resultant blue coloured aqueous colloid of UCNPs-ZnPc@NCs was transparent and no precipitates were observed up to 6 months of storage at room temperature. The monodispersity of the colloid was confirmed by DLS (Table S1 and Fig. S3†).
or P63/m), in excellent agreement with the reference data (JCPDS 27-699). All the nanocapsules formed are well dispersed with an ultrathin but complete silica shell of less than 5 nm thickness. The TEM size of nanocapsules is around 14 nm (ZnPc@NCs), 42 nm (S-UCNPs@NCs), 69 nm (M-UCNPs@NCs), and 35 nm (UCNPs-ZnPc@NCs). DLS analysis (Fig. 1B and Table S1†) reveals their hydrodynamic size (nm) and polydispersity indexes (PDI): ZnPc@NCs, 43/0.4; S-UCNPs@NCs, 93/0.2; M-UCNPs@NCs, 145/0.2; and UCNPs-ZnPc@NCs, 86/0.1. The PDI is an indication of the dispersity with respect to the size distribution. A value within 0.05–0.7 is considered highly monodisperse and acceptable for nanocarriers in biomedical applications.48 The contents of UCNPs and ZnPc in nanocapsules were analysed by ICP-AES (Table S2†). The amounts of nanocapsules used in this study were calculated based on the ICP data. TEM images (Fig. 1A_UCNPs-ZnPc@NCs, Fig. S1E and F†) clearly demonstrate that the UCNPs (dark colour) are centred in the core, and enclosed by the well-defined thin silica shell (∼4 nm thickness) (grey colour). UCNPs, compared with silica, have higher electron density and scatter electrons more strongly, thus appearing darker in colour. The resultant blue coloured aqueous colloid of UCNPs-ZnPc@NCs was transparent and no precipitates were observed up to 6 months of storage at room temperature. The monodispersity of the colloid was confirmed by DLS (Table S1 and Fig. S3†).
The organic/inorganic hybrid structure of the three groups of nanocapsules studied is confirmed by FTIR analysis (Fig. 1C). Characteristic frequencies of F127 triblock copolymers (2890 cm−1, C–H stretching; 1466 cm−1, C–H bending; 1382 cm−1, C–H wagging; and 843 cm−1, C–H rocking) and silica (795 cm−1, Si–O–Si symmetric stretching; 953 cm−1, Si–OH stretching; and 1056 cm−1, Si–O–Si asymmetric stretching) are present in the nanocapsules. The peak at 460 cm−1 is specific to Si–O–Si bending of silica. OA-capped UCNPs (1550 cm−1, OC–OH asymmetric stretching) and ZnPc (668 cm−1, C–H out of plane deformation) are observed within the UCNPs-ZnPc@NCs, whereas only the transmittance for ZnPc is observed for ZnPc@NCs. The characteristic bands of hydroxyl groups (3436 cm−1, O–H stretching; and 1640 cm−1, H–O–H bending) are also observed for the hybrids, indicating the presence of a large number of structural hydroxyl groups and hydroxyl groups from physically adsorbed water. In addition, the absorbance spectrum of UCNPs-ZnPc@NCs and ZnPc@NCs confirmed that ZnPc was successfully encapsulated inside the nanocapsules (Fig. S4†). Fig. 1D shows the corresponding upconversion luminescence spectra of S-UCNPs@NCs and M-UCNPs@NCs following 980 nm laser excitation. In both cases, green emission centered at 525 nm (2H11/2 → 4I15/2) and 545 nm (4S3/2 → 4I15/2), and red emission centered at 660 nm (4F9/2 → 4I15/2) were observed from the Er3+. No significant difference of emission intensity was observed between S-UCNPs@NCs and M-UCNPs@NCs. It is well known that the emission intensity of UCNPs decreases upon dispersion in aqueous media compared to that in hydrophobic solvents. The decrease is attributed to the non-radiative decay of the Ln3+ excited states caused by surface ligands and higher energy vibrational modes of water.49–52 Nonetheless, although decreased in absolute intensity, the upconversion spectra of encapsulated UCNPs (Fig. S5†) revealed no other significant differences compared to those of OA-UCNPs in hexane (Fig. S5†).53
The UCNPs used here, NaGdF4:Er3+, Yb3+, not only serve as energy donors for UC-PDT, but can also be used as a contrast agent for MRI due to the paramagnetic properties of Gd3+.11,54 In fact, we previously reported that ligand-free NaGdF4 UCNPs with sizes between 20 and 60 nm have the ability to enhance negative contrast in T2-weighted MRI.55 To elaborate on the potential of UCNPs@NCs for use in MRI, we investigated their T2 relaxivity at a 7 T magnetic field. Fig. 2A displays the concentration-dependent negative contrast gradient produced by ligand-free UCNPs, S-UCNPs@NCs, and M-UCNPs@NCs, all of which have high r2 values which attest to enhanced negative contrast in T2-weighted images (Fig. 2B). The encapsulated UCNPs demonstrated higher r2 values than ligand-free UCNPs. In UCNPs@NCs the hydrophilic PEO dangling chains facilitate the diffusion of a large amount of water molecules to the vicinity of the NaGdF4 magnetic core, which consequently results in the large T2 relaxation rate.56,57 In addition, the ultrathin porous silica shell synergizes the increase of r2 by decreasing the barrier between Gd3+ and water protons, when compared with a thick silica shell.58 We have also found that M-UCNPs@NCs exhibited slightly higher r2 and a consequent darker T2-weighted image than S-UCNPs@NCs, which could be due to the larger cluster of UCNPs in M-UCNPs@NCs. It has been reported that the r2 values can be greatly increased by clustering T2 contrast agents in reservoirs such as liposomes or micelles, and the increase of r2 is attributed to the intrinsic mechanism of spin–spin relaxation, which relies on the local concentration of the contrast agents.58–60 In this regard, the encapsulation of multiple UCNPs in nanocapsules could be an effective strategy to improve the MRI prowess of Gd3+-based UCNPs. To study the potential diagnostic application of the synthesised UCNPs-ZnPc@NCs, we evaluated their magnetic contrast enhancement by MRI in vitro. BT474 cells were incubated with a maximal benign concentration of UCNPs-ZnPc@NCs (100 μM), while non-treated cells were used as a control. Fig. S6† clearly demonstrates the enhanced negative contrast (darker T2-weighted images) at a 7 T magnetic field, indicating their potential for precisely locating the delivered payloads by MRI.
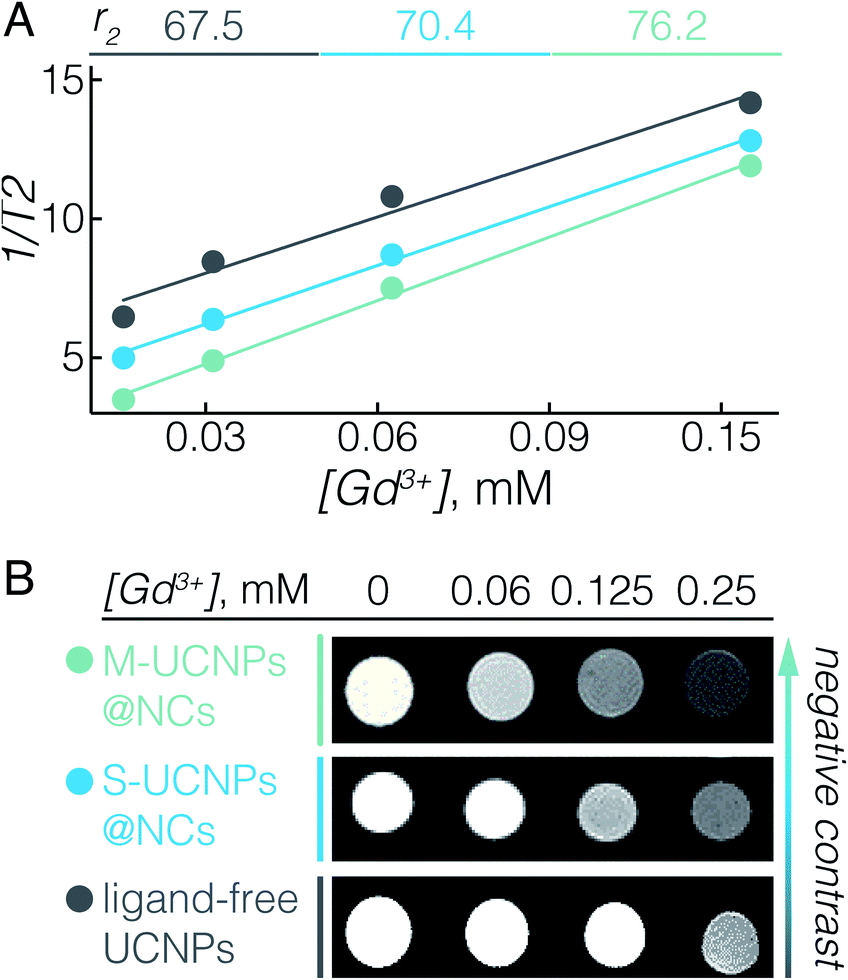 | ||
| Fig. 2 MR relaxivity r2 and T2-weighted images of ligand-free UCNPs (grey), S-UCNPs@NCs (blue) and M-UCNPs@NCs (green). (A) Plot of 1/T2versus Gd3+ concentration, the slope indicates the corresponding specific relaxivities r2. (B) The negative contrast demonstrated on T2-weighted images at a 7 T magnetic field. | ||
2.4. In vitro cytotoxicity and singlet oxygen production by UCNPs-ZnPc@NCs
To study the potential biomedical application of UCNPs-ZnPc@NCs, we first evaluated their cytotoxicity. The BT474 breast cancer cell line was used, which is resistant to chemotherapeutic drugs such as paclitaxel, as the cellular model for the in vitro study.61 In fact, paclitaxel resistance remains a major challenge in the treatment of breast cancer.62 First, the cytotoxicity of UCNPs-ZnPc@NCs, UCNPs@NCs, and ZnPc@NCs was investigated by cell proliferation assay using CCK-8. Fig. 3A demonstrates that there was no significant cell growth inhibition in all three groups up to a 100 μM concentration of UCNPs, which was selected for all further cell studies.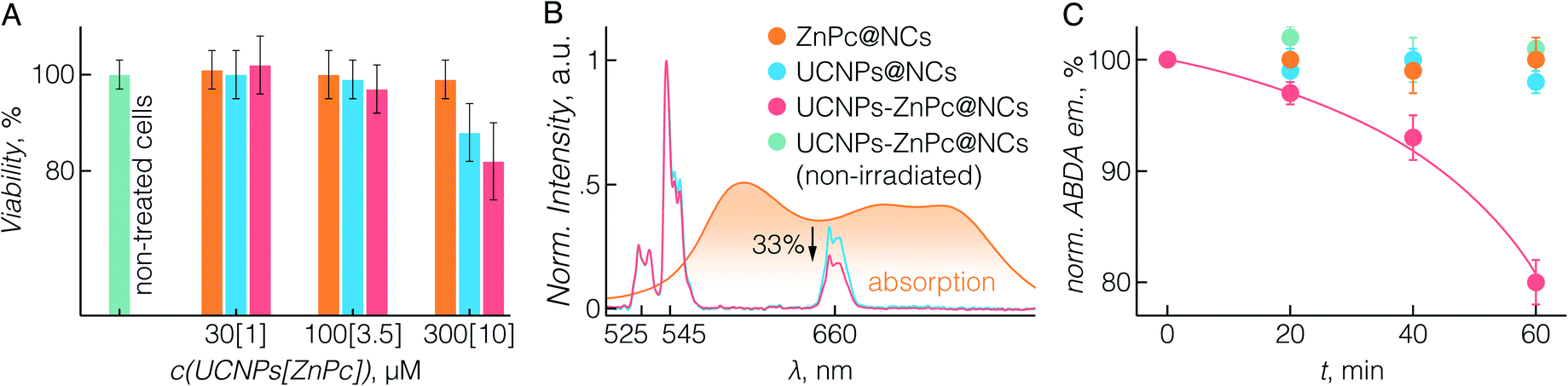 | ||
| Fig. 3 (A) Viability of untreated (green) BT474 cells, and those cultured in the presence of ZnPc@NCs (orange), UCNPs@NCs (blue), and UCNPs-ZnPc@NCs (red) for 72 h. Individual % of cell viability is the mean ± SD from three independent experiments. No significant growth inhibition was observed at or below 100 μM concentration of UCNPs (p > 0.05). (B) Upconversion luminescence spectrum of UCNPs-ZnPc@NCs and UCNPs@NCs upon 980 nm irradiation, and the absorption spectrum of ZnPc@NCs, indicating the partial spectral overlap between the ZnPc absorbance and the UCNP emission. (C) Production of 1O2 (measured using ABDA) upon irradiation for various time periods (0, 20, 40, and 60 min) with a 980 nm laser at 1.5 W cm−2 power density. Only UCNPs-ZnPc@NCs exposed to laser irradiation (red) produced 1O2, while no change in ABDA emission was observed for non-irradiated control UCNPs-ZnPc@NCs (green), or irradiated ZnPc@NCs and UCNPs@NCs. | ||
Further we sought to evaluate the 1O2 generation potential of UCNPs-ZnPc@NCs. As can be seen in Fig. 3B, the emission spectrum of UCNPs, particularly that originating from 4F9/2 → 4I15/2 transition at 660 nm, overlaps with the absorption Q-band of ZnPc. With the presence of ZnPc in the nanocapsules, we observed approximately 33% quenching of the red UC emission around 660 nm compared to significantly lower quenching of the green emission at ca. 545 nm resulting from the lower spectral overlap with the absorption of ZnPc. This indicated that ZnPc molecules were close enough to the surface of UCNPs which then could act as energy donors for ZnPc. The capacity of (1O2) production was assessed using ABDA as a probe molecule.20 As shown in Fig. 3C, three control groups were studied: ABDA incubated with UCNPs-ZnPc@NCs without laser irradiation, ZnPc@NCs with laser irradiation, and UCNPs@NCs with laser irradiation. The emission from ABDA remained constant for these control groups over time, indicating that no 1O2 was generated. However, when incubated with UCNPs-ZnPc@NCs and exposed to laser irradiation, the emission of ABDA significantly decreased demonstrating 1O2 generation by indirectly excited ZnPc molecules (p < 0.05), which indicates that there are sufficient amounts of 1O2 produced by ZnPc.63 Cumulatively, we were able to demonstrate not only the effective and simple co-encapsulation of ZnPc with UCNPs, but also the ability of this system to show photosensitizing behaviour under deep-tissue penetrating NIR excitation.
2.5. In vitro UC-PDT effect of UCNPs-ZnPc@NCs
Upon establishing the biocompatibility and 1O2 generation of UCNPs-ZnPc@NCs, we proceeded to investigate the UC-PDT effect in BT474 cancer cells (Fig. 4). To select the suitable power density for the irradiation, we first investigated the phototoxicity of 980 nm light on live cells considering the potential hyperthermia effect.64 Upon irradiation at 1.5, 3, and 6 W cm−2 power densities for 40 min, the viability of BT474 cells was 100%, 94%, and 70%, respectively (Fig. S7†). No significant damage to cells was observed upon irradiation at 1.5 W cm−2, and therefore this power density was used for subsequent irradiation experiments. Additionally, the chosen value is in the power density range reported in previous studies (usually below 2.5 W cm−2).8,15,20,36,54,65 As a control, non-treated cells and those incubated with UCNPs@NCs and ZnPc@NCs were irradiated (see the Materials and methods section in the ESI†). No significant growth inhibition was observed in these three groups (Fig. 4B). However, cells incubated with UCNPs-ZnPc@NCs and irradiated under 980 nm light had around 31% of growth inhibition at 24 h post irradiation. This indicated that the cell growth inhibition was induced by the UC-PDT effect (Fig. 4C): 1O2 produced by ZnPc caused the oxidative damage of BT474 cells. To further confirm the UC-PDT induced growth inhibition, we analysed the apoptosis induction by staining the cells with a fluorescence assay using Caspase 3/7 green.66–68 This agent is intrinsically non-fluorescent, but in the presence of activated caspase 3/7 (an indicator of apoptosis) produces a bright green-fluorescent signal (λex = 488 nm, λem = 530 nm). Following the same irradiation treatment, cells incubated with UCNPs-ZnPc@NCs, as well as the control groups, were incubated with Caspase 3/7 green, and imaged under a fluorescence confocal microscope (Fig. 4A and S8†). Green luminescence of the apoptosis marker was observed in the cells treated with UCNPs-ZnPc@NCs upon 980 nm laser irradiation compared to the other three groups, revealing the induction of apoptosis. Some green emission was also observed in the cells incubated with UCNPs@NCs. The slight apoptosis induced by UCNPs@NCs may stem from the photothermal damage of UCNPs upon 980 nm irradiation, since there was no apoptosis induction observed in both non-treated cells and those incubated with ZnPc@NCs upon irradiation. Altogether, it proved that UC-PDT can inhibit cell growth through apoptosis, even in the paclitaxel resistant breast cancer cells.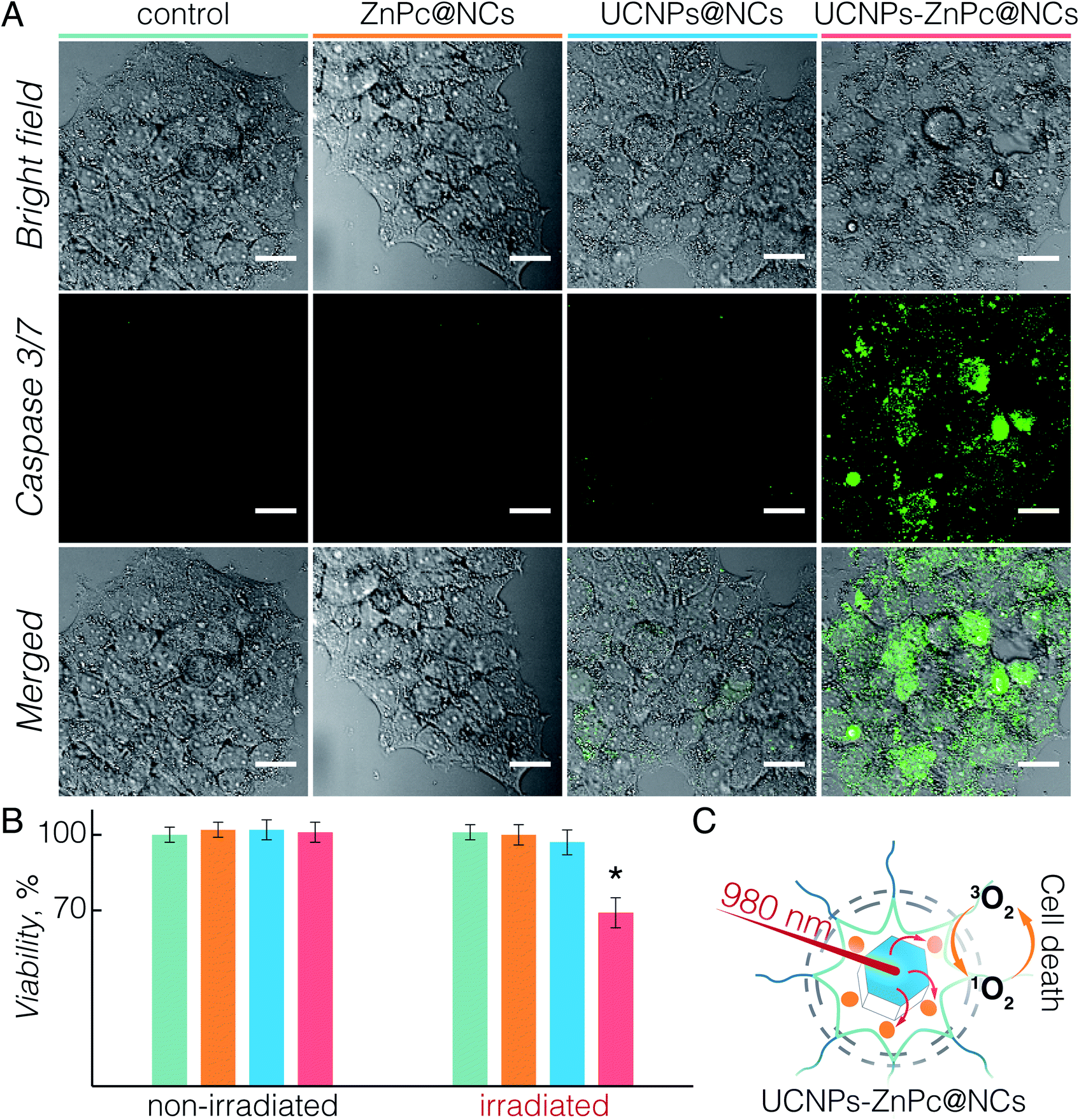 | ||
| Fig. 4 (A) Confocal fluorescence microscopy images of BT474 cells: untreated (control), incubated with ZnPc@NCs (3.5 μM), UCNPs@NCs (100 μM), and UCNPs-ZnPc@NCs (100 μM UCNPs with 3.5 μM ZnPc). Cell nuclei stained with Caspase 3/7 detection reagent (green colour) (λex = 488 nm, λem = 525 nm) indicating the apoptotic cells. Scale bars in all images correspond to 50 μm. (B) Viability of BT474 cells incubated with the studied compounds in the absence of irradiation and post 980 nm irradiation. 31% of cell growth inhibition by UCNPs-ZnPc@NCs post irradiation was observed with a p < 0.05 significance (indicated with an asterisk). Note, colour coding of different compounds in (A) is maintained in (B). (C) Scheme illustrating the UC-PDT mechanism of UCNPs-ZnPc@NCs. | ||
3 Conclusions
We developed a one-pot approach to simultaneously encapsulate Er3+, Yb3+ doped NaGdF4 UCNPs and ZnPc (PS) into polymeric micelle/silica nanocapsules via the interfacial templating condensation approach. Using PEO-PPO-PEO block copolymers as the templating and protecting agent, the encapsulation follows a straightforward microemulsion mechanism which directly proceeds in a near-neutral pH aqueous environment. The surface hydrophobic nature of UCNPs is crucial for the success of encapsulation. To protect the hydrophobic OA ligands of UCNPs from reaction with the silanol groups of hydrated TMOS, we optimized the synthesis procedure by allowing UCNPs to be encapsulated by PEO-PPO-PEO micelles prior to the addition of TMOS. By this means, we have successfully synthesized UCNPs-ZnPc@NCs comprising a single UCNP surrounded by the co-encapsulated ZnPc, and minimized the distance between the two payloads to facilitate the energy transfer from UCNPs to ZnPc. Such an organic/inorganic hybrid nanocapsule demonstrated excellent colloidal stability, biocompatibility, and enhanced negative contrast T2-weighted MRI, as well as a PDT effect in vitro, eradicating BT474 breast cancer cells under NIR excitation. The developed one-pot approach shines light on the co-encapsulation of OA-capped inorganic UCNPs with a hydrophobic PS, and constitutes an important step forward in the surface engineering of UCNPs and UC-PDT systems. The UC-PDT efficacy and the T2 contrast enhancement of the developed NCs will be further expanded upon in future in vivo studies. This strategy could be extended for developing multifunctional nanocarriers to encompass various nanoparticles and therapeutic or diagnostic agents, thus bringing theranostic functionalities such as NIR, MRI or CT imaging guided therapy and controlled drug release to personalized medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. V. acknowledges funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada through the Discovery Grants program and the Discovery Accelerator Supplement (DAS) award, the Canada Foundation for Innovation for infrastructure and its operation, and the Fonds de Recherche du Québec-Nature et technologies (FRQNT). F. R. acknowledges the NSERC for a Discovery Grant and the Canada Research Chairs program for funding and partial salary support. M. W. and A. Y. C. are grateful for the Johns Hopkins Singapore Research Fund provided by a private limited company for supporting their Santa Fe research programme. A. S. is grateful to FRQNT for financial support in the form of a scholarship for doctoral studies. X. L., M. N. and K. B. acknowledge the support of A*STAR, Singapore.References
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, JNCI, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS PubMed.
- G. Yi, S. H. Hong, J. Son, J. Yoo, C. Park, Y. Choi and H. Koo, Quant. Imaging Med. Surg., 2018, 8, 433–443 CrossRef PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2005, 2, 91–106 CrossRef CAS PubMed.
- F. Vatansever and M. R. Hamblin, in Cancer Immunology: Bench to Bedside Immunotherapy of Cancers, ed. N. Rezaei, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 383–399 Search PubMed.
- Q. Sun, F. He, H. Bi, Z. Wang, C. Sun, C. Li, J. Xu, D. Yang, X. Wang, S. Gai and P. Yang, Chem. Eng. J., 2019, 362, 679–691 CrossRef CAS.
- Ł. Lamch, A. Pucek, J. Kulbacka, M. Chudy, E. Jastrzębska, K. Tokarska, M. Bułka, Z. Brzózka and K. A. Wilk, Adv. Colloid Interface Sci., 2018, 261, 62–81 CrossRef PubMed.
- S. Pervaiz and M. Olivo, Clin. Exp. Pharmacol. Physiol., 2006, 33, 551–556 CrossRef CAS PubMed.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- D. Yang, P. a. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- P. Zhang, W. Steelant, M. Kumar and M. Scholfield, J. Am. Chem. Soc., 2007, 129, 4526–4527 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- K. Liu, X. Liu, Q. Zeng, Y. Zhang, L. Tu, T. Liu, X. Kong, Y. Wang, F. Cao, S. A. G. Lambrechts, M. C. G. Aalders and H. Zhang, ACS Nano, 2012, 6, 4054–4062 CrossRef CAS PubMed.
- X. Liu, M. Zheng, X. Kong, Y. Zhang, Q. Zeng, Z. Sun, W. J. Buma and H. Zhang, Chem. Commun., 2013, 49, 3224–3226 RSC.
- H. S. Qian, H. C. Guo, P. C.-L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285–2290 CrossRef CAS PubMed.
- Q. Sun, F. He, C. Sun, X. Wang, C. Li, J. Xu, D. Yang, H. Bi, S. Gai and P. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 33901–33912 CrossRef CAS PubMed.
- Y. Huang, A. Skripka, L. Labrador-Páez, F. Sanz-Rodríguez, P. Haro-González, D. Jaque, F. Rosei and F. Vetrone, Nanoscale, 2018, 10, 791–799 RSC.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS PubMed.
- Y. Liu, X. Meng and W. Bu, Coord. Chem. Rev., 2019, 379, 82–98 CrossRef CAS.
- H. Qiu, M. Tan, T. Ohulchanskyy, J. Lovell and G. Chen, Nanomaterials, 2018, 8, 344 CrossRef PubMed.
- Q. Liu, Y. Sun, T. Yang, W. Feng, C. Li and F. Li, J. Am. Chem. Soc., 2011, 133, 17122–17125 CrossRef CAS PubMed.
- C. Wang, H. Tao, L. Cheng and Z. Liu, Biomaterials, 2011, 32, 6145–6154 CrossRef CAS PubMed.
- G. Chen, J. Shen, T. Y. Ohulchanskyy, N. J. Patel, A. Kutikov, Z. Li, J. Song, R. K. Pandey, H. Ågren, P. N. Prasad and G. Han, ACS Nano, 2012, 6, 8280–8287 CrossRef CAS PubMed.
- C. Wang, L. Cheng, Y. Liu, X. Wang, X. Ma, Z. Deng, Y. Li and Z. Liu, Adv. Funct. Mater., 2013, 23, 3077–3086 CrossRef CAS.
- G. Jalani, R. Naccache, D. H. Rosenzweig, L. Haglund, F. Vetrone and M. Cerruti, J. Am. Chem. Soc., 2016, 138, 1078–1083 CrossRef CAS PubMed.
- N. Liu, R. Marin, Y. Mazouzi, G. O. Cron, A. Shuhendler and E. Hemmer, Nanoscale, 2019, 11, 6794–6801 RSC.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- X.-F. Qiao, J.-C. Zhou, J.-W. Xiao, Y.-F. Wang, L.-D. Sun and C.-H. Yan, Nanoscale, 2012, 4, 4611–4623 RSC.
- G. Yang, D. Yang, P. Yang, R. Lv, C. Li, C. Zhong, F. He, S. Gai and J. Lin, Chem. Mater., 2015, 27, 7957–7968 CrossRef CAS.
- Y. Zhang, B. Y. W. Hsu, C. Ren, X. Li and J. Wang, Chem. Soc. Rev., 2015, 44, 315–335 RSC.
- J. Shan, S. J. Budijono, G. Hu, N. Yao, Y. Kang, Y. Ju and R. K. Prud'homme, Adv. Funct. Mater., 2011, 21, 2488–2495 CrossRef CAS.
- A. Skripka, V. Karabanovas, G. Jarockyte, R. Marin, V. Tam, M. Cerruti, R. Rotomskis and F. Vetrone, Adv. Funct. Mater., 2019, 29, 1807105 CrossRef.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- S. Jiang and Y. Zhang, Langmuir, 2010, 26, 6689–6694 CrossRef CAS PubMed.
- Y. Wang, K. Liu, X. Liu, K. Dohnalová, T. Gregorkiewicz, X. Kong, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2011, 2, 2083–2088 CrossRef CAS.
- M. R. Hamblin, Dalton Trans., 2018, 47, 8571–8580 RSC.
- S. Drozdek, J. Szeremeta, L. Lamch, M. Nyk, M. Samoc and K. A. Wilk, J. Phys. Chem. C, 2016, 120, 15460–15470 CrossRef CAS.
- Y. Wang, G. Yang, Y. Wang, Y. Zhao, H. Jiang, Y. Han and P. Yang, Nanoscale, 2017, 9, 4759–4769 RSC.
- H. Tan, N. S. Liu, B. He, S. Y. Wong, Z.-K. Chen, X. Li and J. Wang, Chem. Commun., 2009, 6240–6242 RSC.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS PubMed.
- A. A. Atia, A. M. El-Nahas, A. M. Marie and L. D. A. Mahdy, Adsorpt. Sci. Technol., 2006, 24, 797–814 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- H. J. Wang, R. Shrestha and Y. Zhang, Part. Part. Syst. Charact., 2014, 31, 228–235 CrossRef CAS.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- R. Naccache, F. Vetrone, V. Mahalingam, L. A. Cuccia and J. A. Capobianco, Chem. Mater., 2009, 21, 717–723 CrossRef CAS.
- A. Skripka, R. Marin, A. Benayas, P. Canton, E. Hemmer and F. Vetrone, Phys. Chem. Chem. Phys., 2017, 19, 11825–11834 RSC.
- M. Kraft, C. Würth, V. Muhr and T. Hirsch, Nano Res., 2018, 11, 6360–6374 CrossRef CAS.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed.
- J.-C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS PubMed.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. a. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed.
- M. Wang, Y. Zhang, Q. Yao, M. Ng, M. Lin, X. Li, K. K. Bhakoo, A. Y. Chang, F. Rosei and F. Vetrone, Chem. Mater., 2019, 31, 5160–5171 CrossRef CAS.
- P. A. Hardy and R. M. Henkelman, Magn. Reson. Imaging, 1989, 7, 265–275 CrossRef CAS PubMed.
- J. Qin, S. Laurent, Y. S. Jo, A. Roch, M. Mikhaylova, Z. M. Bhujwalla, R. N. Muller and M. Muhammed, Adv. Mater., 2007, 19, 1874–1878 CrossRef CAS.
- X. Zhang, B. Blasiak, A. J. Marenco, S. Trudel, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2016, 28, 3060–3072 CrossRef CAS.
- H. Ai, C. Flask, B. Weinberg, X.-T. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. Gao, Adv. Mater., 2005, 17, 1949–1952 CrossRef CAS.
- F. Yang, A. Skripka, M. S. Tabatabaei, S. H. Hong, F. Ren, A. Benayas, J. K. Oh, S. Martel, X. Liu, F. Vetrone and D. Ma, ACS Nano, 2019, 13, 408–420 CrossRef CAS PubMed.
- Z. Li, T. Tian, X. Hu, X. Zhang, F. Nan, Y. Chang, F. Lv and M. Zhang, Biochem. Biophys. Res. Commun., 2013, 441, 538–543 CrossRef CAS PubMed.
- B. T. McGrogan, B. Gilmartin, D. N. Carney and A. McCann, Biochim. Biophys. Acta, Rev. Cancer, 2008, 1785, 96–132 CrossRef CAS PubMed.
- T. Entradas, S. Waldron and M. Volk, J. Photochem. Photobiol., B, 2020, 204, 111787 CrossRef CAS PubMed.
- Q. Zhan, J. Qian, H. Liang, G. Somesfalean, D. Wang, S. He, Z. Zhang and S. Andersson-Engels, ACS Nano, 2011, 5, 3744–3757 CrossRef CAS PubMed.
- J. Xu, R. Lv, S. Du, S. Gai, F. He, D. Yang and P. Yang, J. Mater. Chem. B, 2016, 4, 4138–4146 RSC.
- J. S. Khalili, X. Yu, J. Wang, B. C. Hayes, M. A. Davies, G. Lizee, B. Esmaeli and S. E. Woodman, Clin. Cancer Res., 2012, 18, 4345–4355 CrossRef CAS PubMed.
- M. Miyata, M. Kambe, O. Tajima, S. Moriya, H. Sawaki, H. Hotta, Y. Kondo, H. Narimatsu, T. Miyagi, K. Furukawa and K. Furukawa, Cancer Sci., 2011, 102, 2139–2149 CrossRef CAS PubMed.
- T. C. Huang, J. F. Lee and J. Y. Chen, Mar. Drugs, 2011, 9, 1995–2009 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details including synthesis and characterization procedures, as well as additional experimental data. See DOI: 10.1039/d0sc01033b |
| This journal is © The Royal Society of Chemistry 2020 |