
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA traceless linker for aliphatic amines that rapidly and quantitatively fragments after reduction†
Maomao
He‡
 a,
Jie
Li‡
a,
Hesong
Han
a,
Clarissa Araujo
Borges
b,
Gabriel
Neiman
a,
Joachim Justad
Røise
ac,
Piotr
Hadaczek
d,
Rima
Mendonsa
e,
Victoria R.
Holm
e,
Ross C.
Wilson
e,
Krystof
Bankiewicz
d,
Yumiao
Zhang
af,
Corinne M.
Sadlowski
a,
Kevin
Healy
a,
Lee W.
Riley
b and
Niren
Murthy
*ae
a,
Jie
Li‡
a,
Hesong
Han
a,
Clarissa Araujo
Borges
b,
Gabriel
Neiman
a,
Joachim Justad
Røise
ac,
Piotr
Hadaczek
d,
Rima
Mendonsa
e,
Victoria R.
Holm
e,
Ross C.
Wilson
e,
Krystof
Bankiewicz
d,
Yumiao
Zhang
af,
Corinne M.
Sadlowski
a,
Kevin
Healy
a,
Lee W.
Riley
b and
Niren
Murthy
*ae
aDepartment of Bioengineering, University of California Berkeley, University Avenue, Berkeley, CA 94720, USA. E-mail: nmurthy@berkeley.edu
bDepartment of Public Health, University of California Berkeley, University Avenue, Berkeley, CA 94720, USA
cDepartment of Chemistry, University of California Berkeley, University Avenue, Berkeley, CA 94720, USA
dDepartment of Neurological Surgery, The Ohio State University, Columbus, OH 43210, USA
eInnovative Genomics Institute, University of California, Berkeley, CA 94704, USA
fSchool of Chemical Engineering and Technology, Tianjin University, 300350, China
First published on 12th August 2020
Abstract
Reduction sensitive linkers (RSLs) have the potential to transform the field of drug delivery due to their ease of use and selective cleavage in intracellular environments. However, despite their compelling attributes, developing reduction sensitive self-immolative linkers for aliphatic amines has been challenging due to their poor leaving group ability and high pKa values. Here a traceless self-immolative linker composed of a dithiol-ethyl carbonate connected to a benzyl carbamate (DEC) is presented, which can modify aliphatic amines and release them rapidly and quantitatively after disulfide reduction. DEC was able to reversibly modify the lysine residues on CRISPR–Cas9 with either PEG, the cell penetrating peptide Arg10, or donor DNA, and generated Cas9 conjugates with significantly improved biological properties. In particular, Cas9–DEC–PEG was able to diffuse through brain tissue significantly better than unmodified Cas9, making it a more suitable candidate for genome editing in animals. Furthermore, conjugation of Arg10 to Cas9 with DEC was able to generate a self-delivering Cas9 RNP that could edit cells without transfection reagents. Finally, conjugation of donor DNA to Cas9 with DEC increased the homology directed DNA repair (HDR) rate of the Cas9 RNP by 50% in HEK 293T cell line. We anticipate that DEC will have numerous applications in biotechnology, given the ubiquitous presence of aliphatic amines on small molecule and protein therapeutics.
Introduction
Reduction sensitive linkers (RSLs) have great potential as reagents for developing drug delivery vehicles and are ubiquitously used in drug delivery and pharmaceutical sciences.1–7 RSLs are selectively cleaved in the intracellular reducing environment of the cell and have played an important role in the development of antibody drug conjugates (ADCs), protein conjugates and nucleic acid delivery vehicles.1,8,9 In addition, RSLs are slowly cleaved in the serum and have been used for developing controlled release reservoirs and for enhancing the circulation half-life of biomolecules. Finally, RSLs have compelling synthetic and product development attributes because they are relatively stable and can survive multi-step synthetic procedures without self-hydrolyzing.3An important application of RSLs is for the reversible modification of lysine residues on proteins. For example, a wide number of compounds have been developed that can improve the therapeutic efficacy of proteins, such as PEG or cell penetrating peptides.10–12 These adjuvants need to be conjugated to proteins in order to enhance protein delivery. Lysine residues are ubiquitously present on proteins and can be easily modified under mild aqueous conditions because of their high nucleophilicity, and are therefore an attractive target for conjugating adjuvants to proteins. However, lysine residues are also frequently essential for protein activity and extensive modification usually compromises protein activity. Therefore, RSLs that can reversibly modify lysine residues have the potential to overcome this limitation and thus service as a promising strategy for protein delivery.13
Two major classes of RSLs have been developed to modify aliphatic amines, which are disulfide carbamates (DCBs)14,15 and dithiobenzyl carbamates (DTBs).16–18 Even though DCBs and DTBs (Fig. S1†) have demonstrated the great potential of RSLs in prodrug design, they have not found widespread applications for protein delivery due to their slow self-immolation rates or incomplete release. There is consequently great interest in developing new amine-modifying RSLs that can modify the lysine residues of proteins in a traceless manner.13,19
In this report, we present a traceless self-immolative linker, which is composed of a dithiol-ethyl carbonate linker combined with a benzyl carbamate linker, termed DEC (Fig. 1a). DEC is composed of two rapidly hydrolyzing self-immolative linkers connected in series, and this unique design allows it to overcome the challenges associated with making RSLs for aliphatic amines. The two linkers embedded in DEC are a thiol-ethyl carbonate linker and a 1,6-elimination linker.20 These two linkers were selected for incorporation in DEC because they individually release alcohols instantaneously and amines after activation. Specifically, the thiol-ethyl carbonate linker rapidly releases a cyclic thiol ether after disulfide reduction, and the 1,6-elimination linker rapidly releases aliphatic amines. Our hypothesis is that by combining the thiol-ethyl carbonate linker with the 1,6-elimination linker, we would generate a novel RSL which rapidly releases amines through a two-step release mechanism after reduction (Fig. 1a). In step 1, the thiol-ethyl carbonate linker of DEC is reduced, generating a free thiol which spontaneously cyclizes into a cyclic ether, causing the hydrolysis of the carbonate in DEC and the release of a phenol. In step 2 the phenol intermediate triggers the 1,6-elimination process to release the aliphatic amine. In this paper, DEC was used to develop new prodrugs and also modify the genome editing enzyme CRISPR–Cas9 (ref. 21) with PEG, CPPs and donor DNA (Fig. 1b). We anticipate that DEC has numerous potential applications in biotechnology given the ubiquitous presence of aliphatic amines on small molecules and protein therapeutics.22
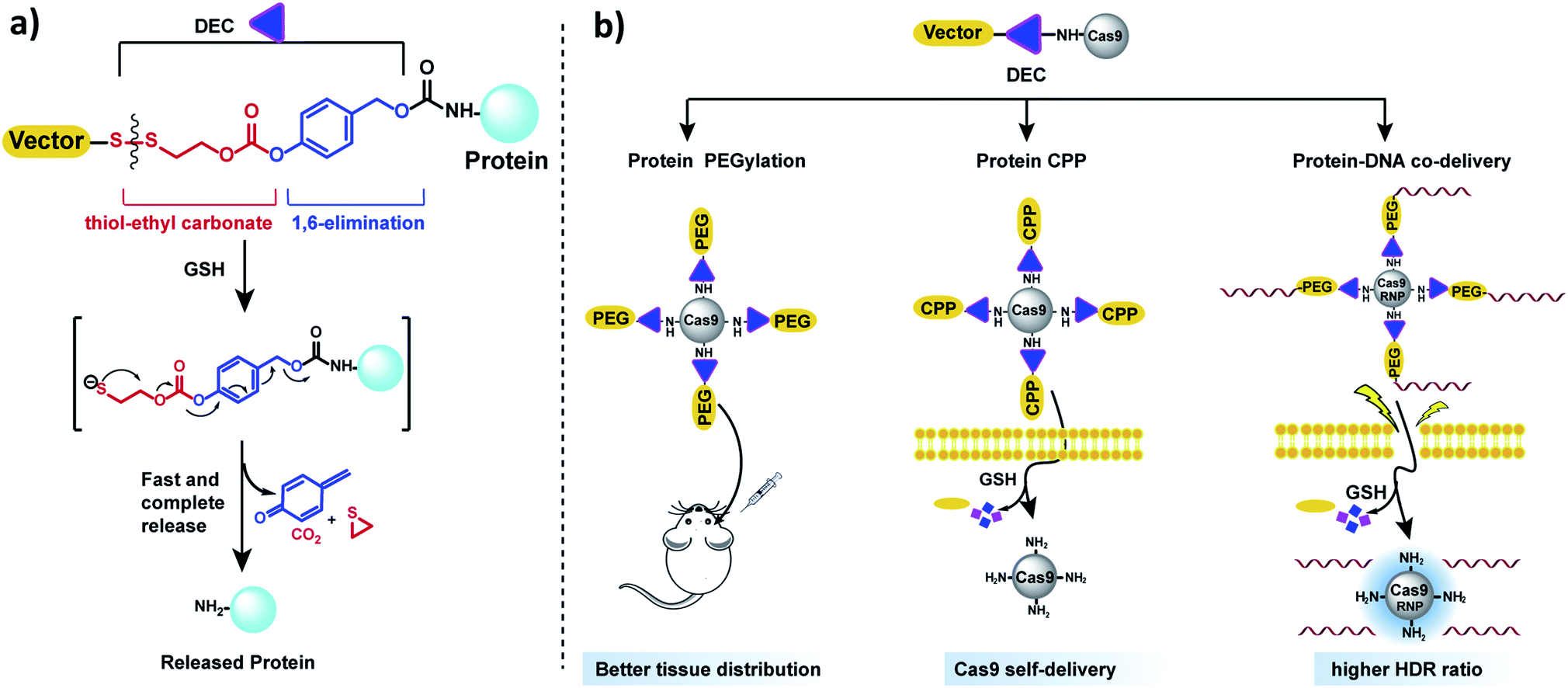 | ||
| Fig. 1 DEC is a new reduction sensitive linker that can modify aliphatic amines and release them in a traceless manner. (a) DEC is composed of two rapidly hydrolyzing self-immolative linkers connected in series, in particular a thiol-ethyl carbonate linker and a 1,6-elimination linker. In the presence of reducing reagents such as GSH, the disulfide bond in DEC is reduced, generating a free thiol which spontaneously cyclizes into a cyclic thioether, causing the release of its phenol (step 1). The phenol intermediate triggers the 1,6-elimination process to release the aliphatic amine (step 2). (b) DEC has the potential to accelerate the development of new prodrugs and protein delivery vehicles. DEC was able to PEGylate Cas9 protein, which was able to diffuse through brain tissue significantly better than free Cas9. In addition, DEC was also used to synthesize a self-delivering Cas9 RNP composed of Cas9 RNP conjugated to the CPP (Arg)10. Finally, conjugation of donor DNA to Cas9 with DEC–PEG dramatically increased the homology directed repair (HDR) rate of the Cas9 RNP. | ||
Results and discussion
To investigate if DEC-modified aliphatic amines can be quantitatively and rapidly released after disulfide reduction, a ciprofloxacin–PEG conjugate was synthesized by modifying the amine on ciprofloxacin with DEC (Fig. 2a) (see Fig. S2† for synthesis). Ciprofloxacin was selected as a model drug for kinetic study of DEC because modification of its amine results in its inactivation.23 In addition, the fluorescence of ciprofloxacin facilitates its analysis by HPLC. As a control, ciprofloxacin was also conjugated to PEG with the DCB and DTB linkers (PEG–DCB–Cipro and PEG–DTB–Cipro, see Fig. S3 and S4† for synthesis). In addition, a methyl substituted DCB linker (DCBMe) was also synthesized (Fig. S5†).9,24 The release kinetics of ciprofloxacin from the four disulfide linkers was investigated in the presence of glutathione (GSH) (10 mM) to mimic the intracellular environment, and was monitored by HPLC (350 nm wavelength) (Fig. S6†). Fig. 2c demonstrates that PEG–DEC–Cipro rapidly and quantitatively released free ciprofloxacin after disulfide reduction, and had a half-life of 5 minutes to regenerate unmodified ciprofloxacin after GSH reduction. In contrast, the release half-life of PEG–DCB–Cipro was 90 minutes after disulfide reduction Interestingly, PEG–DCBMe–Cipro showed much faster release kinetics than the traditional DCB linker (half-life 15 minutes), this data is in accordance with the results recently published by Zhang et al.24PEG–DTB–Cipro had a half-life of 30 minutes. However, PEG–DTB–Cipro was unable to quantitatively regenerate free ciprofloxacin, due to the build-up of a new ciprofloxacin-containing side product (Fig. S6b†). These results demonstrate that DEC can release aliphatic amines rapidly and quantitatively after reduction.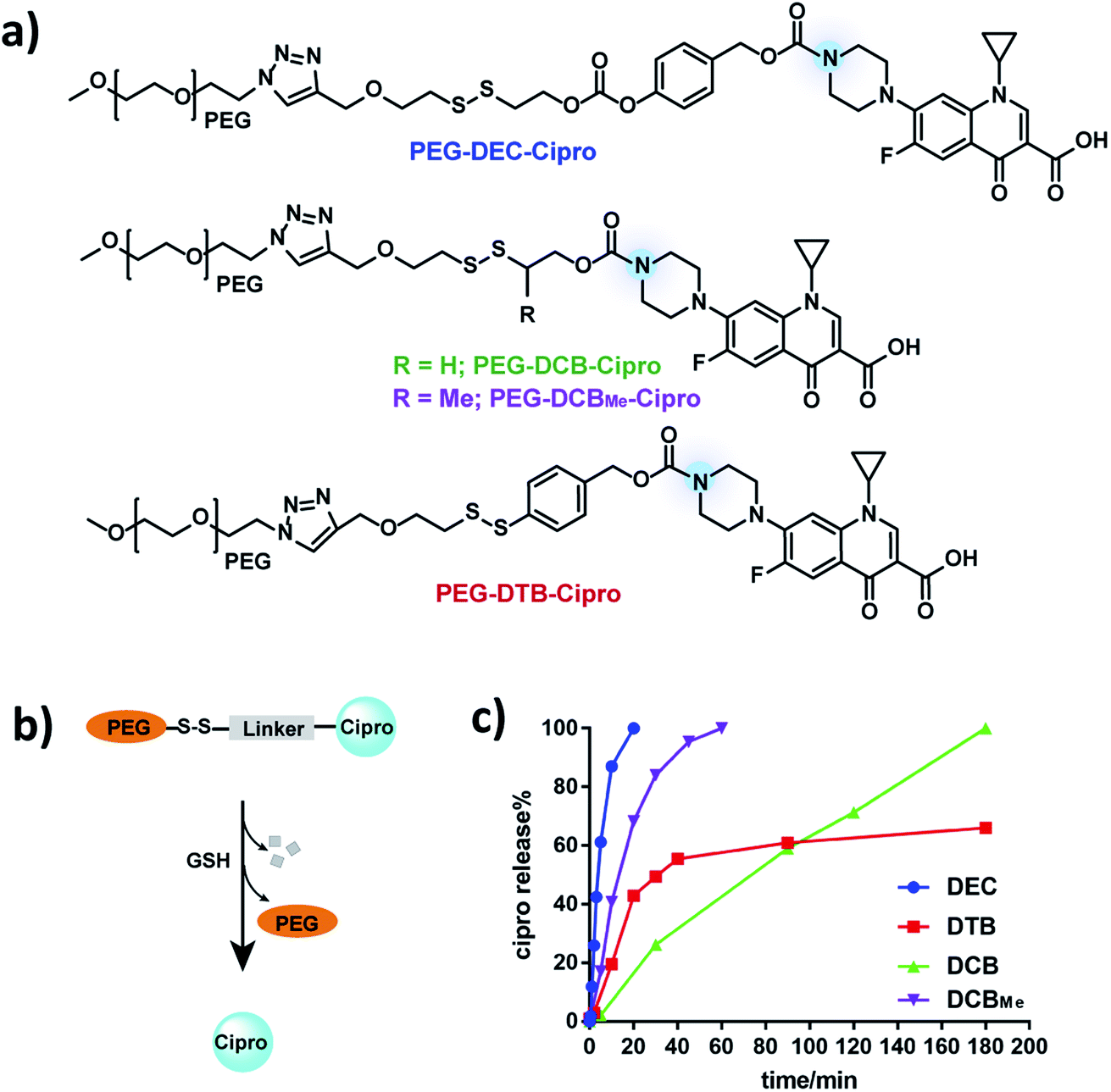 | ||
| Fig. 2 PEG–DEC–Cipro releases ciprofloxacin rapidly and quantitatively after reduction. (a) Chemical structures of PEG–DEC–Cipro, PEG–DCB–Cipro, PEG–DCBMe–Cipro and PEG–DTB–Cipro. (b and c) DEC (blue) releases free ciprofloxacin completely with a t1/2 of 5 minutes in the presence of cellular GSH concentrations (10 mm) at pH 7.4. In contrast, the DTB linker (red) had a t1/2 of 30 minutes and was unable to completely release the free amine and the DCB linker (green) had a t1/2 of 90 minutes. DCBMe linker (purple) has a t1/2 of 15 minutes, which is shorter than the traditional DCB linker. | ||
We performed additional experiments to investigate if the ciprofloxacin released from PEG–DEC–Cipro was biologically active, given that modification of the amine of ciprofloxacin causes its inactivation. The minimum inhibitory concentration (MIC) of PEG–DEC–Cipro was measured in the presence of GSH (10 mM) on E. coli SF207, a clinical strain resistant to a variety of antibiotics, isolated from a patient with bloodstream infection (see ESI page S11† for detail). Table S1† shows that the MIC of PEG–DEC–Cipro was 0.25 μg mL−1 (at an equivalent dose of ciprofloxacin) in the presence of GSH (10 mM), which is the same as free ciprofloxacin, while the MIC is much higher without GSH (>1 μg mL−1).
An important application of amine reactive self-immolative linkers is for the reversible modification of lysine residues on proteins. For example, a wide number of compounds have been developed that can improve the therapeutic efficacy of proteins, such as PEG or cell penetrating peptides.10–12 These adjuvants need to be conjugated to proteins in order to enhance protein delivery. However, this remains challenging due to the lack of efficient linkers that can reversibly modify aliphatic amines. We therefore investigated if DEC could reversibly modify the lysine residues of proteins, and if it exhibited the same benefits over the DTB and DCB linkers, when attached to proteins.
CRISPR–Cas9 was selected as a model protein for investigation and was PEGylated with DEC. Cas9 is an excellent testbed for investigating the self-immolative nature of DEC because it has multiple lysine residues in its active site, and widespread modification of the lysine residues will result in its inactivation.25 In addition, there is currently great interest in PEGylating Cas9, which has the potential to address several of the challenges associated with Cas9 delivery, in particular, its immunogenicity, slow diffusion through tissue and proteolysis in serum.26
mCherryCas9 (a Cas9 variant that is fused to mCherry) was PEGylated using the DEC, DTB or DCB linkers (Fig. 3a and S8†), and the release of PEG and the regeneration of Cas9 nuclease activity was determined after GSH reduction. Fig. S9† and 3b demonstrate that mCherryCas9 was efficiently PEGylated by all three linkers, but only the DEC linker efficiently released PEG after reduction for 1 hour (98% vs. 34% and 29%). Fig. 3c demonstrates that the nuclease activity of mCherryCas9 was significantly inhibited by PEGylation, regardless of the linker used. However, the nuclease activity of Cas9 PEGylated with DEC was significantly increased after reduction with GSH. In contrast, GSH reduction caused minor increases in nuclease activity in mCherryCas9 samples PEGylated with the DTB or DCB linkers, presumably due to incomplete amine release (80% vs. 23% and 19%).
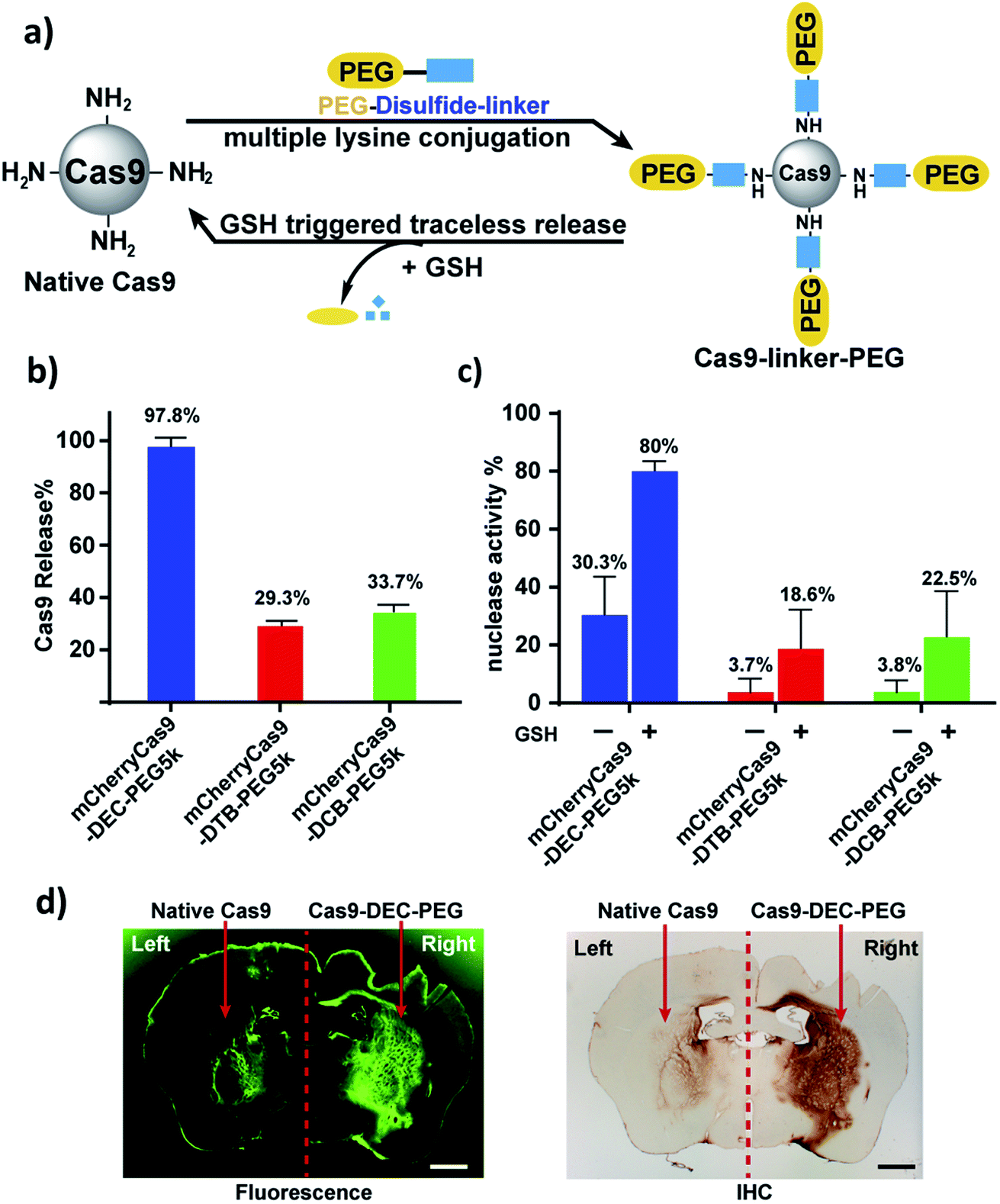 | ||
| Fig. 3 mCherryCas9 can be conjugated to PEG via DEC and regains activity after reduction by GSH. (a) PEGylation of mCherryCas9 via three different disulfide linkers (DEC, DTB and DCB). (b) Release of PEG from mCherryCas9–disulfide–PEG after GSH (10 mm) incubation for 1 hour. mCherryCas9 release was analyzed by SDS-PAGE gel. (c) In vitro nuclease activity of the three mCherryCas9–disulfide–PEG conjugates was evaluated before and after reduction by GSH (10 mm). (d) Striatal distribution in the mouse brain after injection with Alexa488Cas9 (left striatum) or Alexa488Cas9–DEC–PEG5k (right striatum). Immunohistological staining (IHC) against Cas9 protein was visualized with DAB peroxidase substrate kit. Fluorescence microscope (green channel: 488 nm) scale bar: 1 mm. | ||
We evaluated the stability of mCherryCas9–DEC–PEG in plasma, to determine if it had the extracellular stability needed for in vivo applications. Fig. S11† demonstrates that mCherryCas9–DEC–PEG was stable in plasma and had a t1/2 > 20 hours, but was rapidly reduced in the presence of intracellular levels of GSH (t1/2 < 4 hours). PEGylated Cas9 has numerous potential advantages over wild type Cas9, such as lower immunogenicity and improved diffusion through tissue, which is essential for genome editing in a clinical setting. We performed experiments to investigate if Cas9–DEC–PEG could efficiently diffuse through the striatum after an intracranial injection and compared its tissue distribution with unmodified Cas9. We selected the brain as a testbed for Cas9–DEC–PEG because the poor diffusion of Cas9 through brain tissue is a key challenge that needs to be overcome before the clinical potential of Cas9 can be fully realized.27–29 A fluorescently labeled Cas9 (Alexa488Cas9) and Alexa488Cas9–DEC–PEG5k were injected and infused bilaterally into the left and right striatum of mice at a concentration of 1 μg μL−1. The distribution of the injected Alexa488Cas9 or Alexa488Cas9–DEC–PEG5k was then determined based on fluorescent imaging and immunohistological staining (IHC) for Cas9 (Fig. 3d and S13†). Fig. 3d indicates that Alexa488Cas9–DEC–PEG5k diffused through the striatum significantly better than its unmodified counterpart (Alexa488Cas9), and had a volume of distribution (Vd) that was 3.7-fold higher than native Alexa488Cas9 (Table S2†). Collectively these results demonstrate that the DEC linker can reversibly modify the lysine residues of proteins (Cas9) and has the stability in biological fluids needed for in vivo applications.
Next, we investigated if cell penetrating peptides (CPPs) could be conjugated to Cas9 using a DEC linker. CPPs have great potential for enhancing the delivery of Cas9 into cells, and have been extensively explored for Cas9 delivery.30–32 However, linkers that can reversibly conjugate CPPs to Cas9 have not been studied. CPP–DEC was synthesized, which is composed of a fluorescent cell penetrating peptide FITC–Arg10Lys1 connected to the DEC linker, and was then conjugated to Cas9 (Fig. 4a, S14 and S15†).33 Cas9/CPP–DEC conjugation reactions were performed at various molar ratios, ranging from a 10 to 500-fold molar excess of CPP–DEC to Cas9 (Fig. 4b).
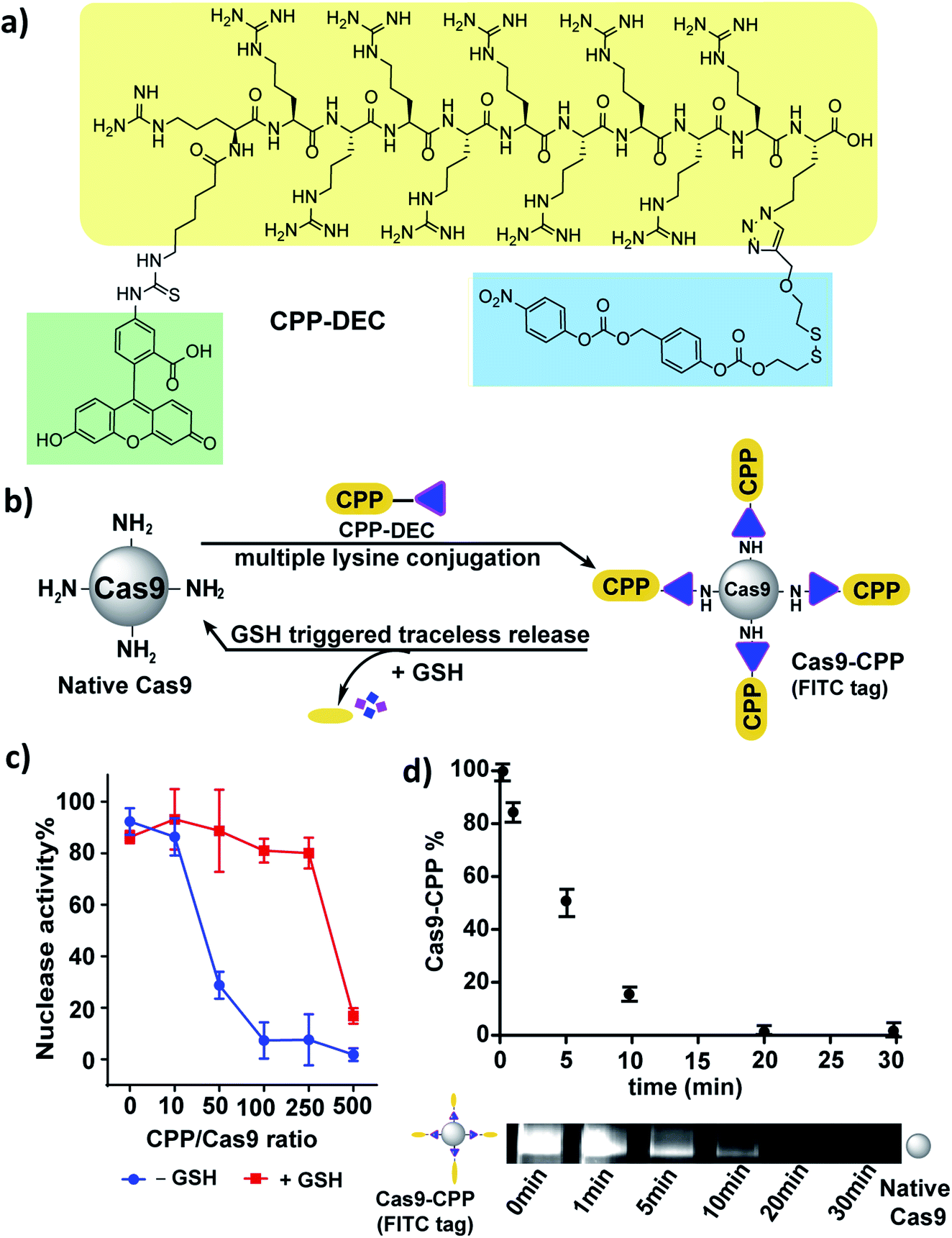 | ||
| Fig. 4 CPP can be conjugated to Cas9 via DEC and releases active Cas9 under cellular concentrations of GSH. (a) Structure of CPP–DEC. (b) CPP–DEC can be conjugated to Cas9 and releases active Cas9 in the presence of cellular concentrations of GSH (10 mM). (c) Nuclease activity of the Cas9 RNP after conjugation with various mole ratios of CPP–DEC (X-axis). Red line represents nuclease activity after incubation with GSH (10 mM). (d) Kinetics of CPP release from Cas9 after addition of 10 mM GSH (0, 1, 5, 10, 20, 30 minutes), the fluorescent bands show the time dependent release of CPP from Cas9–CPP. | ||
An in vitro cleavage assay was performed to test the activity of Cas9–CPP with or without GSH (Fig. 4c and S16†). Fig. 4c shows that Cas9 conjugated with CPP–DEC can regain its nuclease activity after reduction with GSH in a ratio-dependent manner. For example, the nuclease activity of Cas9 was significantly reduced after conjugation with a 50–250 fold excess of CPP–DEC, but was recovered after GSH reduction. However, bioconjugation reactions done at a 500 molar excess of CPP–DEC resulted in only partial activation of the Cas9 RNP under the same conditions, presumably due to denaturation of the protein, incomplete reduction of DEC, or electrostatistic interactions between the conjugated CPPs and the gRNA. Cas9–CPP conjugates made at a 100/1 molar ratio were selected for further investigation. This preparation was analyzed by the BCA assay and fluorescence spectroscopy, and had an average of 40 CPPs per Cas9 protein. The Cas9–CPP conjugate was analyzed by gel electrophoresis to verify that the CPPs could be cleaved from the Cas9 protein after reduction. Fig. S15† demonstrates that CPP–DEC was covalently conjugated to Cas9 or mCherryCas9 and was also released in the presence of cellular concentrations of GSH (10 mM). The release kinetics of the CPPs from Cas9–CPP was determined using gel electrophoresis. Cas9–CPP was mixed with GSH (10 mM) for different durations and release of the CPPs was determined by quantification of the FITC fluorescence on the Cas9–CPP. Fig. 4d demonstrates that the CPPs are released rapidly from Cas9 under intracellular GSH concentrations, with a release half-life of 5 minutes.
The ability of Cas9–CPP to edit cells was investigated. Cas9–CPP (33 μg mL−1) containing a gRNA that cleaves the eGFP gene was incubated with HEK293T eGFP cells. After 72 hours, the genomic DNA was extracted and the eGFP gene was amplified by PCR, and analyzed by Sanger sequencing (Fig. 5a). The HEK293T eGFP cells were also analyzed by flow cytometry (Fig. S17†). As a control, Cas9 RNP was transfected with lipofectamine 2000.34Fig. 5a shows that Cas9–CPP delivered functional Cas9 RNP into cells and edited 5.1% of the eGFP gene in HEK cells, which was comparable to lipofectamine delivered Cas9 RNP (4.1%), as determined by TIDE analysis. Cas9 RNP by itself had nearly no editing activity in cells without transfection reagents.
 | ||
| Fig. 5 CPP–DEC can deliver Cas9 into cells without the addition of transfection reagents. (a) HEK 293T eGFP cells were treated with Cas9–CPP RNP and Cas9 RNP + lipofectamine 2000, or Cas9 RNP by itself. The gRNA was designed to cleave the GFP gene. The cells were isolated and analyzed by sanger sequencing for gene editing (NHEJ). Cas9–CPP edited 5.1% of the cells, which was comparable to lipofectamine treated cells. Gene editing efficiency was quantified by tide analysis. Graph represents mean ± s.d. (n = 3). Statistical analysis was determined using one way anova with post-hoc test (***: p < 0.001; ns: nonsignificant). (b) Flow-cytometry analysis of hela cells treated with mCherryCas9 and mCherryCas9–CPP. The average fluorescence of cells treated with mCherryCas9–CPP was 7 times higher than free mCherryCas9. | ||
Further experiments were performed to study if CPP–DEC could enhance the internalization of Cas9 into cells. mCherryCas9 was conjugated with CPP–DEC with a 100 molar excess ratio. mCherryCas9–CPP was incubated with HeLa cells and the internalization was analyzed by flow cytometry and fluorescent microscopy (Fig. 5b and S18†). As a control, cellular internalization of unmodified mCherryCas9 was investigated. Our results demonstrate that mCherryCas9–CPP was internalized more efficiently than wild-type mCherryCas9 (7 folds higher in HeLa cells). To further determine the tropism of mCherryCas9–CPP, the uptake of mCherryCas9 vs. mCherryCas9–CPP was determined in human cardiomyocytes derived from induced pluripotent stem cells. Fig. S19† demonstrates that mCherryCas9–CPP was also robustly internalized by human cardiomyocytes, suggesting that protein conjugation with CPP–DEC is a broadly applicable strategy for enhancing the internalization of proteins into cells.
Finally, we investigated if DEC can enhance the efficiency of homology-directed repair (HDR) by reversibly conjugating Cas9 RNP to template donor DNA. Generating HDR in cells with Cas9 reagents is a central challenge in genome editing, and is currently one of the key bottlenecks preventing the translation of Cas9 based therapeutics.35 A crucial challenge in promoting efficient HDR is to deliver sufficient quantities of the Cas9 RNP and donor DNA into cells. Recently, chemically tethered Cas9 RNP–donor DNA has been demonstrated to result in significant increases in HDR, due to the simultaneous localization of Cas9 RNP and donor DNA within the same region of the genome.36,37 However, current methods for tethering the Cas9 RNP to donor DNA require making Cas9 fusion proteins, which have been challenging to express and purify.37,38
We therefore investigated if DEC could be used to directly conjugate donor DNA to the Cas9 RNP, in a mix-and-go manner, to generate a reagent that did not require extensive manipulation, and could be easily adopted by the biomedical community. Our synthetic strategy for making Cas9–RNP donor DNA conjugates is shown in Fig. 6a. Cas9 RNP–DEC–azide was first prepared by conjugating Cas9 RNP with DEC–PEG–azide (Fig. S20 and S21†), the resulting conjugate was then reacted with DBCO–DNA (donor DNA with a DBCO tag at 5′) through the strain-promoted click reaction (Fig. S22 and S23†), and directly added to cells.
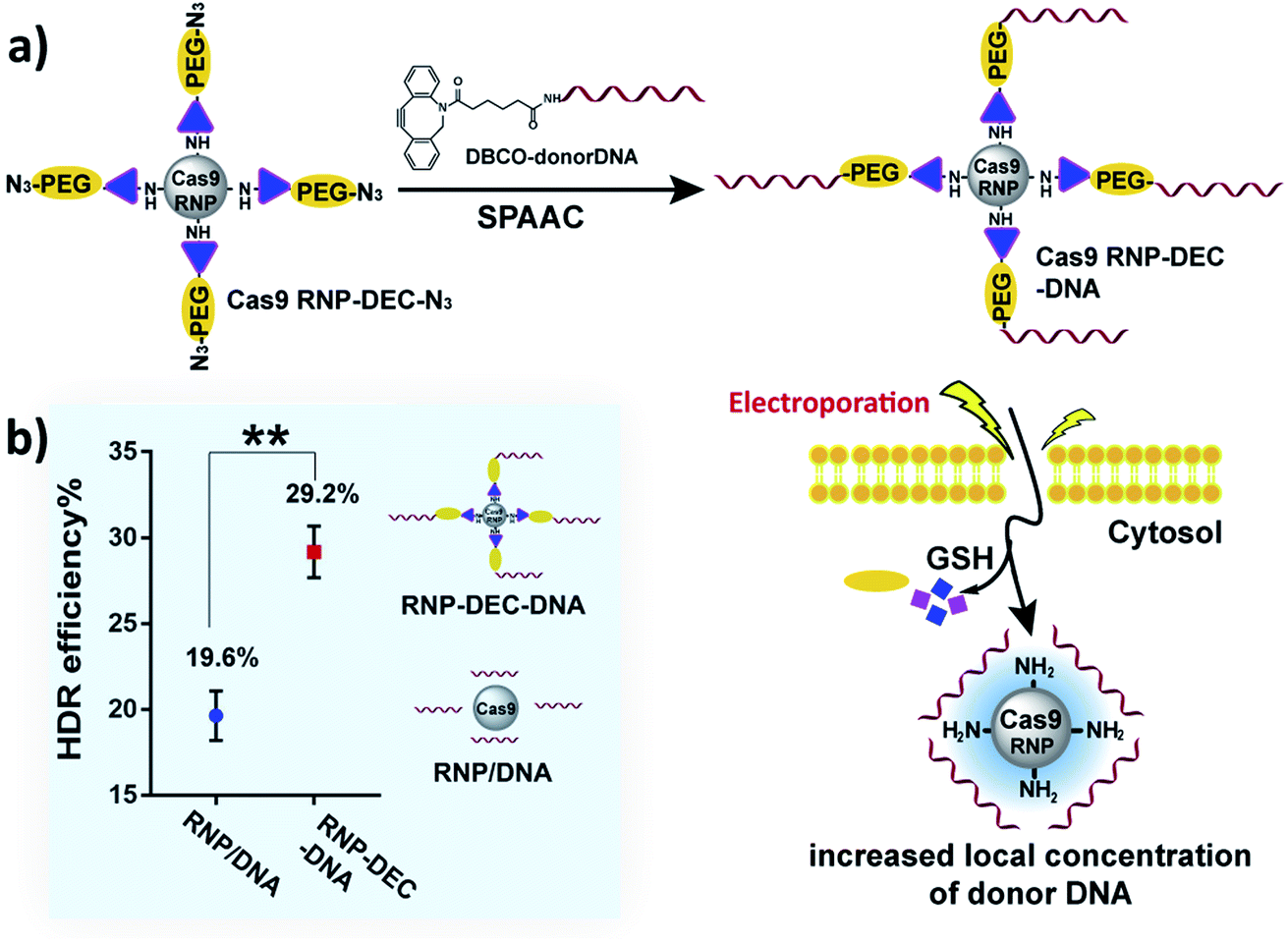 | ||
| Fig. 6 DEC can increase the HDR rate of Cas9 RNP through reversible donor DNA conjugation. (a) Overview of the workflow for conjugating the donor DNA to the Cas9 RNP. The Cas9 RNP is first conjugated to PEG–DEC–N3 to generate Cas9 RNP DEC–N3. Donor DNA with a DBCO handle is then clicked with the Cas9 RNP–DEC–N3 and electroporated into cells. The donor DNA is released from the Cas9 RNP in the reducing environment of the cells, this conjugation strategy ensures that cells with the Cas9 RNP also have donor DNA. (b) Comparison of the HDR efficiency between physically mixed Cas9 RNP/donor DNA and Cas9 RNP–donor DNA conjugate (Cas9 RNP–DEC–DNA) after electroporation. Cas9 RNP–DEC–DNA had a 50% increase in hdr over cells treated with a physical mixture of Cas9 RNP and donor DNA. | ||
To test if Cas9 RNP–DEC–DNA conjugates could increase the HDR rate of the Cas9 RNP, a BFP expressing HEK 293T cell line was used as a reporter system. The BFP gene differs from the GFP gene by a single amino acid, and BFP expressing cells can be transformed into GFP expressing cells by Cas9 mediated cutting of the BFP gene and repair of the double stranded break via HDR from a donor DNA. BFP HEK cells were transfected by electroporation with Cas9 RNP–DEC–DNA, using a 127 nucleotide donor DNA that was designed to convert the BFP gene into the GFP gene. As a control, Cas9 RNP was physically mixed with donor DNA at the same ratio (Cas9 RNP/DNA) and electroporated into cells. The HDR rate was then quantified by determining the percentage of GFP expressing cells via flow cytometry. Fig. 6b and Fig. S24† demonstrate that cells treated with Cas9 RNP–DEC–DNA had a 50% increase in HDR over cells treated with a physical mixture of Cas9 RNP and donor DNA.
Conclusions
In summary, we have developed a new traceless self-immolative linker (DEC), which can modify aliphatic amines and rapidly and quantitatively release them after reduction. We demonstrate here that DEC was able to reversibly modify the lysine residues on Cas9 with PEG, which significantly enhanced the diffusion of Cas9 through brain tissue. In addition, DEC was able to reversibly modify the Cas9 protein via conjugation of its lysine residues with the CPP Arg10. Cas9–CPP was able to successfully edit cells and has great potential as a self-delivering Cas9 RNP formulation. Finally, DEC was also able to increase the HDR rate of the CRISPR system by 50%, by reversibly tethering donor DNA to Cas9 RNP in traceless manner. The DEC linker is a versatile reagent that can be used for multiple applications in protein and small molecule drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Institutes of Health R01AI117064, R33AI119115, UGS NS115599 and RO1EB023776. We also acknowledge support from the Innovative Genomics Institute and the Bakar Fellows program. We thank Dr Jeffrey Pelton at the QB3 NMR Facility for help with the 900 MHz NMR (funded by NIH grant GM68933). We thank Dr Wenbo Yang for help with the ICP measurements.Notes and references
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- F. H. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- M. Sengee, J. J. Eksteen, S. L. Nergard, T. Vasskog and L. K. Sydnes, Bioconjugate Chem., 2019, 30, 1489–1499 CrossRef CAS PubMed.
- G. Leriche, L. Chisholm and A. Wagner, Biorg. Med. Chem., 2012, 20, 571–582 CrossRef CAS PubMed.
- K. Tsuchikama and Z. Q. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS PubMed.
- C. Luo, J. Sun, B. J. Sun and Z. G. He, Trends Pharmacol. Sci., 2014, 35, 12–22 CrossRef PubMed.
- T. H. Pillow, J. D. Sadowsky, D. Zhang, S. F. Yu, G. Del Rosario, K. Xu, J. He, S. Bhakta, R. Ohri, K. R. Kozak, E. Ha, J. R. Junutula and J. A. Flygare, Chem. Sci., 2017, 8, 366–370 RSC.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- S. Jevsevar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef CAS PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS.
- Y. H. Gong, J. C. Leroux and M. A. Gauthier, Bioconjugate Chem., 2015, 26, 1172–1181 CrossRef CAS PubMed.
- H. Zhang, K. Wang, K. Na, D. Li, Z. Li, D. Zhao, L. Zhong, M. Wang, L. Kou, C. Luo, H. Zhang, Q. Kan, H. Ding, Z. He and J. Sun, J. Med. Chem., 2018, 61, 4904–4917 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, M. Wang, H. Peng, Q. Hua, L. Ma, B. Wang and H. Wei, Bioconjugate Chem., 2018, 29, 2239–2247 CrossRef CAS PubMed.
- S. Zalipsky, N. Mullah, C. Engbers, M. U. Hutchins and R. Kiwan, Bioconjugate Chem., 2007, 18, 1869–1878 CrossRef CAS PubMed.
- P. D. Senter, W. E. Pearce and R. S. Greenfield, J. Org. Chem., 1990, 55, 2975–2978 CAS.
- S. Zalipsky, M. Qazen, J. A. Walker, N. Mullah, Y. P. Quinn and S. K. Huang, Bioconjugate Chem., 1999, 10, 703–707 CrossRef CAS PubMed.
- J. P. Krise and R. Oliyai, in Prodrugs: Challenges and Rewards Part 1, ed. V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, H. Maag and J. W. Tilley, Springer New York, New York, NY, 2007, pp. 801–831, DOI:10.1007/978-0-387-49785-3_22.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Y. Guan, J. Med. Chem., 1999, 42, 3657–3667 CAS.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discov. Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- M. Schmidt, S. Harmuth, E. R. Barth, E. Wurm, R. Fobbe, A. Sickmann, C. Krumm and J. C. Tiller, Bioconjugate Chem., 2015, 26, 1950–1962 CrossRef CAS PubMed.
- D. L. Zhang, T. H. Pillow, Y. Ma, J. dela Cruz-Chuh, K. R. Kozak, J. D. Sadowsky, G. D. L. Philips, J. Guo, M. Darwish, P. Fan, J. T. Chen, C. R. He, T. Wang, H. Yao, Z. J. Xu, J. H. Chen, J. Wai, Z. H. Pei, C. E. C. A. Hop, S. C. Khojasteh and P. S. Dragovich, ACS Med. Chem. Lett., 2016, 7, 988–993 CrossRef CAS PubMed.
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed.
- L. B. Harrington, D. Paez-Espino, B. T. Staahl, J. S. Chen, E. B. Ma, N. C. Kyrpides and J. A. Doudna, Nat. Commun., 2017, 8, 1424 CrossRef PubMed.
- B. Lee, K. Lee, S. Panda, R. Gonzales-Rojas, A. Chong, V. Bugay, H. M. Park, R. Brenner, N. Murthy and H. Y. Lee, Nat. Biomed. Eng., 2018, 2, 497–507 CrossRef CAS PubMed.
- D. B. T. Cox, R. J. Platt and F. Zhang, Nat. Med., 2015, 21, 121–131 CrossRef CAS.
- J. van Haasteren, J. Li, O. J. Scheideler, N. Murthy and D. V. Schaffer, Nat. Biotechnol., 2020, 38, 845–855 CrossRef CAS PubMed.
- I. Lostale-Seijo, I. Louzao, M. Juanes and J. Montenegro, Chem. Sci., 2017, 8, 7923–7931 RSC.
- S. Ramakrishna, A. B. Kwaku Dad, J. Beloor, R. Gopalappa, S. K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed.
- H. X. Wang, Z. Song, Y. H. Lao, X. Xu, J. Gong, D. Cheng, S. Chakraborty, J. S. Park, M. Li, D. Huang, L. Yin, J. Cheng and K. W. Leong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4903–4908 CrossRef CAS PubMed.
- M. Vazdar, J. Heyda, P. E. Mason, G. Tesei, C. Allolio, M. Lund and P. Jungwirth, Acc. Chem. Res., 2018, 51, 1455–1464 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z. Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS.
- Z. Y. Mao, M. Bozzella, A. Seluanov and V. Gorbunova, DNA Repair, 2008, 7, 1765–1771 CrossRef CAS.
- J. Carlson-Stevermer, A. A. Abdeen, L. Kohlenberg, M. Goedland, K. Molugu, M. Lou and K. Saha, Nat. Commun., 2017, 8, 1711 CrossRef.
- N. Savic, F. C. A. S. Ringnalda, H. Lindsay, C. Berk, K. Bargsten, Y. Z. Li, D. Neri, M. D. Robinson, C. Ciaudo, J. Hall, M. Jinek and G. Schwank, Elife, 2018, 7, 33761 CrossRef PubMed.
- E. J. Aird, K. N. Lovendahl, A. St. Martin, R. S. Harris and W. R. Gordon, Commun. Biol., 2018, 1, 54 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00929f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |