
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation promoted self-assembly of π-conjugated polymers†
Garion E. J.
Hicks
 ,
Charles N.
Jarrett-Wilkins
,
Jenny R.
Panchuk
,
Joseph G.
Manion
and
Dwight S.
Seferos
*
,
Charles N.
Jarrett-Wilkins
,
Jenny R.
Panchuk
,
Joseph G.
Manion
and
Dwight S.
Seferos
*
Lash Miller Chemical Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dseferos@chem.utoronto.ca
First published on 6th April 2020
Abstract
Self-assembly is an attractive strategy for organizing molecules into ordered structures that can span multiple length scales. Crystallization Driven Self-Assembly (CDSA) involves a block copolymer with a crystallizable core-forming block and an amorphous corona-forming block that aggregate into micelles with a crystalline core in solvents that are selective for the corona block. CDSA requires core- and corona-forming blocks with very different solubilities. This hinders its use for the self-assembly of purely π-conjugated block copolymers since blocks with desirable optoelectronic properties tend to have similar solubilities. Further, this approach is not readily reversible, precluding stimulus-responsive assembly and disassembly. Here, we demonstrate that selective oxidative doping of one block of a fully π-conjugated block copolymer promotes the self-assembly of redox-responsive micelles. Heteroatom substitution in polychalcogenophenes enables the modulation of the intrinsic polymer oxidation potential. We show that oxidized micelles with a narrow size distribution form spontaneously and disassemble in response to a chemical reductant. This method expands the scope of π-conjugated polymers that can undergo controlled self-assembly and introduces reversible, redox-responsive self-assembly of π-conjugated polymers.
Introduction
The self-assembly of block copolymer nanostructures1 has been explored extensively in recent decades2–4 for a wide range of applications.5–11 Unique properties and functions emerge in self assembled nanostructures, due to precise molecular arrangements over a range of length scales and control over assembly and disassembly in response to distinct stimuli.1–6,10 Crystallization-Driven Self-Assembly (CDSA) is one technique for controlled block copolymer self-assembly in solution.2 It involves the crystallization of a core block into nanostructures, driven by solvent interactions, while an amorphous corona block remains solvated for solution stability.12,13 A preference for core crystallization drives the formation of micelle morphology.14 These micelles can be fragmented by sonication into shorter ‘seed’ structures, which have a low dispersity in size. Adding a solvated block copolymer, or a unimer, to the seeds preferentially causes micelle growth to occur epitaxially from the exposed crystal faces at the termini of the micelles. This control allows a range of complex micelle architectures to be formed such as block co-micelles,15 branched micelles,16 and larger hierarchical structures.17CDSA has been used with block copolymers containing π-conjugated cores and amorphous coronas.18–23 π-conjugated polymers have a wide range of applications in optoelectronic devices,24–26 where their properties are heavily impacted by their nanoscale morphology.27,28 Using CDSA, one-dimensional (1D) nanofibers18–22 and two-dimensional (2D) platelets23 can be prepared with control over length and area, allowing applications in transistors29 and as platforms for photophysical studies.30 Investigating nanostructure formation in π-conjugated polymers is necessary to expand the scope of functional organic materials.
However, nanostructures formed by CDSA have significant limitations. First, micelles formed by CDSA are kinetically trapped by crystallization and only assemble in response to solvent or temperature changes.12,13 Photo-responsive self-assembly has been demonstrated in specific π-conjugated polymers,31 but is irreversible. Expanding the scope of application requires nanostructures that assemble and disassemble in response to a diverse range of stimuli.5,6,10 Second, the core and corona blocks must possess significant solubility differences to allow selective crystallization. Unfortunately, π-conjugated polymers with desirable properties for optoelectronic applications, such as regioregularity, tend to have similar solubilities (i.e. they are primarily soluble in halogenated solvents) such that selective crystallization is very difficult.24,32–34 One approach to address, rather than differentiating the blocks, is to eliminate the corona block and instead add bulky aliphatic side chains to a crystallizable homopolymer.35,36 However, this approach requires extensive sidechain engineering and has only been demonstrated on a unique homopolymer; a general approach capable of working even with minimal block differentiation would be of greater use. Thus, expanding the range of stimuli to promote the self-assembly and scope of polymers that can be assembled is an important challenge.
Polychalcogenophenes are a well-studied class of π-conjugated polymers37 often utilized in CDSA19,21,38 whose properties can be predictably modified through heteroatom substitution.26,39 S- and Te-containing polychalcogenophenes are called poly(thiophene)s (PThs) and poly(tellurophene)s (PTes), respectively. Relative to PThs, PTes possess slightly reduced solubility in halogenated solvents,40 while their planarity and energetic barriers to backbone rotation are increased.41 In addition, PTes have an oxidation potential ∼0.2 eV higher than PThs, allowing the polymer to be oxidized more readily than other polychalcogenophenes.42 Polychalcogenophenes possess other attractive features; regioregular polychalcogenophenes will crystallize,43,44 which narrows their electronic band gap and increases their oxidation potential.45,46 Polychalcogenophene oxidation, or oxidative doping, results in cations delocalized along the polymer backbone, which improves their charge-transport properties in functional materials.42,47 Importantly, π-conjugated polymer oxidative doping also induces aggregation and precipitation from solution,48,49 a feature which we realized could be exploited to promote self-assembly.
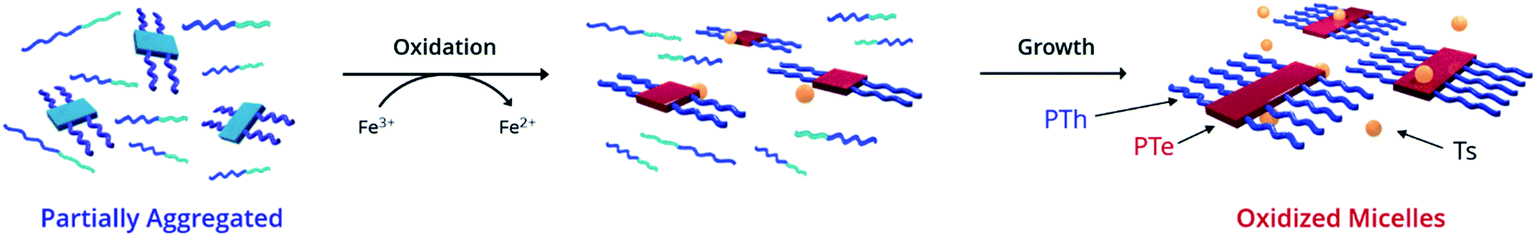
We hypothesized that in a π-conjugated block copolymer containing blocks with different oxidation potentials, selective oxidation would promote polymer self-assembly (Scheme 1). The oxidized block would aggregate and form a crystalline core, while the unoxidized corona block would promote colloidal stability, a process which we have termed Oxidation-Promoted Self-Assembly (OPSA). We took inspiration from supramolecular polymerization, wherein a chemical input can act as a fuel for assembly or disassembly to allow stimulus-responsive, dynamic, or transient systems.50–52 OPSA uses a chemical oxidant as a fuel for self-assembly and can therefore use a reductant fuel for disassembly. Oxidation has previously been shown to promote self-assembly in non-conjugated systems, but never in π-conjugated polymers.53,54 This process allows the reversible, stimulus-responsive nanostructure formation promoted by oxidative doping and provides the potential for a general method to promote self-assembly in π-conjugated block copolymers.
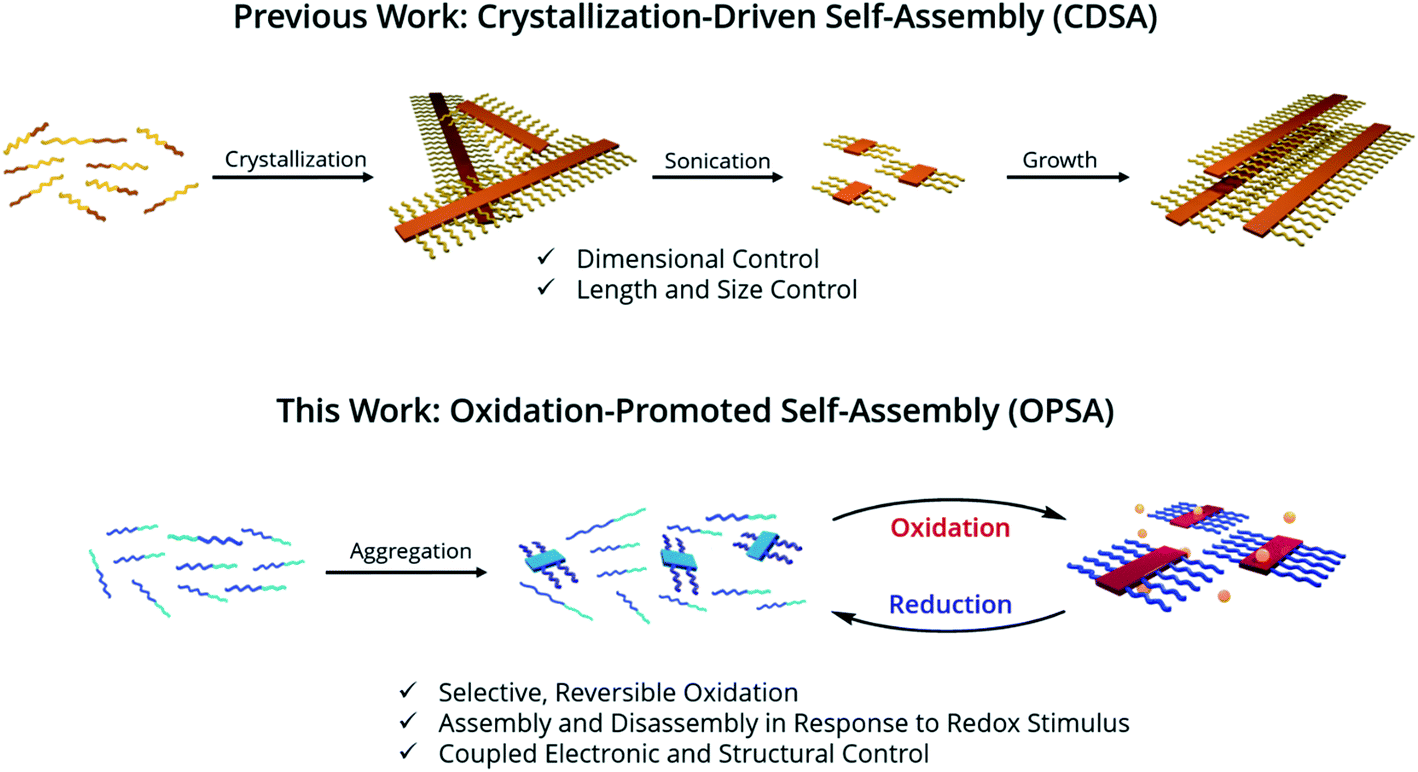 | ||
| Scheme 1 Strategy for 1D structural control in π-conjugated polymers. 1D CDSA is shown for comparison, in which crystallization drives the formation of a single micelle morphology. Sonication induces fragmentation of the polymer crystals, resulting in much smaller ‘seed’ micelles. An additional unimer preferentially crystallizes on the exposed crystal faces, resulting in length-controlled structures. OPSA utilizes selective oxidative doping of a single block of a π-conjugated polymer to drive self-assembly. Redox-active micelles with uniform lengths form which are reversibly disassembled upon reduction. | ||
Results and discussion
To test our hypothesis, poly((3-(3,7-dimethyloctyl)thiophene)-b-(3-(3,7-dimethyloctyl)tellurophene)) (PTh80-b-PTe60) (Scheme 2) was selected as a model system. A 3,7-dimethyloctyl side chain was chosen to enable the controlled polymerization of the PTe block to a high molecular weight.41 PTh80-b-PTe60 was prepared by Kumada catalyst-transfer polycondensation (KCTP) using previously reported conditions,55 and characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC) (Scheme 2) (see the ESI, Fig. S1–S4† for characterization and the Experimental section for synthetic details). The PTh and PTe blocks are 80 and 60 repeat units long, respectively, with a block copolymer dispersity (Đ) of 1.3. Heteroatom substitution allows us to prepare a block copolymer in which the blocks differ only in oxidation potential and solubility, allowing us to sequentially study the effects of solvent environment and oxidation on self-assembly.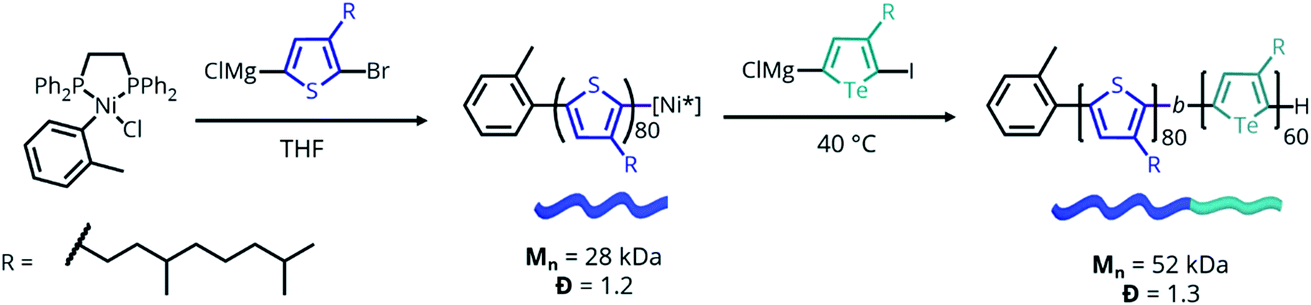 | ||
| Scheme 2 Synthesis of PTh80-b-PTe60. Mn and Đ determined by GPC performed at 1.0 mg mL−1 in 1,2,4-trichlorobenzene at 140 °C. | ||
Ultraviolet-Visible-Near Infrared Absorption Spectroscopy (UV-Vis-NIR) was used to understand the state of solvation and oxidation of the polymer. Solvated PTh80-b-PTe60 in chloroform displays a major absorption peak at 450 nm characteristic of the π–π* transition of PTh, with a minor absorption shoulder at ∼600 nm corresponding to the π–π* transition of PTe (Fig. 1b, black trace). After ageing for 18 h in a 95% dichloromethane (DCM)/5% chloroform solution, a fraction of PTh80-b-PTe60 aggregates into micelles with a PTe core to form a partially aggregated solution. Partially aggregated PTh80-b-PTe60 (blue trace) retains the PTh signal, with new peaks appearing from 650–900 nm which are associated with solid-state PTe that we attribute to PTe aggregation (consistent with solid-state absorption data; see the ESI, Fig. S5†). The observed vibronic structure indicates molecular alignments previously attributed to polymer crystallization.44,56–58 Once this spectral change was observed, we added the oxidant iron(III) p-toluenesulfonate (FeTs3), which has been previously used to oxidatively dope PTe and is soluble in organic solvents.42 Upon the addition of iron(III) p-toluenesulfonate (FeTs3) (50 μM), we observe broad absorption in the near-IR region (red trace, >900 nm) indicative of (bi)polarons.59 Polarons and bipolarons arise from radical cations and dications on the polymer backbone, compensated by the p-toluenesulfonate counter ion, and therefore demonstrate oxidation. The PTh absorption peak at 450 nm is unchanged while the PTe signal at ∼600 nm decreases, indicating that the PTe block is selectively oxidized. A further increase of FeTs3 concentration (see the ESI, Fig. S6†) is correlated with the decreasing intensity of the crystalline PTe absorption from 650–900 nm and increasing intensity of the (bi)polaron signal. This inverse relationship strongly suggests that the degree of oxidation can be modulated, and that oxidation is selective for PTe.47 Electron Paramagnetic Resonance (EPR) spectroscopy (Fig. 1d) confirms the presence of a radical cation species in oxidized PTh80-b-PTe60, with a singlet centered at a g-factor of 2.0023 consistent with an organic radical cation and identical to that of a free electron.60 The evidence demonstrates that the PTe block has been selectively oxidized and suggests it crystallizes as well.
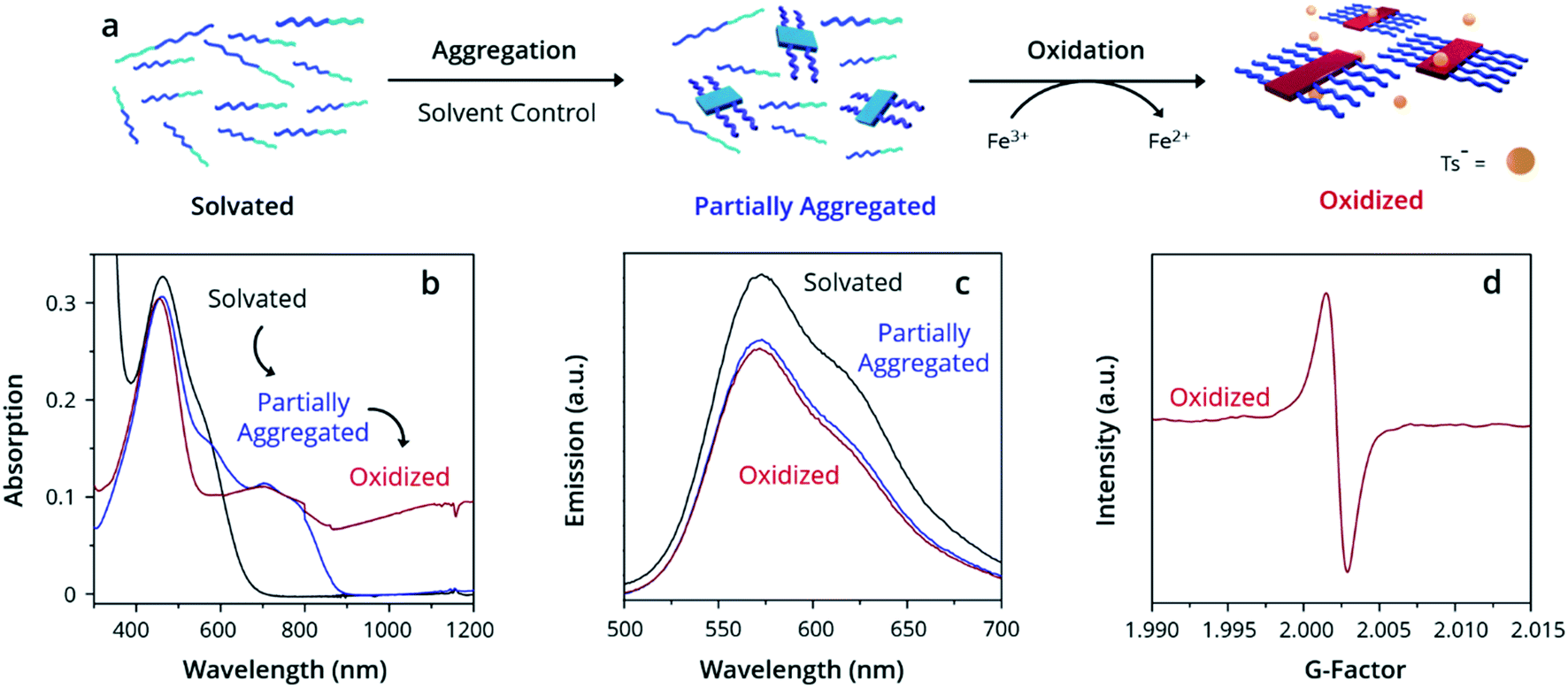 | ||
| Fig. 1 Oxidation-promoted self-assembly. (a) Schematic depicting the aggregation and oxidation of PTh80-b-PTe60 by FeTs3 to oxidized micelles. Demonstrated by (b) UV-Vis-NIR spectra, (c) PL spectra, and (d) EPR spectra. All solutions prepared at 0.05 mg mL−1 PTh80-b-PTe60. | ||
While PTe is selectively crystallized and oxidized, PTh remains fully solvated. Solvated poly(thiophene)s exhibit strong photoluminescence (PL) that is quenched by polymer crystallization.61 By contrast, poly(tellurophene)s are only marginally photoluminescent, displaying emission three orders of magnitude weaker than poly(thiophene)s.62 Emission observed from PTh80-b-PTe60 must be due to the PTh block as any PTe emission is negligible; PL spectroscopy is therefore an excellent tool for exclusively assessing PTh solvation. PL spectroscopy of solvated PTh80-b-PTe60 shows strong emission from 525–700 nm upon excitation at 450 nm (Fig. 1c) as we expect. Photoluminescence is slightly reduced in both partially aggregated and oxidized PTh80-b-PTe60 due to quenching by crystallized PTe. However, the majority retention of photoluminescence is strong evidence for the neutral, solvated state of PTh. Both UV-Vis-NIR and PL spectroscopy indicate PTh solvation in the solvated, partially aggregated, and oxidized samples.
By examining the structure of the partially aggregated sample, we observe the limits of solvent-driven assembly. First, Dynamic Light Scattering (DLS) was used to measure the apparent hydrodynamic radius (Rhapp) of polymer aggregates in solution. There are two size distributions in the partially aggregated sample (Fig. 2a, blue trace); a major distribution (Rhapp ∼ 20 nm) attributed to an aggregate species and a minor distribution (Rhapp ∼ 5 nm) attributed to a unimer. Partially aggregated PTh80-b-PTe60 can therefore best be described as a mixture of small aggregates and the unimer, which is consistent with the UV-Vis-NIR peak attributed to crystalline PTe (blue trace, Fig. 1b). This characterization is consistent with Scanning Transmission Electron Microscopy (STEM) images of the drop-cast, partially aggregated solution (see the ESI, Fig. S7†). We observe primarily amorphous features attributed to the unimer and a broad length distribution of 1D fibers, including ∼10–20 nm long aggregates. The longer fibres likely result from unimer deposition on aggregates during drying.33 This demonstrates the presence of the unimer and small aggregates in the partially aggregated sample, and it highlights that the solvent conditions are insufficient to control the self-assembly of PTh80-b-PTe60. This may be due to similar solubilities, poor PTe crystallization limiting a crystallization-driven approach, or both.
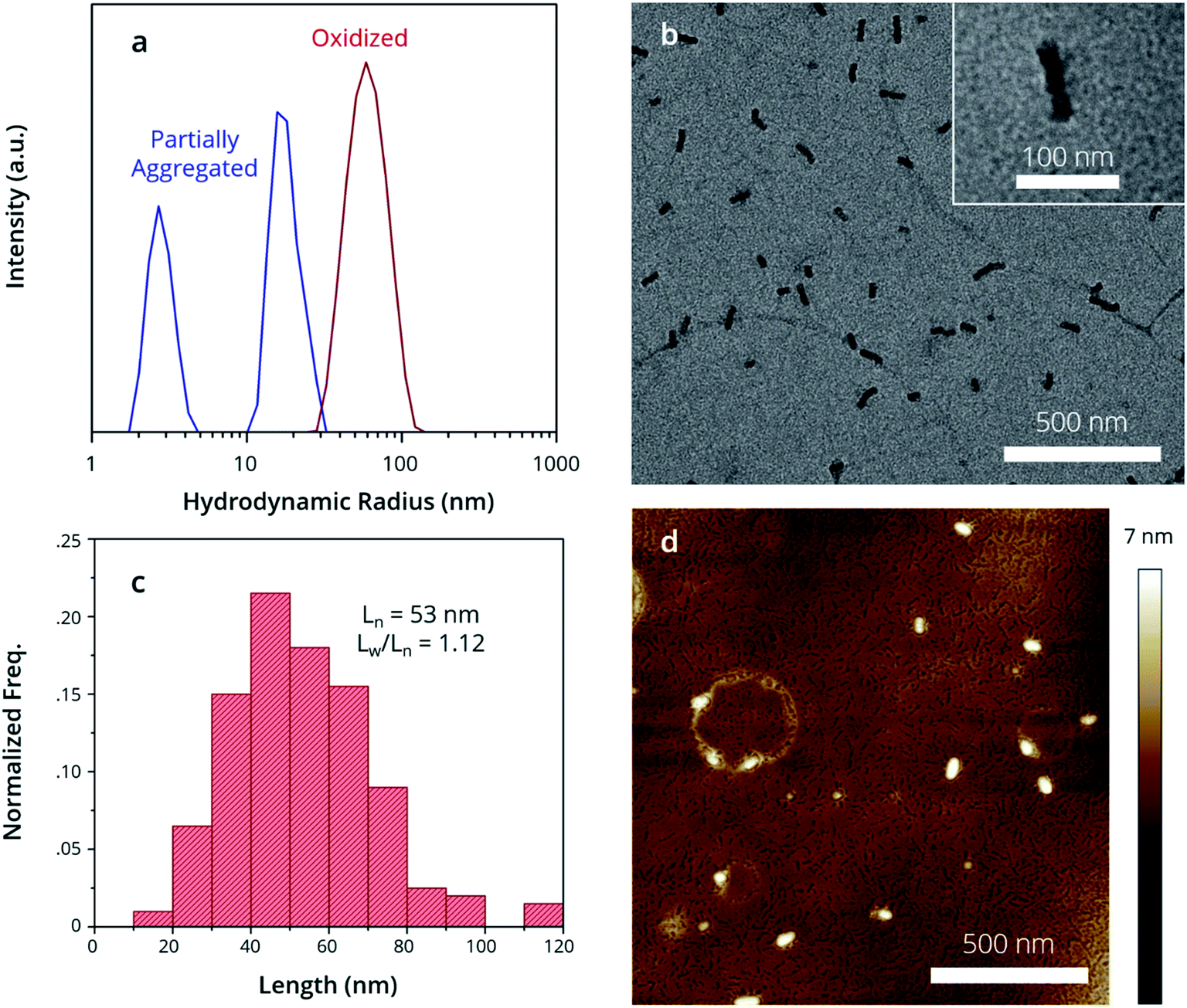 | ||
| Fig. 2 Structure of oxidized micelle seeds. (a) DLS demonstrates oxidized micelle structure in solution, while morphologies are observed by (b) TEM images, (c) contour length distribution of micelle seeds in (a) and (d) AFM images. | ||
Oxidation, conversely, promotes complete self-assembly into micelles. DLS indicates a single size distribution (Fig. 2a, red trace) which is larger (Rhapp ∼ 60 nm) than partially aggregated PTh80-b-PTe60. By Transmission Electron Microscopy (TEM), we observe exclusively 1D nanostructures after oxidation (Fig. 2b) with uniform widths of 20 ± 2 nm. The crystalline core-forming PTe block has an average contour length of approximately 17 nm,63 suggesting that the micelle widths result from fully extended PTe upon crystallization. An average micelle contour length (Ln) of 53 nm (Fig. 2c) with an Ln/Lw of 1.12 is consistent with DLS measurements, corroborating that this is the species we observe in solution. The absence of a detectable unimer in both DLS and TEM suggests that oxidation promotes complete aggregation. To measure the widths of the full polymer micelles, i.e. of both the PTe core and PTh corona, we used Atomic Force Microscopy (AFM). The full micelle width is expected to be equal to the PTe core block width plus a large contribution from PTh,64 which has a contour length of 22 nm.63 We measure micelle widths of 49 ± 4 nm by AFM (Fig. 2d), which is consistent with a crystalline PTe core with the PTh corona being present on both sides of the core (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism for oxidation promoted self-assembly. | ||
To rationalize the narrow oxidized micelle size distribution, we propose this mechanism for OPSA. First, the PTe aggregates formed by solvent conditions become preferentially oxidized due their higher intrinsic oxidation potential (Scheme 3).43,65 Next, growth occurs as the electron-rich unimer is attracted to the oxidized aggregates, while oxidized aggregates are repelled from one another. Finally, larger micelles are formed as the unimer is added epitaxially to the exposed crystal faces and becomes oxidized.43,48,65 In this way, the process is promoted by both electrochemical induction, which causes the electron-rich unimer to be attracted to the oxidized aggregates, as well as the greater stablization of the radical cations by delocalization over a larger crystalline polymer domain.66,67 This mechanism is consistent with the increase in size and the loss of the unimer observed upon oxidation. The small size and narrow distribution of the oxidized micelles originates from the even smaller size of the initially aggregated PTe nuclei (Fig. 2a) which appear to only add the unimer and do not aggregate with each other.
The micelles in their oxidized form are colloidally stable, contain a crystalline core, and are likely kinetically trapped. Long-term colloidal stability was assessed by aging the PTh80-b-PTe60 oxidized micelles under an inert atmosphere for 2 months in a sealed container. DLS and STEM measurements of the oxidized micelles after 2 months display no change in micelle size or morphology (see the ESI, Fig. S11 and S12†). This indicates that, in the absence of an external stimulus, the micelles remain assembled and colloidally stable. It further suggests that the micelles are kinetically trapped in their oxidized state much like micelles formed by CDSA.12,13,19 Kinetic trapping implies that there is a large energetic barrier to dissociation, potentially due to a crystalline core, which we investigated by Wide-Angle X-ray Scattering (WAXS). The oxidized micelles display a broad scattering peak with a d-spacing of 24.2 Å (see the ESI, Fig. S13†), wider than the previously reported homopolymer PTh and PTe d-spacings of 22.8 and 19.0 Å, respectively.68 This wider spacing between scattering planes suggests the intercalation of counter ions between the PTe crystalline lamellae. Given the need to charge balance the positively charged, oxidized PTe, it is expected that the presence of counter ions would increase the d-spacing. This is consistent with the oxidation-induced increase in d-spacing previously observed by our group in PTe homopolymers.42 Importantly, it demonstrates that a change in crystallization is associated with oxidation, and is consistent with micelles that are kinetically trapped in their oxidized form.
Perhaps the most striking feature of this process, however, is its reversible nature. Unique to OPSA, the addition of a chemical reductant causes disassembly of PTh80-b-PTe60 oxidized micelles back to neutral aggregates and free unimers. Bis(pentamethyl-cyclopentadienyl)cobalt(II) [Co(Cp*)2], a strong reducing agent with a high oxidation potential of 3.2 eV,69 was added to the oxidized PTh80-b-PTe60 at 100 μM to return the polymer to its neutral state (Fig. 3a). By DLS, the reduced PTh80-b-PTe60 displays two size distributions (Fig. 3b, blue trace) which are nearly identical to the partially aggregated PTh80-b-PTe60 size distributions prior to oxidation (Fig. 2a, blue trace). No (bi)polaron states are observed by UV-Vis-NIR spectroscopy (see the ESI, Fig. S14†), with an absorption spectrum identical to the nucleated PTh80-b-PTe60 pre-oxidation (Fig. 1b), confirming the reduction to a neutral polymer. This evidence demonstrates that oxidized micelles reversibly assemble and disassemble in response to redox stimuli and are only kinetically trapped when oxidized.
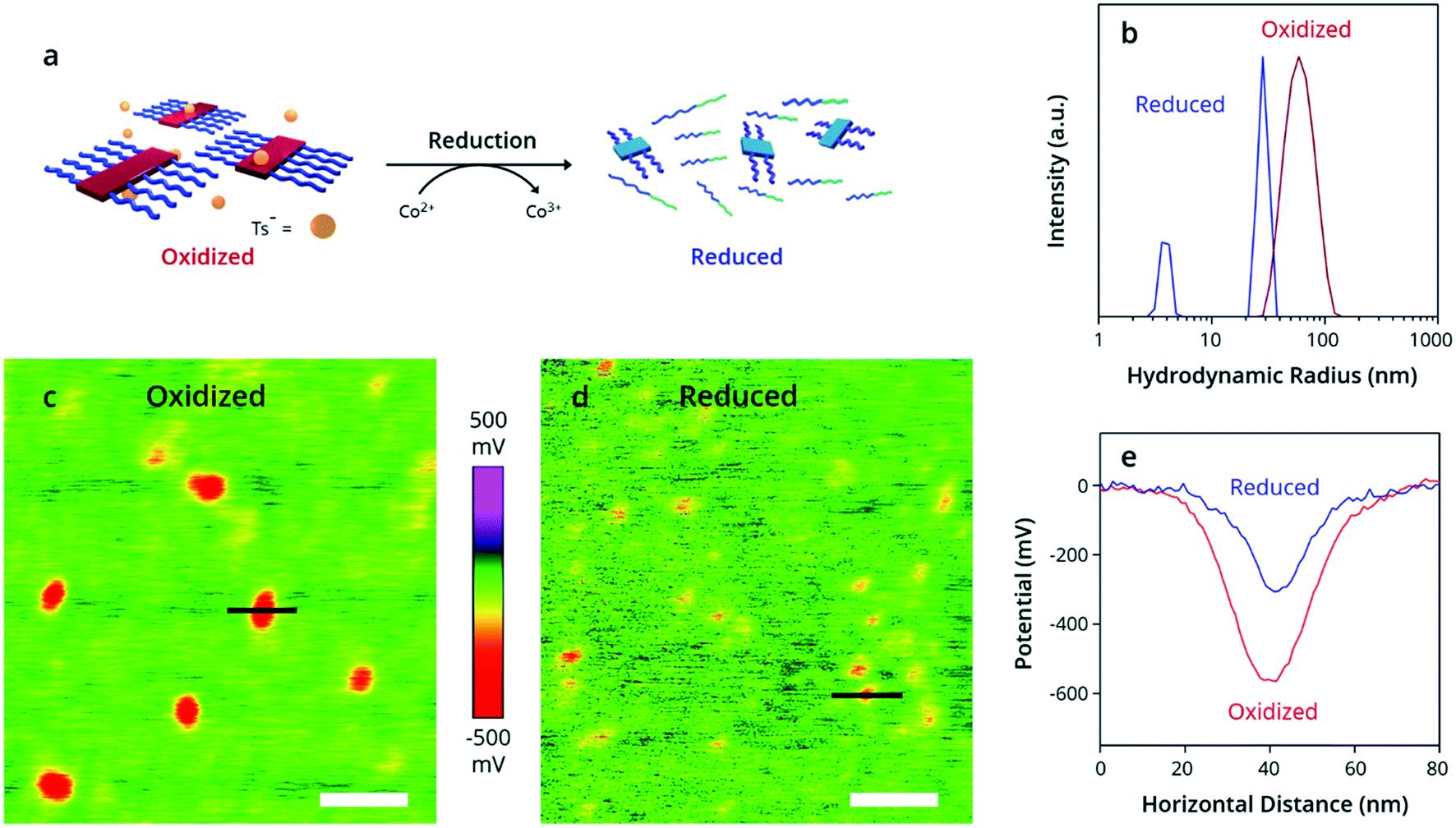 | ||
| Fig. 3 Reduction of oxidized micelles to a neutral polymer. (a) Schematic depicting addition of Co(Cp*)2 to oxidized PTh80-b-PTe60, resulting in reduced PTh80-b-PTe60. (b) DLS demonstrates the change in size distributions from oxidized (red trace) to reduced (blue trace) PTh80-b-PTe60. (c–e) Kelvin probe force microscopy images of the (c) oxidized and (d) reduced polymer, with potential measurements across indicated black lines plotted in (e). White scale bars = 100 nm. | ||
Kelvin Probe Force Microscopy (KPFM) (Fig. 3c–e) was used to measure the change in work function and enable the estimation of the doping level. KPFM allows spatial mapping of the chemical potential and work function (φ) across a surface70 referenced to the background substrate (φSi ∼ 4.6 eV). We measured a lower potential and higher work function in the oxidized micelles (Fig. 3e). For comparison, we prepared PTe films dip-doped at 1, 5, and 10 mM FeTs3. The difference in work function between the neutral and oxidized samples is 270 ± 90 meV for the micelles, which agrees with the 240 ± 50 meV difference in work function for the 1 mM dip-doped PTe film (Table S1†). Because a 1 mM dip-doped PTe film has a doping level of 10–30%,42,47 we estimate a 10–30% doping level for the oxidized micelles.71,72
Conclusion
This work demonstrates oxidation-promoted self-assembly. Oxidized micelles of a narrow size distribution reversibly form due to the selective chemical oxidation of a single block of a π-conjugated block copolymer. This selectivity is enabled by the 0.2 eV higher oxidation potential of PTe relative to PTh, such that self-assembly can be achieved with only heteroatom substitution differentiating the polymer blocks. The high similarity of the blocks in this study demonstrates the potential for a general method that can be used in a wide range of π-conjugated block copolymers as long as the blocks differ in oxidation potential. We hypothesized that the growth of the oxidized micelles and their narrow size-distribution are the consequence of a mechanism wherein the PTe nuclei are oxidized first in solution.Critically, the structural dependence of the micelles on the polymer oxidation state allows oxidized micelles to be responsive to a redox stimulus. Beyond this proof-of-concept, OPSA could be employed to fabricate stimulus-responsive functional materials composed of all-regioregular π-conjugated polymers. Nanostructures formed by OPSA possess further utility as a platform to study the oxidative doping process. The demonstration of OPSA in a larger range of π-conjugated polymers and in a range of solvents for specialized applications is a subject of future work.
Experimental section
Materials
All reagents were purchased from Sigma-Aldrich and used as received unless otherwise noted. Monomers and block copolymer PTh80-b-PTe60 were synthesized according to the literature procedures.55 [1,2-Bis(diphenylphosphino)ethane](o-tolyl)chloronickel(II) was synthesized according to a literature procedure.73 Iron(III) p-toluenesulfonate hexahydrate (462861) was dried under vacuum at 75 °C to eliminate hydration adducts prior to use. DCM and chloroform were dried over calcium choride, distilled from phosphorus pentoxide, then stored over 4 Å molecular sieves in darkness in an atmosphere of dry N2. THF was dried over sodium metal overnight, then cannulated into a sealed bomb and stored over 4 Å molecular sieves in darkness in an atmosphere of dry N2.Instrumentation
NMR spectra were recorded on a 500 MHz Agilent DD2. Chemical shifts are reported in ppm at ambient temperature. 1H chemical shifts are referenced to the residual protonated chloroform peak at 7.26 ppm. Polymer molecular weights were determined with a Viscotek HT-GPC (1,2,4-trichlorobenzene, 140 °C, and 1 mL min−1 flow rate) using Tosoh Bioscience LLC TSK-GEL GMHHR-HT mixed-bed columns and narrow molecular weight distribution polystyrene standards. A UV detector was used to detect the eluted polymer with respect to elution volume. UV-Vis-NIR absorption spectra were recorded on a Varian Cary 5000 UV-vis-NIR spectrophotometer using Hellma quartz cuvettes with a 5 mm path length. Baseline correction was performed by subtracting the spectrum of the pure solvent. Light scattering experiments were carried out using a Malvern Zetasizer Nano-ZS instrument equipped with a He–Ne laser (λ = 633 nm). The TEM imaging was performed on a Hitachi HT7700 microscope at an acceleration voltage of 80 kV. Scattered electrons were blocked with an objective aperture in the focal plane of the objective lens. The STEM imaging was performed on a QUANTA FEG 250 microscope at an acceleration voltage of 20 kV. Dimension measurements were obtained by direct measurement using ImageJ of 200 individual structures on different parts of the grid. Carbon-coated copper grids were exposed to UV-radiation for 30 minutes prior to use. One drop of the sample solution was dropped onto the grids and allowed to dry overnight. AFM and KPFM were conducted using a Bruker Dimension Icon atomic force microscope. AFM measurements were performed using a Bruker NCHV-A antimony (n) doped silicon tip with a nominal tip radius of 8 nm in tapping mode, with width measurements obtained on >25 different structures. KPFM measurements were performed using PQFNE-AL silicon nitride tips in Peakforce KPFM mode; micelle samples were prepared by immersing a silicon wafer in the sample for 30 s, then washing twice in pure DCM for 1 min. Films of PTe were spin-coated from 10 mg mL−1 chlorobenzene solutions onto indium tin oxide (ITO) on glass. Films were dip-doped into acetonitrile solutions of FeTs3 for 3 minutes then washed with neat acetonitrile to remove any residual FeTs3. WAXS measurements were performed using an Anton Paar SAXSpace. All self-assembly was performed under a dry N2 atmosphere.Synthesis of PTh80-b-PTe60
Isopropyl magnesium chloride (0.97 eq.) was added to a solution of 2,5-dibromo-3-(3,7-dimethyloctyl)thiophene (173 mg, 0.45 mmol) in dry THF, and the mixture was stirred for 15 min at rt. External initiator Ni(dppe)(o-tolyl)Cl (2.57 mg, 4.4 μmol) was added in THF, and the mixture was stirred at rt for 30 min, after which an aliquot (1.0 mL) was removed and quenched with dilute HCl followed by extraction with chloroform. The organic layer was dried over MgSO4 and concentrated under reduced pressure. In a separate flask, isopropyl magnesium chloride (0.97 eq.) was added to a solution of 2,5-diiodo-2-(3,7-dimethyloctyl)tellurophene (194 mg, 0.34 mmol), which was then added to the polymerization flask. The polymerization mixture was immersed in an oil bath at 40 °C and allowed to stir for an additional 1 h before quenching with dilute HCl. The polymer was precipitated into methanol and purified by sequential Soxhlet extraction (methanol, hexane, and chloroform). The chloroform fraction was further purified by column chromatography in chloroform before being concentrated under reduced pressure to yield a dark purple solid (54.2 mg, 27%). GPC profiles (see the ESI, Fig. S4†) and 1H NMR spectra of the monomers and block copolymer (see the ESI, Fig. S1–S3) are provided in the ESI.†General procedure for self-assembly of PTe–PTh
1.0 mg of the block copolymer poly((3-(3,7-dimethyloctyl)thiophene)-b-(3-(3,7-dimethyloctyl)tellurophene)) was dissolved in 20.0 mL of 95% DCM and 5% chloroform in a vial to form a 0.05 mg mL−1 red solution. The polymer solution was then allowed to age for 18 h, becoming orange-yellow in colour. Iron(III) p-toluenesulfonate (0.57 mg, 1.0 μmol) was added in a 0.5 mM standard solution in THF to a separate vial, and the solvent was removed under reduced pressure. The polymer solution was added to iron(III) p-toluenesulfonate (50 μM), after which the solution immediately turned bright yellow. This solution was allowed to age 18 h prior to characterization.Procedure for disassembly of PTe–PTh
20.0 mL of a 0.05 mg mL−1 solution of the block copolymer in 95% DCM and 5% chloroform containing iron(III) p-toluenesulfonate (50 μM) was prepared as described above. Bis(pentamethyl-cyclopentadienyl)cobalt(II) (0.66 mg, 2.0 μmol) was added in 0.1 mL DCM, after which the solution changed from bright yellow to orange. The solution was allowed to age 1 h prior to characterization.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the University of Toronto, the NSERC, the CFI, and the Ontario Research Fund. D. S. S. is grateful to the Connaught Foundation for the McLean Award and the NSERC for the E. W. R. Steacie Memorial Fellowship. We are grateful to Nimrat K. Obhi and Mitchell A. Winnik for editing and advice.References
- Y. Mai and A. Eisenberg, Self-assembly of block copolymers, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, 50th Anniversary Perspective: Functional Nanoparticles from the Solution Self-Assembly of Block Copolymers, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, Getting into Shape: Reflections on a New Generation of Cylindrical Nanostructures' Self-Assembly Using Polymer Building Blocks, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- G. M. Whitesides, Self-Assembly at All Scales, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Design of polymeric nanoparticles for biomedical delivery applications, Chem. Soc. Rev., 2012, 41, 2545 RSC.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Polymeric Nanostructures for Imaging and Therapy, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- N. Haberkorn, S. Kim, K.-S. Kim, M. Sommer, M. Thelakkat, B.-H. Sohn and P. Theato, Template-Assisted Fabrication of Highly Ordered Interpenetrating Polymeric Donor/Acceptor Nanostructures for Photovoltaic Applications, Macromol. Chem. Phys., 2011, 212, 2142–2150 CrossRef CAS.
- R. Li, H. Wang, Y. Song, Y.-N. Lin, M. Dong, Y. Shen, S. Khan, S. Zhang, J. Fan, F. Zhang, L. Su and K. L. Wooley, In Situ Production of Ag/Polymer Asymmetric Nanoparticles via a Powerful Light-Driven Technique, J. Am. Chem. Soc., 2019, 141, 19542–19545 CrossRef CAS PubMed.
- J. Yuan, Y. Xu, A. Walther, S. Bolisetty, M. Schumacher, H. Schmalz, M. Ballauff and A. H. E. Müller, Water-soluble organo-silica hybrid nanowires, Nat. Mater., 2008, 7, 718–722 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Emerging applications of stimuli-responsive polymer materials, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- G. M. Whitesides, The ‘right’ size in nanobiotechnology, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS PubMed.
- R. C. Hayward and D. J. Pochan, Tailored Assemblies of Block Copolymers in Solution: It Is All about the Process, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Block Copolymer Assembly via Kinetic Control, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, Self-Assembly of Organometallic Block Copolymers: The Role of Crystallinity of the Core-Forming Polyferrocene Block in the Micellar Morphologies Formed by Poly(ferrocenylsilane-b-dimethylsiloxane) in n-Alkane Solvents, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Cylindrical block copolymer micelles and co-micelles of controlled length and architecture, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- H. Qiu, Y. Gao, V. A. Du, R. Harniman, M. A. Winnik and I. Manners, Branched Micelles by Living Crystallization-Driven Block Copolymer Self-Assembly under Kinetic Control, J. Am. Chem. Soc., 2015, 137, 2375–2385 CrossRef CAS PubMed.
- X. Li, Y. Gao, C. E. Boott, M. A. Winnik and I. Manners, Non-covalent synthesis of supermicelles with complex architectures using spatially confined hydrogen-bonding interactions, Nat. Commun., 2015, 6, 8127 CrossRef PubMed.
- J. Qian, G. Guerin, Y. Lu, G. Cambridge, I. Manners and M. A. Winnik, Self-Seeding in One Dimension: An Approach To Control the Length of Fiberlike Polyisoprene-Polyferrocenylsilane Block Copolymer Micelles, Angew. Chem., Int. Ed., 2011, 50, 1622–1625 CrossRef CAS PubMed.
- U. Tritschler, J. Gwyther, R. L. Harniman, G. R. Whittell, M. A. Winnik and I. Manners, Toward Uniform Nanofibers with a π-Conjugated Core: Optimizing the “Living” Crystallization-Driven Self-Assembly of Diblock Copolymers with a Poly(3-octylthiophene) Core-Forming Block, Macromolecules, 2018, 51, 5101–5113 CrossRef CAS.
- D. Tao, C. Feng, Y. Cui, X. Yang, I. Manners, M. A. Winnik and X. Huang, Monodisperse Fiber-like Micelles of Controlled Length and Composition with an Oligo(p-phenylenevinylene) Core via “Living” Crystallization-Driven Self-Assembly, J. Am. Chem. Soc., 2017, 139, 7136–7139 CrossRef CAS PubMed.
- E. L. Kynaston, O. E. C. Gould, J. Gwyther, G. R. Whittell, M. A. Winnik and I. Manners, Fiber-Like Micelles from the Crystallization-Driven Self-Assembly of Poly(3-heptylselenophene)-block-Polystyrene, Macromol. Chem. Phys., 2015, 216, 685–695 CrossRef CAS.
- J. Qian, Y. Lu, A. Chia, M. Zhang, P. A. Rupar, N. Gunari, G. C. Walker, G. Cambridge, F. He, G. Guerin, I. Manners and M. A. Winnik, Self-Seeding in One Dimension: A Route to Uniform Fiber-like Nanostructures from Block Copolymers with a Crystallizable Core-Forming Block, ACS Nano, 2013, 7, 3754–3766 CrossRef CAS PubMed.
- L. Han, M. Wang, X. Jia, W. Chen, H. Qian and F. He, Uniform two-dimensional square assemblies from conjugated block copolymers driven by π–π interactions with controllable sizes, Nat. Commun., 2018, 9, 865 CrossRef PubMed.
- L. Ying, F. Huang and G. C. Bazan, Regioregular narrow-bandgap-conjugated polymers for plastic electronics, Nat. Commun., 2017, 8, 14047 CrossRef CAS PubMed.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Organic and solution-processed tandem solar cells with 17.3% efficiency, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- J. G. Manion, J. R. Panchuk and D. S. Seferos, Applying Heteroatom Substitution in Organic Photovoltaics, Chem. Rec., 2019, 19, 1113–1122 CrossRef CAS PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, A general relationship between disorder, aggregation and charge transport in conjugated polymers, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- J. Vogelsang, T. Adachi, J. Brazard, D. A. Vanden Bout and P. F. Barbara, Self-assembly of highly ordered conjugated polymer aggregates with long-range energy transfer, Nat. Mater., 2011, 10, 942–946 CrossRef CAS PubMed.
- X. Li, P. J. Wolanin, L. R. MacFarlane, R. L. Harniman, J. Qian, O. E. C. Gould, T. G. Dane, J. Rudin, M. J. Cryan, T. Schmaltz, H. Frauenrath, M. A. Winnik, C. F. J. Faul and I. Manners, Uniform electroactive fibre-like micelle nanowires for organic electronics, Nat. Commun., 2017, 8, 15909 CrossRef CAS PubMed.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Long-range exciton transport in conjugated polymer nanofibers prepared by seeded growth, Science, 2018, 360, 897–900 CrossRef CAS PubMed.
- S. Shin, F. Menk, Y. Kim, J. Lim, K. Char, R. Zentel and T.-L. Choi, Living Light-Induced Crystallization-Driven Self-Assembly for Rapid Preparation of Semiconducting Nanofibers, J. Am. Chem. Soc., 2018, 140, 6088–6094 CrossRef CAS PubMed.
- T. M. Swager, 50th Anniversary Perspective: Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- N. E. Persson, P.-H. Chu, M. McBride, M. Grover and E. Reichmanis, Nucleation, Growth, and Alignment of Poly(3-hexylthiophene) Nanofibers for High-Performance OFETs, Acc. Chem. Res., 2017, 50, 932–942 CrossRef CAS PubMed.
- G. L. Schulz and S. Ludwigs, Controlled Crystallization of Conjugated Polymer Films from Solution and Solvent Vapor for Polymer Electronics, Adv. Funct. Mater., 2017, 27, 1603083 CrossRef.
- I. Choi, S. Yang and T.-L. Choi, Preparing Semiconducting Nanoribbons with Tunable Length and Width via Crystallization-Driven Self-Assembly of a Simple Conjugated Homopolymer, J. Am. Chem. Soc., 2018, 140, 17218–17225 CrossRef CAS PubMed.
- S. Yang, S. Kang and T. Choi, Morphologically Tunable Square and Rectangular Nanosheets of a Simple Conjugated Homopolymer by Changing Solvents, J. Am. Chem. Soc., 2019, 141, 19138–19143 CrossRef CAS PubMed.
- J. P. Lutz, M. D. Hannigan and A. J. McNeil, Polymers synthesized via catalyst-transfer polymerization and their applications, Coord. Chem. Rev., 2018, 376, 225–247 CrossRef CAS.
- J. Qian, X. Li, D. J. Lunn, J. Gwyther, Z. M. Hudson, E. Kynaston, P. A. Rupar, M. A. Winnik and I. Manners, Uniform, High Aspect Ratio Fiber-like Micelles and Block Co-micelles with a Crystalline π-Conjugated Polythiophene Core by Self-Seeding, J. Am. Chem. Soc., 2014, 136, 4121–4124 CrossRef CAS PubMed.
- E. I. Carrera and D. S. Seferos, Semiconducting Polymers Containing Tellurium: Perspectives Toward Obtaining High-Performance Materials, Macromolecules, 2015, 48, 297–308 CrossRef CAS.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. Buchaca Domingo, C. Hellmann, Y. Lee and D. S. Seferos, Poly(3-alkyltellurophene)s Are Solution-Processable Polyheterocycles, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed.
- S. Ye, L. Janasz, W. Zajaczkowski, J. G. Manion, A. Mondal, T. Marszalek, D. Andrienko, K. Müllen, W. Pisula and D. S. Seferos, Self-Organization and Charge Transport Properties of Selenium and Tellurium Analogues of Polythiophene, Macromol. Rapid Commun., 2019, 40, 1800596 CrossRef PubMed.
- J. R. Panchuk, A. W. Laramée, J. G. Manion, S. Ye and D. S. Seferos, Heavy atom substitution—A strategy for improving conductivity in conjugated polymers, Synth. Met., 2019, 253, 57–61 CrossRef CAS.
- K. Tang, F. M. McFarland, S. Travis, J. Lim, J. D. Azoulay and S. Guo, Aggregation of P3HT as a preferred pathway for its chemical doping with F 4-TCNQ, Chem. Commun., 2018, 54, 11925–11928 RSC.
- F. C. Spano and C. Silva, H- and J-aggregate behavior in polymeric semiconductors, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- T. Eder, J. Vogelsang, S. Bange, K. Remmerssen, D. Schmitz, S. Jester, T. J. Keller, S. Höger and J. M. Lupton, Interplay between J- and H-type coupling in aggregates of π-conjugated polymers: a single-molecule perspective, Angew. Chem., Int. Ed., 2019, 58, 18898–18902 CrossRef CAS PubMed.
- H. Yamagata and F. C. Spano, Interplay between intrachain and interchain interactions in semiconducting polymer assemblies: The HJ-aggregate model, J. Chem. Phys., 2012, 136, 184901 CrossRef CAS PubMed.
- S. A. Gregory, A. K. Menon, S. Ye, D. S. Seferos, J. R. Reynolds and S. K. Yee, Effect of heteroatom and doping on the thermoelectric properties of poly(3-alkylchalcogenophenes), Adv. Energy Mater., 2018, 8, 1802419 CrossRef.
- F. M. McFarland, C. M. Ellis and S. Guo, The aggregation of poly(3-hexylthiophene) into nanowires: with and without chemical doping, J. Phys. Chem. C, 2017, 121, 4740–4746 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Electrochemistry of Conducting Polymers—Persistent Models and New Concepts†, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Transient assembly of active materials fueled by a chemical reaction, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- J. Leira-Iglesias, A. Tassoni, T. Adachi, M. Stich and T. M. Hermans, Oscillations, travelling fronts and patterns in a supramolecular system, Nat. Nanotechnol., 2018, 13, 1021–1027 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Non-equilibrium steady states in supramolecular polymerization, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed.
- J.-C. Eloi, D. A. Rider, G. Cambridge, G. R. Whittell, M. A. Winnik and I. Manners, Stimulus-responsive self-assembly: reversible, redox-controlled micellization of polyferrocenylsilane diblock copolymers, J. Am. Chem. Soc., 2011, 133, 8903–8913 CrossRef CAS PubMed.
- T. Yoshizawa, M. Onoda, T. Ueki, R. Tamate, A. M. Akimoto and R. Yoshida, Fabrication of self-oscillating micelles with a built-in oxidizing agent, Angew. Chem., Int. Ed., 2020, 59, 3871–3875 CrossRef CAS PubMed.
- S. Ye, M. Steube, E. I. Carrera and D. S. Seferos, What limits the molecular weight and controlled synthesis of poly(3-alkyltellurophene)s?, Macromolecules, 2016, 49, 1704–1711 CrossRef CAS.
- J. Clark, C. Silva, R. H. Friend and F. C. Spano, Role of intermolecular coupling in the photophysics of disordered organic semiconductors: aggregate emission in regioregular polythiophene, Phys. Rev. Lett., 2007, 98, 206406 CrossRef PubMed.
- M. Baghgar, J. Labastide, F. Bokel, I. Dujovne, A. Mckenna, A. M. Barnes, E. Pentzer, T. Emrick, R. Hayward and M. D. Barnes, Probing inter-and intrachain exciton coupling in isolated poly(3-hexylthiophene) nanofibers: effect of solvation and regioregularity, J. Phys. Chem. Lett., 2012, 3, 37 Search PubMed.
- N. J. Hestand and F. C. Spano, Molecular aggregate photophysics beyond the kasha model: novel design principles for organic materials, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, Polarons, Bipolarons, And Absorption Spectroscopy of PEDOT, ACS Appl. Polym. Mater., 2019, 1, 83–94 CrossRef CAS.
- M. M. Roessler and E. Salvadori, Principles and applications of EPR spectroscopy in the chemical sciences, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- B. Xu and S. Holdcroft, Molecular control of luminescence from poly(3-hexylthiophenes), Macromolecules, 1993, 26, 4457–4460 CrossRef CAS.
- R. D. Pensack, Y. Song, T. M. McCormick, A. A. Jahnke, J. Hollinger, D. S. Seferos and G. D. Scholes, Evidence for the Rapid Conversion of Primary Photoexcitations to Triplet States in Seleno- and Telluro-Analogues of Poly(3-hexylthiophene), J. Phys. Chem. B, 2014, 118, 2589–2597 CrossRef CAS PubMed.
- Y. Han, Y. Guo, Y. Chang, Y. Geng and Z. Su, Chain Folding in Poly(3-hexylthiophene) Crystals, Macromolecules, 2014, 47, 3708–3712 CrossRef CAS.
- D. W. Hayward, D. J. Lunn, A. Seddon, J. R. Finnegan, O. E. C. Gould, O. Magdysyuk, I. Manners, G. R. Whittell and R. M. Richardson, Structure of the Crystalline Core of Fiber-like Polythiophene Block Copolymer Micelles, Macromolecules, 2018, 51, 3097–3106 CrossRef CAS.
- F. M. McFarland, L. R. Bonnette, E. A. Acres and S. Guo, The impact of aggregation on the p-doping kinetics of poly(3-hexylthiophene), J. Mater. Chem. C, 2017, 5, 5764–5771 RSC.
- J. Gao, B. W. Stein, A. K. Thomas, J. A. Garcia, J. Yang, M. L. Kirk and J. K. Grey, Enhanced Charge Transfer Doping Efficiency in J-Aggregate Poly(3-hexylthiophene) Nanofibers, J. Phys. Chem. C, 2015, 119, 16396–16402 CrossRef CAS.
- J. Gao, E. T. Niles and J. K. Grey, Aggregates Promote Efficient Charge Transfer Doping of Poly(3-hexylthiophene), J. Phys. Chem. Lett., 2013, 4, 2953–2957 CrossRef CAS PubMed.
- J. G. Manion, S. Ye, A. H. Proppe, A. W. Laramée, G. R. McKeown, E. L. Kynaston, S. O. Kelley, E. H. Sargent and D. S. Seferos, Examining Structure–Property–Function Relationships in Thiophene, Selenophene, and Tellurophene Homopolymers, ACS Appl. Energy Mater., 2018, 1, 5033–5042 CrossRef CAS.
- C. K. Chan, W. Zhao, S. Barlow, S. Marder and A. Kahn, Decamethylcobaltocene as an efficient n-dopant in organic electronic materials and devices, Org. Electron., 2008, 9, 575–581 CrossRef CAS.
- W. Melitz, J. Shen, A. C. Kummel and S. Lee, Kelvin probe force microscopy and its application, Surf. Sci. Rep., 2011, 66, 1–27 CrossRef CAS.
- H. Shimotani, G. Diguet and Y. Iwasa, Direct comparison of field-effect and electrochemical doping in regioregular poly(3-hexylthiophene), Appl. Phys. Lett., 2005, 86, 22104 CrossRef.
- O. Bubnova and X. Crispin, Towards polymer-based organic thermoelectric generators, Energy Environ. Sci., 2012, 5, 9345 RSC.
- H. A. Bronstein and C. K. Luscombe, Externally Initiated Regioregular P3HT with Controlled Molecular Weight and Narrow Polydispersity, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00806k |
| This journal is © The Royal Society of Chemistry 2020 |