
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiophene-fused polyaromatics: synthesis, columnar liquid crystal, fluorescence and electrochemical properties†
Yifan
Li
,
Alberto
Concellón
 ,
Che-Jen
Lin
,
Nathan A.
Romero
,
Sibo
Lin
and
Timothy M.
Swager
*
,
Che-Jen
Lin
,
Nathan A.
Romero
,
Sibo
Lin
and
Timothy M.
Swager
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02143, USA. E-mail: tswager@mit.edu
First published on 24th April 2020
Abstract
Efficient syntheses that incorporate thiophene units into different extended conjugation systems are of interest as a result of the prevalence of sulfur-rich aromatics in organic electronics. Self-organization by using liquid crystal properties is also desirable for optimal processing of organic electronics and optical devices. In this article, we describe a two-step process to access extended regioisomers of polyaromatics with different shapes. This method involves an efficient single or double benzannulation from an alkyne precursor followed by Scholl cyclization. In spite of their unconventional nondiscoid shape, these materials display stable columnar liquid crystal phases. We examine the photophysical and electrochemical properties and find that structurally very similar thiophene-fused polyaromatics display significant differences in their properties.
Introduction
Extended polycyclic aromatic hydrocarbons (PAHs) with precise structures enable many organic electronic devices, including organic light emitting diodes (OLEDs), organic field-effect transistors (OFETs), and organic photovoltaic devices (OPVs).1 Thiophene-based materials dominate organic semiconducting materials as a result of their versatile/unique chemistry and electronic properties.2 Of particular interest is the fact that thiophene containing PAHs in ordered arrangements can offer enhanced performance in organic electronic devices.2d,3 Liquid crystals self-organize in fluid states for superior processing into highly organized anisotropic self-healing structures.4Disk-shaped molecules consisting of π-conjugated PAH rigid cores and disordered sidechains are the basis of many columnar thermotropic liquid crystals.5 The resultant organizations provide strong intermolecular π–π stacking, resulting in one-dimensional electronic correlations and associated anisotropic charge-carrier transport. The design of new PAH cores is key to extend the electronic performance of liquid crystalline organic materials. To this end, there have been previous investigations of thiophene-containing liquid crystalline semiconductive molecules. Oligothiophene derivatives end-capped with alkyl chains display liquid crystalline properties and self-organize into lamellar (smectic) structures.6 Discotic liquid crystals that contain thiophene-fused units are attractive candidates for organic electronics. However, the number of thiophene-containing columnar liquid crystals is limited when compared to their calamitic liquid crystals relatives.7
Alkynes are a widely used efficient precursor to nanographenes. Our group has a long-standing interest in accessing PAHs via efficient alkyne benzannulation reactions,8 which has been expanded considerably in scope by others in recent years.9 We report herein the design and synthesis of columnar self-assembling systems based on thiophene-fused PAHs via alkyne benzannulation followed by oxidative coupling reactions. All the compounds are fluorescent, and despite their non-discoid shapes, stable enantiotropic columnar liquid crystal phases are observed. The self-assembled columnar structure was studied by X-ray diffraction. Interestingly, small differences in the position and orientation of the thiophene moieties in the molecules has significant impact on their photoluminescence in solution and in the solid state and their electrochemical behaviour.
Results and discussion
Synthesis
Our initial efforts focused on the oxidative annulation of biaryl acetylene (1a) to directly access dithienochrysene (3a).8d Commonly used oxidative conditions such as SbCl5, FeCl3, MoCl5 and DDQ/TFA only led to uncontrolled decomposition of starting materials. As a result, we turned our attention to a stepwise procedure, fusing one thienyl moiety per step. Several gold and platinum catalysts10 as well as strong Brønsted acids8a were tested for the first benzannulation, giving desired intermediate 2 in moderated yields, but these conditions also gave an inseparable unidentified impurity. However intermediate 2 is successfully produced in pure form in 88% yield by treatment with 20 mol% of InCl3 in toluene at 100 °C for 12 hours.9b Reaction of 2 with a mixture of FeCl3 in dichloromethane (DCM)/CH3NO2 gave dithienochrysene (3a) in 94% yield after 15 minutes (Scheme 1). When the thiophenes are linked via C3 positions in the starting biaryl acetylene 1b, 10 mol% PtCl2 proves to be the optimal reagent for the alkyne annulation; after Scholl reaction, isomeric dithienochrysene 3b was obtained in 86% overall yield. This high-yielding transformation allows us to access an extended, thiophene fused conjugated system. | ||
| Scheme 1 Stepwise transformation of alkynes to thiophene-fused polyaromatics. | ||
To obtain oligomeric fused systems, we subsequently applied this two-step transformation to di-acetylene precursors with both C2 and C3 connectivity to the thiophenes (Scheme 2 and see the ESI† for detailed syntheses). The initial InCl3-catalyzed benzannulation for di-alkynes gives a mixture of regioisomers. However, these new rings are desired connections for the same final products after the second cyclization. For the thiophene C2 linked precursors, the yields of annulation are generally moderate using 0.2 eq. of InCl3. The efficiency of this step can be significantly improved by using 0.4 eq. of catalyst.11 For the dual benzannulation of the C3-linked thiophene starting materials, 10 mol% of PtCl2 delivered cyclized products in 67–83% yield. Submitting the isolated regioisomeric mixtures to oxidative Scholl reaction conditions affords thiophene fused polyaromatics (4a–6a and 4b–6b) in good overall yield. Starting from para-, meta-, and ortho-substituted dialkynylbenzene cores and C2- or C3-attached thiophene regioisomers, we obtained rapid access to a library of thiophene-fused conjugated systems with different shapes.
 | ||
| Scheme 2 Thiophene-fused conjugation system (note that overall yields are given). | ||
Liquid crystal properties
Thermal stability is required for melt processing and was studied by thermogravimetric analysis (TGA). In most of the cases the 5% weight loss temperature (T5%) was detected more than 100 °C above the isotropization point (vide infra) (Table 1). The thermal transitions and liquid crystal properties were studied by polarizing optical microscopy (POM) and differential scanning calorimetry (DSC) as summarized in Table 1. Compounds 4a, 4b, 5a, and 5b exhibited stable enantiotropic mesophases over a wide temperature range. These mesophases were assigned as hexagonal columnar on the basis of the mosaic-like textures observed by POM (Fig. S1†).12 Compounds 4a and 4b were crystalline materials at room temperature but exhibited liquid crystal properties at elevated temperatures. Consequently, their DSC curves showed two peaks associated to the Cr-to-Colh and Colh-to-I transitions (Fig. 2). In addition, the high clearing temperature at the isotropization results in some decomposition as evidenced in TGA and DSC experiments. In contrast, the DSC curves of 5a and 5b were reproducible through multiple thermal cycles, and cooling cycles showed only one identifiable peak corresponding to the clearing point in addition to a glass transition, which freezes the mesomorphic order at room temperature (Fig. 1). The DSC of 5a reveals that the mesophase undergoes a cold crystallization during the second heating process. Compounds 3a and 3b are crystalline materials that melt to give an isotropic liquid phase without showing liquid crystalline properties. Compounds 6a and 6b are amorphous materials and DSC curves only show a baseline step corresponding to the glass transition. The steric interactions between thiophene–C6H13 groups in 6a and 6b results in a twisted configuration of the aromatic core that decreases the efficiency of π–π stacking interactions that are necessary for the formation of liquid crystal mesophases (see the ESI† for detailed geometry optimization).| T 5% (°C) | Phase transitionsb | Structural parametersc | |
|---|---|---|---|
| a Temperature at which 5% mass lost is detected in the thermogravimetric curve. b DSC data of the second heating process at a rate of 10 °C min−1. Temperatures (°C) are read at the onset of the corresponding peaks, and enthalpies (kJ mol−1) are in brackets. Cr: crystal phase, G: glass, I: isotropic liquid, Colh: hexagonal columnar mesophase, Colh(G): glassy hexagonal columnar mesophase, dec: decomposition. c Structural parameters obtained by XRD. a: lattice constant of the columnar phase, c: mean stacking distance. | |||
| 3a | 269 | Cr1 34 (5.6) Cr2 63 (36.3) I | |
| 3b | 282 | Cr 97 (47.6) I | |
| 4a | 294 | Cr 72 (22.1) Colh 140 (6.2) I | a = 22.5 Å; c = 3.5 Å |
| 4b | 236 | Cr1 110 (3.4) Cr2 145 (26.1) 148 Colh 215 Idec | a = 22.5 Å; c = 3.5 Å |
| 5a | 389 | Colh(G) 18 Colh 84 (−11.8) Cr 135 (18.5) I | a = 20.5 Å; c = 4.0 Å |
| 5b | 343 | Colh(G) 65 Colh 194 (4.9) I | a = 20.3 Å; c = 4.1 Å |
| 6a | 398 | G 75 I | |
| 6b | 426 | G 202 I | |
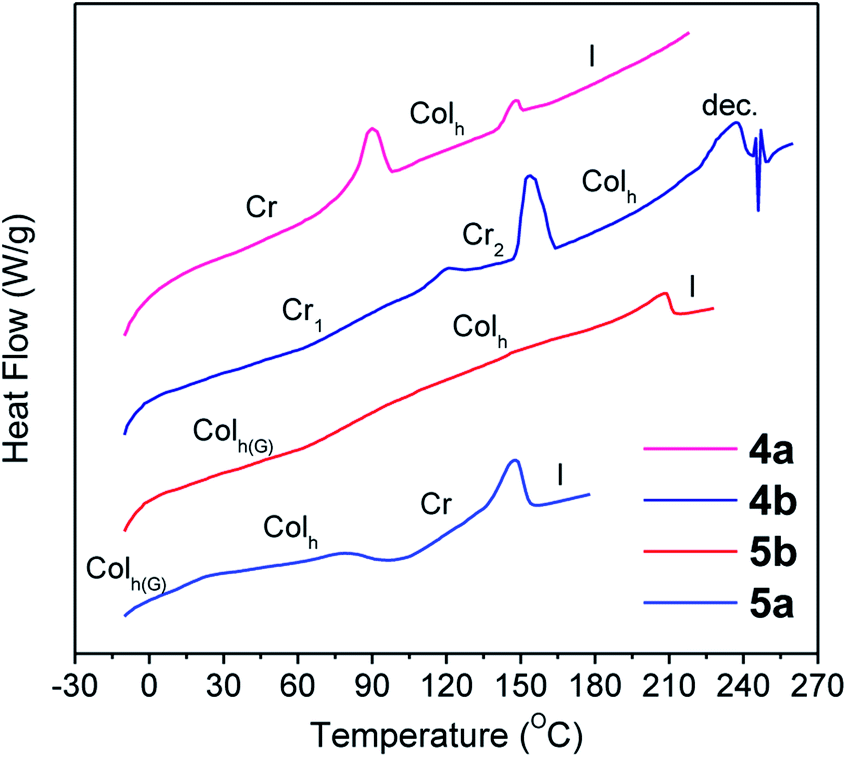 | ||
| Fig. 1 DSC traces corresponding to the second heating scan (10 °C min−1, exo down). (The first heating scan is shown for 4b.) | ||
Although the textures observed by POM were suggestive of hexagonal columnar mesophases, the absolute assignment and the determination of the lattice parameters were achieved by X-ray diffraction (XRD). XRD experiments were carried out on samples that were cooled from the isotropic liquid. As mention before, 4b showed some decomposition at the transition to the isotropic liquid, and thus the experiments were carried out at lower temperatures wherein the stability was confirmed by TGA and DSC experiments.
The XRD patterns of all the LC compounds contain three or four small-angle region reflections with d-spacings in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/√3:1/√4:1/√7 arising from the (1 0 0), (1 1 0), (2 0 0), and (2 1 0) reflections of the two-dimensional hexagonal lattice in a columnar hexagonal LC arrangement (Fig. 2 and S2†). The lattice parameters (a) are gathered in Table 1 and we also observe broad diffuse scattering in the high-angle region (d = 4.5 Å), which is characteristic of the liquid-like order of the hydrocarbon chains. Additionally, a more defined broad reflection at 3.5–4.1 Å was observed, which is indicative the periodic correlations of the cores along the columns (parameter c). The significant shortening in the π–π stacking distance (c) is found in 4a and 4b, when compared to 5a and 5b, and is consistent with our expectations. Specifically, the proximity of thiophene–C6H13 units in 5a and 5b induces a moderate twisting of the aromatic cores, whereas 4a and 4b have a planar core. This feature is at the origin of the tighter packing of the units within the columns.
:1/√3:1/√4:1/√7 arising from the (1 0 0), (1 1 0), (2 0 0), and (2 1 0) reflections of the two-dimensional hexagonal lattice in a columnar hexagonal LC arrangement (Fig. 2 and S2†). The lattice parameters (a) are gathered in Table 1 and we also observe broad diffuse scattering in the high-angle region (d = 4.5 Å), which is characteristic of the liquid-like order of the hydrocarbon chains. Additionally, a more defined broad reflection at 3.5–4.1 Å was observed, which is indicative the periodic correlations of the cores along the columns (parameter c). The significant shortening in the π–π stacking distance (c) is found in 4a and 4b, when compared to 5a and 5b, and is consistent with our expectations. Specifically, the proximity of thiophene–C6H13 units in 5a and 5b induces a moderate twisting of the aromatic cores, whereas 4a and 4b have a planar core. This feature is at the origin of the tighter packing of the units within the columns.
 | ||
| Fig. 2 (a) 1D XRD profiles of 4a, and (b) proposed arrangement of the hexagonal columnar mesophase. | ||
The hexagonal symmetry implies that columns have an average cylindrical cross-section with a uniform distribution of aliphatic chains along their periphery (aliphatic continuum). However, these LC compounds have elliptical shapes rather than the classical disk shape characteristic of many columnar liquid crystals. Therefore, these elliptic mesogens must adopt a distribution of orientations along the stacking directions and may have dynamics that likely include tilting inside and between columns to on average fill the column cross-section (Fig. 2), similar to what is observed for other non-discotic mesogens in hexagonal columnar phases.7e,f,13 Such molecular organizations are largely driven by the nanosegregation between the aromatic cores and the aliphatic continuum, and is similar each of the mesophases examined here.
Additional support for this structural model was obtained by simple calculations based on the parameters measured by XRD. The relationship between the density (ρ) of the compounds in the mesophase and the measured XRD parameters is given by the equation ρ = M × Z/NA × V, where M is the molar mass, Z is the number of molecules in the unit cell, NA is Avogadro's number, and V is the unit cell volume (V = a2 × √3/2 × c × 10−24). If we consider one molecule per disk (Z = 1), the density of the compounds was calculated to be close to 1.0 g cm−3 (Table S1†), which is a reasonable value for organic liquid crystalline materials. Therefore, these calculations are in good agreement with the proposed model for the packing in each column stratum of the hexagonal columnar mesophase.
Photophysical and electrochemical characteristics
The photophysical properties of this set of thienoacenes reveal subtle electronic variations arising from thienyl group isomerism and differential acene-backbone configurations. Thienoacenes 3a/b exhibit strong absorptions near 300 nm, along with less intense bands on the low energy side (Fig. 3). The absorption spectra for 4a/b–6a/b show similar transitions shifted bathochromically by roughly 60 nm, consistent with more extensive π-conjugation in 4–6 relative to 3. While the band edges are at similar energies for 4a/b–6a/b, the extinction coefficients (ε) for the low energy transitions are significantly larger in 3b–5b as compared to their respective isomers 3a–5a, but are similar to 6a and 6b. These characteristics are in accord with TD-DFT calculations on model structures 3a′/b′–6a′/b′, for which calculated oscillator strengths (f) for S0 → S1 transitions are larger in 3b′–5b′ than the corresponding isomers 3a′–5a′ and again 6a′ and 6b′ are similar in magnitude to 3b′–5b′ (see ESI† for full computational details). Solution state photoluminescence spectra reveal small Stokes shifts and relatively narrow, structured emission bands for all compounds with the exception of 3b, which shows a broadened emission relative to 3a (Fig. 3). While emission energies in 4a/b and 6a/b appear to be unaffected by thienyl substitution, the emission maximum for 5b exhibits a 15 nm bathochromic shift compared to 5a. Emission quantum yields (Φem) are higher for isomers 3b–6b than their counterparts 3a–6a. This observation again reveals that the orientation of thienyl group affects the efficiency of radiative transition (Table 2). | ||
| Fig. 3 Absorption spectra (dashed lines) and normalized emission spectra (solid lines) of 3a–6a and 3b–6b in dichloromethane. | ||
| λ abs (nm) | ε (Lmol−1 cm−1) | λ em (nm) | Φ em (nm) | λ em | E p1 (V vs. Fc+/0)e | |
|---|---|---|---|---|---|---|
| a Measured at 10 −6 M. b Emission maxima in DCM solution (0–1 band was shown in parenthesis). c Emission quantum yield in DCM solution obtained from integrated sphere under air. d Emission maxima in thin film at different temperatures (0–1 band was shown in parenthesis and the phase is shown in brackets). e Lowest onset peak oxidation potentials determined by cyclic voltammetry. See Fig. 5 and ESI for details. | ||||||
| 3a | 305 | 13000 |
402 (421) | 0.6% | 412 (430) [Cr] | +0.84 |
| 4a | 360 | 40500 |
470 (499) | 3.5% | 522 [Cr]/528 [Colh] | +0.49 |
| 5a | 360 | 40800 |
452 (477) | 4.4% | 498 [Colh] | +0.57 |
| 6a | 351 | 73500 |
455 (480) | <0.1% | 460 (489) [G] | +0.74 |
| 3b | 309 | 6000 | 412 | 4.4% | 449 [Cr] | +0.79 |
| 4b | 348 | 33400 |
467 (495) | 14.8% | 552 [Cr]/567 [Colh] | +0.45 |
| 5b | 347 | 38600 |
467 (495) | 9.3% | 559 [Colh] | +0.36 |
| 6b | 358 | 75400 |
452 (475) | 0.2% | 455 (485) [G] | +0.71 |
Solid-state fluorescence spectra of 3a, 3b, 6a, and 6b were recorded at room temperature in their solid quenched glass phases obtained by cooling from the isotropic liquid. As was performed for XRD experiments, the emission spectra for 4a, 4b, 5a, and 5b were recorded at temperatures at wherein the molecules display hexagonal columnar mesophases. Although the emission is clearly observable, the determination of quantum yields from these samples was below the limits of detection of the integrating sphere device used in this study. (Fig. 4a). The dominant emission bands were red-shifted and broadened in the solid state relative to solution measurements (Table 2, Fig. 4 and S3†). The broad emission band combined with the significant red shift in the maxima of the solid-state emission of these films are indicative of the emission arising from the aggregated state of these molecules in the solid, liquid crystalline phase.14 Another distinctive feature of the solid-state emission spectra is a very long and relatively unstructured tail extending to 700 nm and beyond.
 | ||
| Fig. 4 (a) Pictures of the emission of the films under illumination by a 365 nm UV lamp. (b) Normalized emission spectra of 4a in DCM and thin film at different temperatures (I: isotropic liquid, Colh: hexagonal columnar mesophase). (c) Normalized emission spectra of 6b in DCM and thin film at different temperatures (G: glass phase). | ||
The electrochemical characteristics of thienoacenes 3a–6a and 3b–6b were investigated by cyclic voltammetry (Fig. 5, Tables 2 and S3;† potentials referenced to Fc+/0). All compounds undergo oxidation at relatively low potentials and exhibit no discernible reduction down to −2 V. The thienoacenes show somewhat complicated redox behaviour, with multiple overlapping oxidation events. Overall, there is a general resemblance in electrochemical behaviour on the forward (oxidative) sweep within each pair of thienoacene isomers, consistent with a previous investigation on the electrochemistry of acenedithiophenes which suggested the nature of this thienyl isomerism does not greatly impact electronic structure.9c The earliest onset of oxidation occurs at marginally lower potentials for compounds 3b–6b compared to their respective isomers 3a–6a, in correlation with DFT calculations which estimate HOMO levels in 3b′–6b′ at slightly higher energies than their counterparts 3a′–6a′ (Fig. S4†). A larger difference in redox potential was observed for compound 5b, which is oxidized at a ca. 200 mV lower potential than its isomer 5a. This correlates with a larger disparity in photophysical properties (e.g., λem, Table 2) between 5a and 5b than for the other pairs of thienoacenes. Evidently, the electronic nature of the aromatic core in 5a/b is more sensitive to heteroarene substitution pattern than the other acene structures.
 | ||
| Fig. 5 Cyclic voltammograms of (a) compounds 3a and 3b, (b) compounds 4a and 4b, (c) compounds 5a and 5b, and (d) compounds 6a and 6b. Experiments were conducted at a scan rate of 100 mV s−1 with a glassy carbon working electrode, a platinum wire counter electrode, and an Ag/AgNO3 pseudo-reference electrode. Analysis of compounds 3a–6a, 3b and 5b–6b was conducted in 100 mM [Bu4N][PF6] in DCM, and in 80 mM [Bu4N][PF6] in o-DCB for 4b. Potentials referenced to ferrocene as an external standard. | ||
Compounds 3a–6a display greater redox-reversibility than their respective isomers 3b–6b, which show decreased cathodic current on the reductive sweep of the voltammogram. This behaviour is particularly evident for thienoacene 3a, which exhibits two clear quasi-reversible oxidation events at +0.80 and +1.32 V in stark contrast to isomer 3b (Fig. 5a). The diminished electrochemical reversibility of 3b–6b likely arises from irreversible follow-up chemical reactions upon oxidation, which we attribute to oxidative coupling at the 3-position (i.e., β-carbon) of the thienyl groups, in accord with prior literature on oxidative homocoupling of 2-substituted benzothiophenes.15 Whereas the thienyl β-carbons in compounds 3a–6a reside in sterically encumbered cove regions, compounds 3b–6b all possess β-carbons located in more accessible bay regions and are likely more prone to oxidative oligomerization. These steric differences are apparent in the computed structures 3a′–6a′/3b′–6b′. Thus, steric crowding in compounds 3a–6a may enhance the kinetic stability of higher oxidation states and could account for their improved electrochemical reversibility relative to isomers 3b–6b.
Conclusions
In conclusion, we have developed an efficient two-step process to access thiophene-fused polyaromatics from easily accessible starting materials in high overall yield. The practicality of the process is reflected by the efficient synthesis of a library of eight PAHs. These elliptical shaped molecules display hexagonal columnar liquid crystal phases. Two have low clearing temperatures and organized alignment at room temperature. All the PAHs are fluorescent at solid state. The UV-absorption and emission in solution, frontier molecular orbitals and electrochemical properties were studied, showing the role of thiophene connectivity in determining these properties. These synthetic methods have utility for the production of thiophene-fused graphene nanoribbons, and these studies are ongoing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Y. L. is grateful to the Swiss National Science Foundation (SNSF) for a postdoctoral mobility fellowship (P2ELP2_165170). C. L. thanks the Minister of Science and Technology, Taiwan, for postdoc fellowship (106-2917-I-564-058). N. A. R. acknowledges support from a National Institutes of Health postdoctoral fellowship (F32 GM126643). This research was supported the Air Force Office of Scientific Research AFOSR Grant # 17RT0904, FA9550-18-1-0341.Notes and references
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed; (b) M. Watanabe, K.-Y. Chen, Y. Chang and T. Chow, Acc. Chem. Res., 2013, 46, 1606 CrossRef CAS PubMed; (c) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef CAS PubMed; (d) M. Emi Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef PubMed; (e) T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 1116, 1139 CrossRef PubMed.
- (a) I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-Based Materials,Wiley-VCH, Weinheim, 2009 CrossRef; (b) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC; (c) L. Zhang, C.-A. Di, G. Yu and Y. Liu, J. Mater. Chem., 2010, 20, 7059 RSC; (d) H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211 RSC; (e) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347 CrossRef CAS PubMed.
- (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed; (b) W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC.
- (a) T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355 CrossRef CAS PubMed; (b) T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef; (c) A. Concellón, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Angew. Chem., Int. Ed., 2017, 56, 1259 CrossRef PubMed; (d) M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566 CrossRef PubMed.
- (a) W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634 CrossRef CAS PubMed; (b) B. Zhao, B. Liu, R. Q. Png, K. Zhang, K. A. Lim, J. Luo, J. Shao, P. K. H. Ho, C. Chi and J. Wu, Chem. Mater., 2010, 22, 435 CrossRef CAS; (c) W. Yuan, X. Ren, M. Li, H. Guo, Y. Han, M. Wu, Q. Wang, M. Li and Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 6161 CrossRef CAS PubMed.
- (a) A. J. J. M. van Breemen, P. T. Herwig, C. H. T. Chlon, J. Sweelssen, H. F. M. Schoo, S. Setayesh, W. M. Hardeman, C. A. Martin, D. M. de Leeuw, J. J. P. Valeton, C. W. M. Bastiaansen, D. J. Broer, A. R. Popa-Merticaru and S. C. J. Meskers, J. Am. Chem. Soc., 2006, 128, 2336 CrossRef CAS PubMed; (b) K. Oikawa, H. Monobe, K. Nakayama, T. Kimoto, K. Tsuchiya, B. Heinrich, D. Guillon, Y. Shimizu and M. Yokoyama, Adv. Mater., 2007, 19, 1864 CrossRef CAS; (c) Y. Shimizu, K. Oikawa, K. Nakayama and D. Guillon, J. Mater. Chem., 2007, 17, 4223 RSC; (d) F. Lincker, B. Heinrich, R. D. Bettignies, P. Rannou, J. Pécaut, B. Grévin, A. Pron, B. Donnio and R. Demadrille, J. Mater. Chem., 2011, 21, 5238 RSC; (e) L. Mazur, A. Castiglione, K. Ocytko, F. Kameche, R. Macabies, A. Ainsebaa, D. Kreher, B. Heinrich, B. Donnio, S. Sanaur, E. Lacaze, J. Fave, K. Matczyszyn, M. S. Jeong, W. Wu, A. Attias, J. Ribierre and F. Mathevet, Org. Electron., 2014, 15, 943 CrossRef CAS; (f) Y. Funatsu, A. Sonoda and M. Funahashi, J. Mater. Chem. C, 2015, 3, 1982 RSC; (g) A. Richard, V. Lemaur, Y. Olivier, J. Cornil, A. R. Kennedy, Y. Diao, W. Lee, S. Mannsfeld, Z. Bao and Y. H. Geerts, J. Mater. Chem. C, 2015, 3, 674 RSC.
- (a) T. Yasuda, H. Ooi, J. Morita, Y. Akama, K. Minoura, M. Funahashi, T. Shimomura and T. Kato, Adv. Funct. Mater., 2009, 19, 411 CrossRef CAS; (b) T. Yasuda, T. Shimizu, F. Liu, G. Ungar and T. Kato, J. Am. Chem. Soc., 2011, 133, 13437 CrossRef CAS PubMed; (c) L. Chen, S. R. Puniredd, Y. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869 CrossRef CAS PubMed; (d) Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268 CrossRef CAS PubMed; (e) C. Liu, H. Wang, J. Du, K. Zhao, P. Hu, B. Wang, H. Monobe, B. Heinrich and B. Donnio, J. Mater. Chem. C, 2018, 6, 4471 RSC; (f) K.-C. Zhao, J.-Q. Du, H.-F. Wang, K.-Q. Zhao, P. Hu, B.-Q. Wang, H. Monobe, B. Heinrich and B. Donnio, Chem.–Asian J., 2019, 14, 462 CrossRef CAS PubMed.
- (a) M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895 CrossRef CAS; (b) M. B. Goldfinger, K. B. Crowfold and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578 CrossRef CAS; (c) M. B. Goldfinger, K. B. Crowfold and T. M. Swager, J. Org. Chem., 1998, 63, 1676 CrossRef CAS; (d) S. Yamaguchi and T. M. Swager, J. Am. Chem. Soc., 2001, 123, 12087 CrossRef CAS PubMed; (e) J. M. W. Chan, J. R. Tischler, S. E. Kooi, V. Bulovic and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 5659 CrossRef CAS PubMed; (f) T. Ikai, T. Yohida, K. Shinohara, T. Taniguchi, T. Y. Wada and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 4696 CrossRef CAS PubMed.
- (a) S. Yamaguchi, C. Xu and K. Tamao, J. Am. Chem. Soc., 2003, 125, 13662 CrossRef CAS PubMed; (b) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556 CrossRef CAS PubMed; (c) S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024 CrossRef CAS PubMed; (d) L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 10351 CrossRef CAS PubMed; (e) W. Yang, J. Monteiro, A. de Bettencourt-Dias, V. J Catalano and W. A. Chalifoux, Angew. Chem., Int. Ed., 2016, 55, 10427 CrossRef CAS PubMed; (f) R. K. Mohamed, S. Mondal, J. V. Guerrera, T. M Eaton, T. E. Albrecht-Schmitt, M. Shatruk and I. V. Alabugin, Angew. Chem., Int. Ed., 2016, 55, 12054–12058 CrossRef CAS PubMed; (g) Y. Li, G. Gryn’ova, F. Saenz, X. Jeanbourquin, K. Sivula, C. Corminboeuf and J. Waser, Chem.–Eur. J., 2017, 23, 8058 CrossRef CAS PubMed; (h) W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102 CrossRef CAS PubMed; (i) H. Lin, N. Mitoma, L. Meng, Y. Segawa, A. Wakamiya and K. Itami, Mater. Chem. Front., 2018, 2, 275 RSC.
- (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- W. Yang, R. R. Kazemi, N. Karunathilake, V. J. Catalano, M. A. Alpuche-Aviles and W. A. Chalifoux, Org. Chem. Front., 2018, 5, 2288 RSC.
- (a) K. Zhao, L. An, X. Zhang, W. Yu, P. Hu, B. Wang, J. Xu, Q. Zeng, H. Monobe, Y. Shimizu, B. Heinrich and B. Donnio, Chem.–Eur. J., 2015, 21, 10379 CrossRef CAS PubMed; (b) V. Iguarbe, A. Concellón, R. Termine, A. Golemme, J. Barberá and J. L. Serrano, ACS Macro Lett., 2018, 7, 1138 CrossRef CAS.
- B. Donnio, B. Heinrich, H. Allouchi, J. Kain, S. Diele, D. Guillon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 15258 CrossRef CAS PubMed.
- (a) S. Varghese, N. S. S. Kumar, A. Krishna, D. S. S. Rao, S. K. Prasad and S. Das, Adv. Funct. Mater., 2009, 19, 2064 CrossRef; (b) D. D. Prabhu, N. S. S. Kumar, A. P. Sivadas, S. Varghese and S. Das, J. Phys. Chem. B, 2012, 116, 13071 CrossRef CAS PubMed.
- (a) T. Wirtanen, M. K. Mäkelä, J. Sarfraz, P. Ihalainen, S. Hietala, M. Melchionna and J. Helaja, Adv. Synth. Catal., 2015, 357, 3718 CrossRef CAS; (b) L. Bering, F. m. Paulussen and A. P. Antonchick, Org. Lett., 2018, 20, 1978 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional information, and characterization data. See DOI: 10.1039/d0sc00714e |
| This journal is © The Royal Society of Chemistry 2020 |