
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–metal cooperative bond activation by heterobimetallic alkyl, aryl, and acetylide PtII/CuI complexes†
Shubham
Deolka
 a,
Orestes
Rivada-Wheelaghan‡
*a,
Sandra L.
Aristizábal
a,
Robert R.
Fayzullin
b,
Shrinwantu
Pal
c,
Kyoko
Nozaki
c,
Eugene
Khaskin
a and
Julia R.
Khusnutdinova
*a
a,
Orestes
Rivada-Wheelaghan‡
*a,
Sandra L.
Aristizábal
a,
Robert R.
Fayzullin
b,
Shrinwantu
Pal
c,
Kyoko
Nozaki
c,
Eugene
Khaskin
a and
Julia R.
Khusnutdinova
*a
aCoordination Chemistry and Catalysis Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, 904-0495, Okinawa, Japan. E-mail: juliak@oist.jp; orestes.rivada@u-paris.fr
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, 8 Arbuzov Street, Kazan, 420088, Russian Federation
cDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 2nd May 2020
Abstract
We report the selective formation of heterobimetallic PtII/CuI complexes that demonstrate how facile bond activation processes can be achieved by altering the reactivity of common organoplatinum compounds through their interaction with another metal center. The interaction of the Cu center with the Pt center and with a Pt-bound alkyl group increases the stability of PtMe2 towards undesired rollover cyclometalation. The presence of the CuI center also enables facile transmetalation from an electron-deficient tetraarylborate [B(ArF)4]− anion and mild C–H bond cleavage of a terminal alkyne, which was not observed in the absence of an electrophilic Cu center. The DFT study indicates that the Cu center acts as a binding site for the alkyne substrate, while activating its terminal C–H bond.
Introduction
Metal–metal cooperation plays a crucial role in small molecule activation in enzymes and synthetic systems, including homogeneous and heterogeneous catalysts.1 Many classical catalytic systems rely on the thorough optimization of the ligand environment to induce the desired reactivity at a single metal center. However, there is currently a growing realization that catalysts’ reactivities can be significantly altered through close communication with another metal center, either by design or via unexpected bimetallic processes, thus enabling new approaches to bond activation.2The second metal may facilitate substrate binding and pre-activation or even stabilize the bond activation product (Scheme 1). The adoption of the bimetallic approach has led to many recent advances in stoichiometric and catalytic bond activation processes.2,3 Bimetallic cooperation is often proposed in many C–C coupling processes, e.g. the Cu-to-Pd transmetalation step in the Sonogashira coupling.4 The accelerating effect of metal additives (e.g. Cu salts) in Pd-catalyzed C–C coupling reactions such as Stille and Suzuki coupling is commonly referred to as “the copper effect”.5
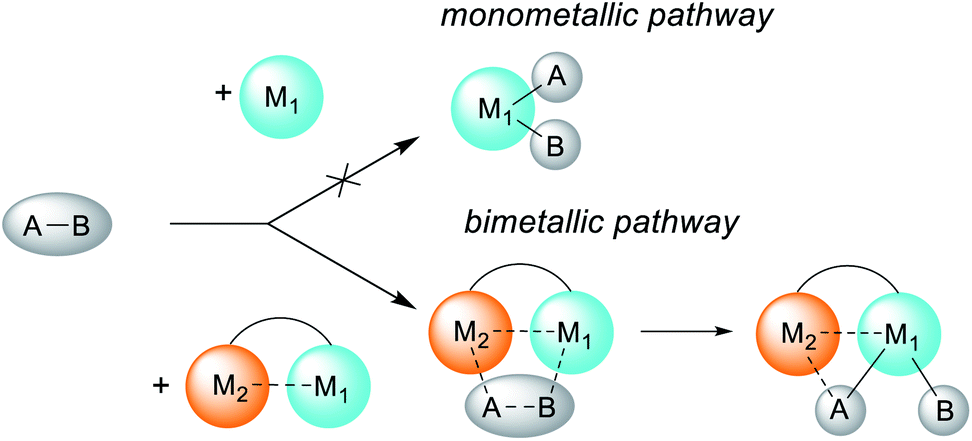 | ||
| Scheme 1 Schematic representation of the altering reactivity of a single metal center through heterobimetallic complex formation. | ||
However, a precise understanding of how the reactivity at the single metal center can be affected by communication with a second metal is often lacking due to the synthetic challenges in selective synthesis of such reactive heterobimetallic complexes with a well-defined structure.
While multiple symmetrical ligand platforms have been developed for the construction of homobimetallic complexes,6 examples of ligand scaffolds that could selectively support metal–metal interactions between two different metals and at the same time contain available reactive sites, are exceedingly rare.7 This is especially the case when the combination of a 1st row and a late 2nd or 3rd row transition metal is targeted. Among known heterobimetallic complexes, rigid ligand design often blocks access to coordination sites suitable for cooperative substrate binding.8 Although several other classes of binucleating bridging ligands have been developed, many of these ligands do not allow for close metal–metal interactions in homo- or heteropolymetallic systems.9
In this work, we report a new bifunctional soft/hard unsymmetrical ligand scaffold, which selectively incorporates both PtII and CuI centers. The close proximity between the two metals allows for coordination of alkyl, aryl, or acetylide ligands to both metal centers, and for the dialkyl complexes, enables metal–metal interaction. These heterobimetallic complexes allow us to directly observe the effect of the second metal center on the reactivity of Pt, which is interesting given that Pt complexes with d10 metal additives are widely used in C–H bond activation10 and studied as models for Pd-catalyzed cross-coupling.11 Our findings demonstrate that even subtle interactions between two different metals alter the solution reactivity of common organometallic species, particularly in the C–H bond activation and boron-to-metal transmetalation reactions, and ligand architecture that induces proximity significantly affects reactivity.
Results and discussion
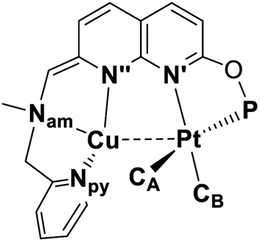
Ligand design and synthesis of monometallic and heterobimetallic complexes
We have previously reported the reversible stepwise formation of homomultimetallic CuI linear chain complexes using an unsymmetrical napthyridinone-based ligand L0 (Scheme 2).12 Simple functionalization of the O-atom of this ligand by reaction with chlorobis(tert-butyl)phosphine leads to a new ligand L, in which two well-defined coordination sites are created: a hard-donor site containing a picolylamine arm and a soft-donor site containing a phosphinite arm. We first tested if ligand L shows differentiation between soft and hard Lewis acids using (NBD)PtIIMe2 (NBD = norbornadiene) and [PtIVMe3I]4 precursors, respectively. As expected, the specific binding of the PtII center to the soft phosphinite site only is observed, giving complex 1. The PtIV center, on the other hand, specifically binds to the hard N-donor site (complex 2). Interestingly, upon reacting complex 1 with methyl iodide, immediate migration of the Pt center from the soft to the hard site is observed, presenting an alternative pathway for the formation of 2. The mono-metallic reactivity thus confirms a proof-of-concept for using this ligand platform further in designing heterobimetallic systems selectively via the soft/hard Lewis acid concept.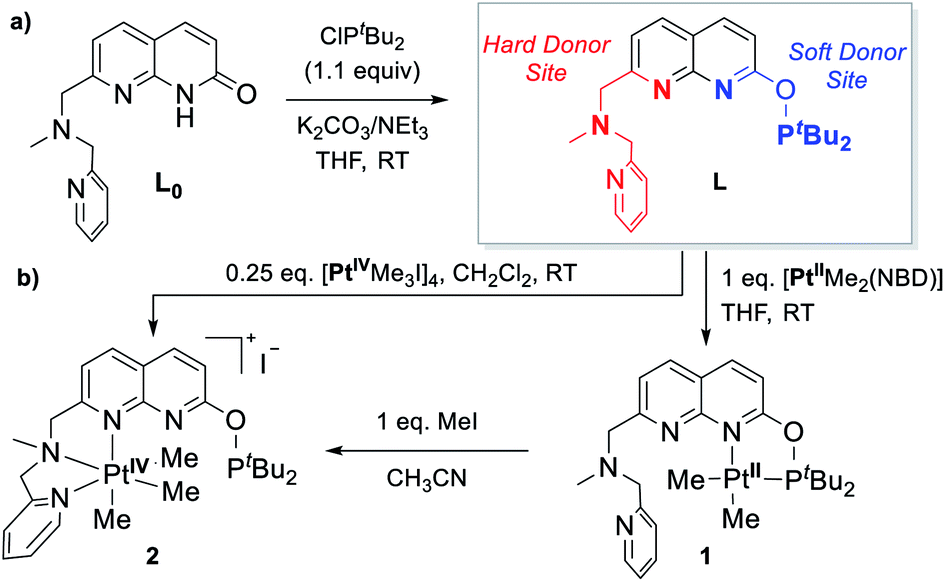 | ||
| Scheme 2 (a) Synthesis of ligand L; (b) selective binding to PtII and PtIV centers via hard and soft sites. | ||
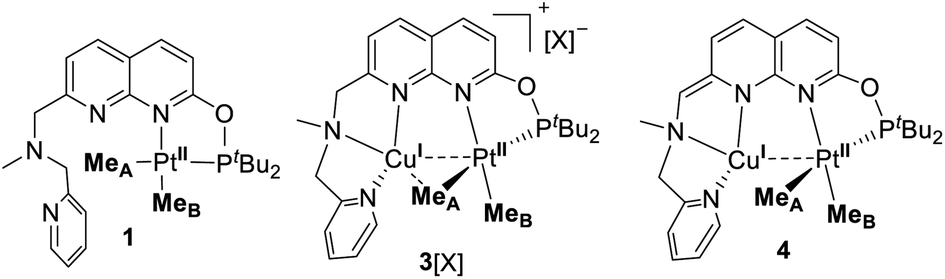
Next we targeted the formation of heterobimetallic complexes using PtII and CuI precursors as this combination is known to show metallophilic closed-shell d8–d10 interactions.13 First, treatment of complex 1 with 2 equiv. of CuICl led to the formation of the heterobimetallic PtIIMe2/CuI complex 3[CuICl2] (Scheme 3). By comparison, the reaction with 1 equiv. of CuICl gave a mixture of products with the major species characterized by an 1H NMR spectrum similar to that of complex 3[CuICl2] indicative of the formation of a similar CuI/PtII core; however, the reaction was generally less clean and did not proceed with high yield. To avoid the presence of a potentially non-innocent counter anion, [CuI(MeCN)4][X] (X = BF4 or B(ArF)4; B(ArF)4 = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) was used leading to complexes 3[BF4] and 3[B(ArF)4], respectively.
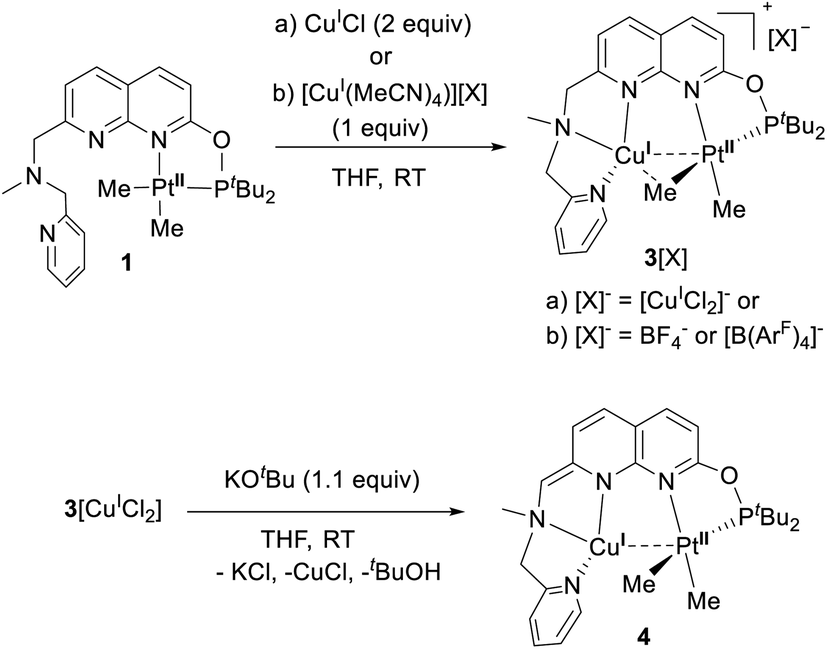 | ||
| Scheme 3 Formation of cationic heterobimetallic complexes 3[X] and a dearomatized heterobimetallic complex 4. | ||
All complexes were isolated in 65–71% yields, characterized by X-ray diffraction (XRD) (vide infra), NMR, IR, and UV-vis spectroscopies, ESI-MS, and elemental analysis.
Noting that the ligand platform also contains an acidic benzylic CH2 position and the previously reported ability of the mononucleating PNN pincer ligands to undergo an N-bound CH2-arm deprotonation coupled with dearomatization,14 we attempted the dearomatization of our binucleating P,N-donor ligand L using a strong base.
Gratifyingly, treatment of 3[CuCl2] with KOtBu resulted in a deep red solution, from which 4 could be isolated cleanly. Complex 4 features a dearomatized naphthyridine ring owing to the deprotonation of its CH2 arm and thus is the first example of a dearomatized heterobimetallic complex, resembling dearomatization in pincer-based mononucleating ligands utilized for metal–ligand cooperation catalysis.15 Dearomatization of the naphthyridine-based binucleating PNNP (“expanded pincer”) ligand has also been recently reported by Broere and co-workers in a homobimetallic Cu2 complex.6f
Characterization of heterobimetallic complexes in the solid state and solution and analysis of metal–metal interactions
The structures of complexes 3[X] and 4 were confirmed by single crystal XRD studies (Fig. 1).16 The geometries of the Pt centers in 3[X] are distorted square planar, with τ4 and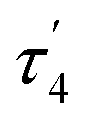 values17 in the range of 0.10–0.13 and 0.07–0.09, respectively (the value for ideal square planar geometry is 0). This indicates that the oxidation state of the Pt center is unlikely to change upon the formation of a heterobimetallic species and it can be formally assigned as +2, consistent with solution NMR studies (vide infra). The PtII⋯CuI distances (2.6119(3)–2.6486(3) Å) are shorter than the sum of covalent radii (2.68 Å),18 but slightly longer than in the PtIIMe2/CuI complex reported by Chen et al. (2.5275(7) Å).11 Interestingly, CuI in 3[X] has close interaction with the carbon of the proximal Me group with Cu1⋯C1 distances of 2.160(3)–2.362(9) Å. Thus, complexes 3[X] are the examples of a solution-stable heterobimetallic complex with an unsymmetrical bridging Me group.19
values17 in the range of 0.10–0.13 and 0.07–0.09, respectively (the value for ideal square planar geometry is 0). This indicates that the oxidation state of the Pt center is unlikely to change upon the formation of a heterobimetallic species and it can be formally assigned as +2, consistent with solution NMR studies (vide infra). The PtII⋯CuI distances (2.6119(3)–2.6486(3) Å) are shorter than the sum of covalent radii (2.68 Å),18 but slightly longer than in the PtIIMe2/CuI complex reported by Chen et al. (2.5275(7) Å).11 Interestingly, CuI in 3[X] has close interaction with the carbon of the proximal Me group with Cu1⋯C1 distances of 2.160(3)–2.362(9) Å. Thus, complexes 3[X] are the examples of a solution-stable heterobimetallic complex with an unsymmetrical bridging Me group.19
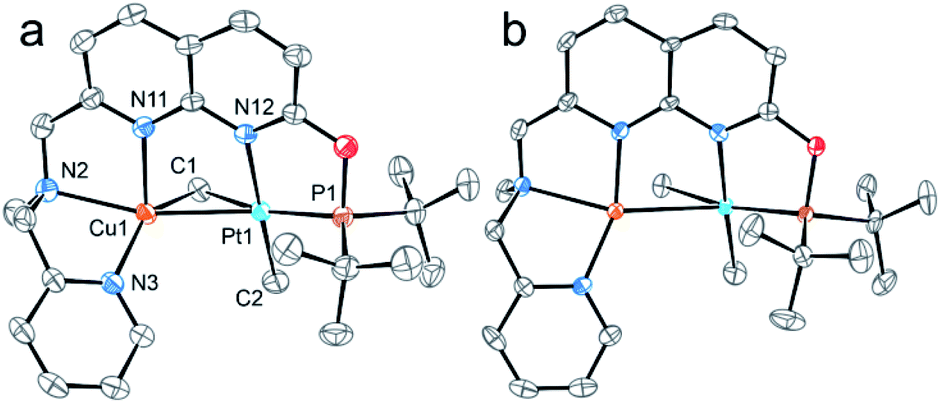 | ||
| Fig. 1 ORTEP of 3[B(ArF)4] (a) and 4 (b) at the 50% probability level. Hydrogen atoms, the counterions for 3[B(ArF)4], and solvent molecules for 4 are omitted for clarity. In the case of 4, only one of three symmetrically independent molecules is shown. Hereinafter coordination bonds are shown in accordance with AIM analysis for the gas-phase optimized structures. | ||
Interestingly, the dearomatized complex 4, characterized by three molecules in the asymmetric cell, features a noticeably longer interaction between CuI and PtII, 2.6890(5)–2.7459(6) Å, which is not much larger than the sum of the covalent radii.18 The distances from C of the proximal Me group to CuI are longer (2.518(5)–2.559(5) Å) compared to that of complexes 3[X]. These structural changes are ascribed to the loss of electrophilicity at a formally neutral CuI center in 4 leading to weakening interactions of CuI with both PtII and the bridging Me group. The selected interatomic and bond distances in complexes 3[X] and 4 and the τ4 and 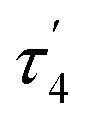 values17 for the Pt centers are summarized in Tables 1 and 2. The comparison between complexes 3[X] and 4 shows that the distances between Pt and the carbon of the bridging Me group are consistently longer in complexes 3[X] demonstrating stronger interaction of the bridging Me group with the CuI center as compared to 4. Dearomatization of the ligand in 4 is evident from X-ray diffraction data featuring double bond character (1.359(6)–1.371(7) Å) in the deprotonated arms as opposed to C11–C12 of 1.512(3) Å in the non-dearomatized complex 3[CuCl2].
values17 for the Pt centers are summarized in Tables 1 and 2. The comparison between complexes 3[X] and 4 shows that the distances between Pt and the carbon of the bridging Me group are consistently longer in complexes 3[X] demonstrating stronger interaction of the bridging Me group with the CuI center as compared to 4. Dearomatization of the ligand in 4 is evident from X-ray diffraction data featuring double bond character (1.359(6)–1.371(7) Å) in the deprotonated arms as opposed to C11–C12 of 1.512(3) Å in the non-dearomatized complex 3[CuCl2].
| Bond distancea (Å) | 3[CuCl2] | 3[BF4] | 3[B(ArF)4] |
|---|---|---|---|
| a Atom numbering is according to Fig. 1a. | |||
| Pt1–C1 | 2.201(2) | 2.251(12) | 2.164(3) |
| Pt1–C2 | 2.050(2) | 2.048(11) | 2.048(3) |
| Pt1–N12 | 2.1823(19) | 2.172(9) | 2.1894(19) |
| Pt1–P1 | 2.2280(6) | 2.228(3) | 2.2180(6) |
| Cu1–N11 | 2.064(2) | 2.038(10) | 2.076(2) |
| Cu1–N2 | 2.320(2) | 2.335(11) | 2.322(2) |
| Cu1–N3 | 1.966(2) | 1.982(10) | 1.953(2) |
| Cu1–C1 | 2.277(2) | 2.362(9) | 2.160(3) |
| Pt1–Cu1 | 2.6486(3) | 2.625(2) | 2.6119(3) |
| τ 4 | 0.10 | 0.10 | 0.13 |
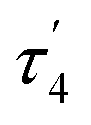
|
0.07 | 0.07 | 0.09 |
|
|
|||
|---|---|---|---|
| Bond distanceb (Å) | 4 | 4 | 4 |
| a For each symmetrically independent molecule. b General scheme for atom labelling in coordination spheres of Pt and Cu is shown above. c From XRD data for the 1st symmetrically independent molecule. d From XRD data for the 2nd symmetrically independent molecule. e From XRD data for the 3rd symmetrically independent molecule. | |||
| Pt–CA | 2.109(5) | 2.127(5) | 2.124(5) |
| Pt–CB | 2.062(5) | 2.050(5) | 2.060(5) |
| Pt–N′ | 2.156(3) | 2.174(4) | 2.171(3) |
| Pt–P | 2.2371(11) | 2.2428(11) | 2.2427(11) |
| Cu–N′′ | 1.933(4) | 1.939(4) | 1.939(4) |
| Cu–Nam | 2.346(4) | 2.317(4) | 2.301(4) |
| Cu–Npy | 1.925(4) | 1.937(4) | 1.941(4) |
| Cu–CA | 2.558(5) | 2.518(5) | 2.559(5) |
| Pt–Cu | 2.6890(5) | 2.7459(6) | 2.7201(5) |
| τ 4 | 0.12 | 0.10 | 0.11 |
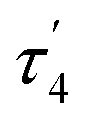
|
0.07 | 0.07 | 0.07 |
ESI-MS analysis also confirmed that the bimetallic cationic PtII/CuI species 3+ is present in polar solvents (MeCN or THF), confirming its stability.
NMR spectra of complexes 3[BF4] and 3[B(ArF)4] exhibit well-resolved, sharp proton resonances. Diagnostic features of the NMR spectra corresponding to the proximity of a CuI center to the Pt–Me group in 3[X] (X = BF4 and B(ArF)4) are compared to those of 1 and 4 in Tables 3 and S1.† The position of the Me groups was determined by selective nuclear Overhauser effect (NOE) experiments. The MeA group located between the PtII and CuI atoms shows a significant downfield shift of the 13C signal by ca. 21 ppm compared to the analogous MeA group located trans to the phosphinite in the Cu-free analogue 1. In comparison, almost no change in chemical shift was observed for the MeB group distal from the CuI center of 3 as compared to 1. The latter observation is also consistent with a PtII formal oxidation state assignment in complexes 3[X] and 4 despite the presence of metal–metal interactions. This is also in line with the previous studies by Chen and co-workers who described d8–d10 interactions between an electron-rich PtII center and a Lewis acidic d10 metal, which have a significant donor–acceptor character and are described as Pt→M dative bonds.13 Moreover, considerably smaller Pt–H and Pt–C coupling constants were observed for the MeA group of complexes 3[X] compared to 1, while only minor changes are seen in the distal MeB. As expected from crystallographic data, neutral complex 4 features a MeA group with the 13C chemical shift and coupling constant values that are intermediate between those observed for complexes 3[X] and 1, consistent with a weaker Cu/Pt–Me interaction when compared to 3[X]. Coordination of the CuI center also leads to an upfield shift of the 195Pt signal, which shows a larger coupling constant to the P-atom when short PtII⋯CuI contacts are present. At the same time, 195Pt chemical shifts for complexes 1, 3[X] and 4 are significantly upfield shifted as compared to the characterized PtIV complex 2 (δPt −2320.9) supporting the assigned formal PtII oxidation state in these complexes and consistent with the earlier literature reports.20
|
|
|||||
|---|---|---|---|---|---|
| Complex | δ H (2JH,Pt, Hz) | δ C (1JC,Pt, Hz) | δ Pt (1JP,Pt, Hz) | ||
| MeA | MeB | MeA | MeB | Pt | |
| a Not determined due to low intensity caused by insufficient solubility; the corresponding 1JC,Pt for MeA in CD3CN solution was determined to be 505 Hz (see Table S1). b Not determined due to low intensity caused by insufficient solubility. | |||||
| 1 | 0.93 (66) | 0.96 (94) | 15.7 (662) | −21.9 (805) | −3894 (2006) |
| 3[B(ArF)4] | 1.10 (44) | 1.23 (86) | −5.9 (491) | −21.3 (711) | −3971 (2877) |
| 3[BF4] | 1.04 (36) | 1.18 (82) | −5.6 (n.d.)a | −21.3 (719) | −3971 (2866) |
| 4 | 0.82 (56) | 0.88 (84) | 3.0 (n.d.)b | −20.2 (n.d.)b | −3980 (2467) |
Dearomatization of the naphthyridine ring in complex 4 was also observed in the 1H NMR spectrum, showing a significant upfield shift for the naphthyridine protons compared to 3[X] and 1 (Fig. 2 and S77†), and the presence of a CH group singlet at 4.83 ppm in THF-d8 solution.
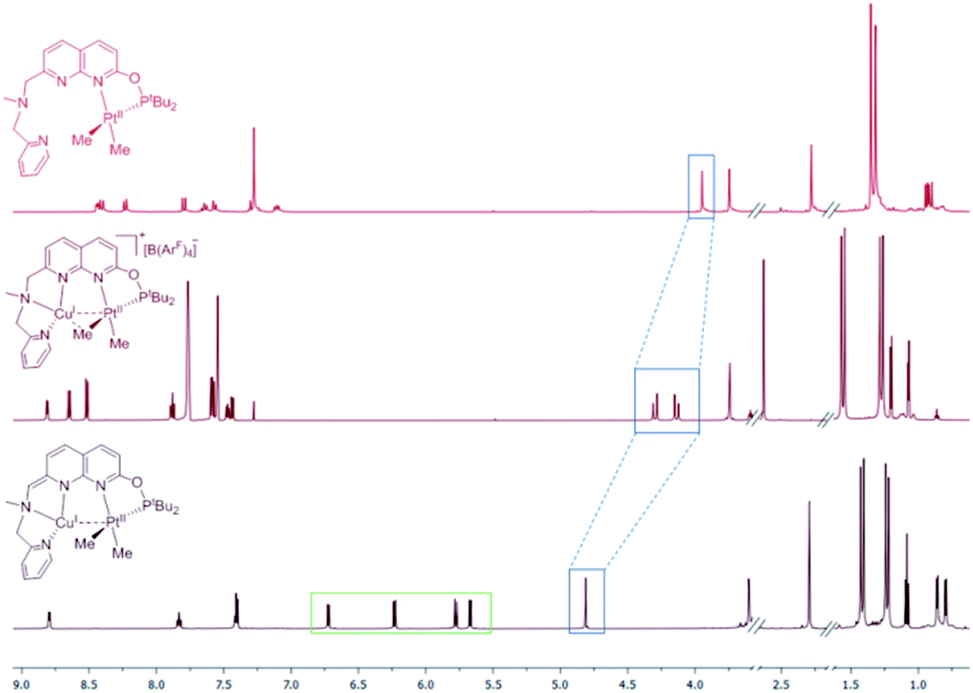 | ||
| Fig. 2 1H NMR spectra (THF-d8, 25 °C) of isolated complexes 1 (top), 3[B(ArF)4] (middle) and dearomatized complex 4 (bottom). Blue rectangles highlight the naphthyridine-CH2 signal or CH for 4. Green rectangle highlights the naphthyridine Csp2–H signals of 4. | ||
Atoms in Molecules (AIM) analyses for DFT-optimized structures of complexes 3 and 4 revealed that bond critical points (bcp) were located between Pt and Cu atoms (Fig. 3) with characteristics typical for closed-shell, metal–metal interactions (positive value for ∇2ρb, low ρb, negative Vb and Hb, with Hb value close to zero).21 Interestingly, the bcp was also located between Cu and carbon of the proximal MeA group in complex 3 with characteristics indicative of metal–ligand interactions (ρb 0.059 a.u.; ∇2ρb 0.218 a.u.), but not in complex 4, consistent with longer Cu⋯C distances observed by XRD. The characteristics for the bond critical points for all complexes are listed in Tables S2–S5 in the ESI.† The comparison between bond distances obtained in the geometry-optimized structures used for QTAIM analysis and XRD parameters show reasonable agreement and the expected trend in PtII⋯CuI and CuI⋯MeA contacts (Tables S6–S9†), showing longer PtII⋯CuI and CuI⋯MeA distances in complex 4 as compared to 3.
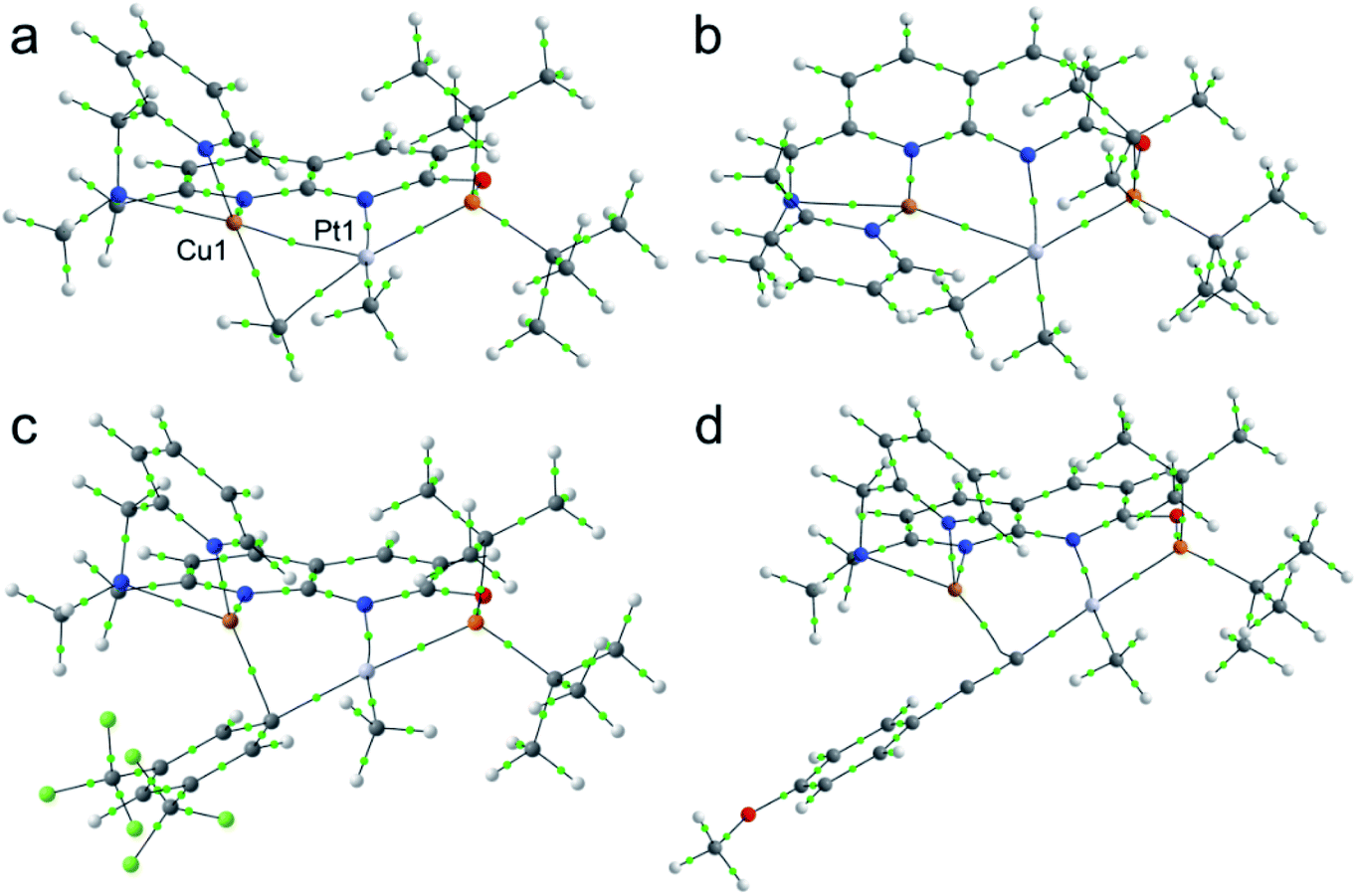 | ||
| Fig. 3 Molecular graphs for “gas-phase” DFT-optimized complexes 3+ (a), 4 (b), 6+ (c), and 7+ (d). Bond critical points (3, −1) with a threshold of ∇ρb > 0.025 a.u. and corresponding bond paths are shown with green dots and black lines, respectively. | ||
NBO analysis also shows that complex 3 exhibits strong electron density donation from the proximal Pt–MeA fragment to an s-type orbital on Cu [σ(Pt–CA(sp3)→Cu(s)); E(2) = 89.68 kcal mol−1] (Table S11 and Fig. S153†). The metal–metal interactions are manifested in a moderate donation from a d-type orbital on the Pt center to an s-orbital on Cu [Pt(d)→Cu(s); E(2) = 24.49 kcal mol−1]. Interestingly, the distal Pt–MeB fragment also shows a donation to a Cu center [σ(Pt–CB(sp3)→Cu(s)); E(2) = 25.67 kcal mol−1], albeit much weaker compared to the donation from Pt–MeA. A weak back-donation is also found from the d-type orbitals on Cu to a Pt–MeA [E(2) = 6.0 kcal mol−1] and a Pt–MeB fragments [E(2) = 3.0 kcal mol−1].
The predominant interaction of the proximal Pt–MeA fragment with a Cu center is also evident from the comparison of the Natural Binding Index (NBI): an NBI between Cu and a carbon atom of MeA is 0.3476, as compared to a much lower NBI between Cu and a carbon atom of MeB (0.0946) (Table S12†). The Pt→Cu interaction results in an NBI of 0.2687 between the Pt and Cu centers. The comparison of complex 3 and copper-free complex 1 shows a strong effect of the coordination of the Cu center with a bridging MeA ligand resulting in the elongation of the Pt–MeA bond from 2.08 Å in 1 to 2.15 Å in 3. At the same time, the Pt–MeB bond length remains essentially unchanged (at 2.05 Å) in both complexes, showing that the effect of Cu on the Pt–MeB bond length is negligible. Accordingly, the NBI between Pt–MeA is noticeably lower in complex 3 (0.6760) compared to complex 1 (0.7444), while only minor changes are seen in the NBI for the Pt–MeB fragment (0.8403 and 0.8301 in complexes 3 and 1, respectively) (Tables S10 and S12†).
This analysis confirms that the three-center two-electron binding in complex 3 can be best described as a donor–acceptor interaction between a bridging Pt–MeA and the Lewis-acidic Cu center that is further supported by the Pt→Cu interaction. The distance between Cu and carbon of the MeA fragment remains significantly longer (Tables 1 and S6†) compared to that in the symmetrical Me-bridged dicopper complex with a naphthyridine-based ligand reported by Tilley and co-workers (2.06–2.08 Å), which showed a three-center, two-electron bond with essentially equivalent binding of the bridging Me to both Cu centers.22 The donor–acceptor type σ(Pt–MeA)→Cu(s) three-center, two-electron interaction in 3 is perhaps unsurprisingly unsymmetrical due to the heterobimetallic nature of the complex. However, it resembles the binding interaction in the unsymmetrical donor–acceptor-type Me-bridged [Cu(PPh3)2(μ-Me)CuMe] dicopper complex reported by Steffen and co-workers that is also stabilized by metal–metal interactions.23
Compared to 3, NBO analysis shows that complex 4 exhibits only moderate donation from a Pt–MeA fragment to Cu [E(2) = 44.23 kcal mol−1] and similar metal–metal interactions manifest in the donation from a d-type orbital on Pt to an s-orbital of Cu [E(2) = 32.13 kcal mol−1] (Table S13 and Fig. S155†). Weaker donation from the Pt–MeB fragment to Cu is also observed [E(2) = 17.5 kcal mol−1]. The NBI between the carbon of the distal MeA and Cu is 0.1985 in 4, significantly less than in 3, while NBI between the Pt and Cu centers remain similar (0.2498) (Table S14†).
The detailed analysis of orbital contributions to the Pt→Cu interaction in complexes 3 and 4 shows that the donation from Pt occurs predominantly from the filled dz2-type orbital in both complexes. This is also consistent with the general description of the dative Pt→Cu bonding in heterobimetallic Pt/Cu complexes reported by Chen and co-workers.13
Reactivity of PtII/CuI complexes and comparison with monometallic Pt complexes
We first studied the solution-state stability of complexes 3[X] compared with their monometallic counterpart 1. The common decomposition pathway for dimethyl Pt complexes with N,P-donor ligands involves rollover cyclometalation leading to undesired C–H bond activation of the ligand.24 Heating monometallic complex 1 at 40 °C in THF for 12 h (or 3 days in benzene) led to the expected cyclometalation to form complex 5 characterized by XRD (Scheme 4a). In contrast, bimetallic complex 3[BF4] was considerably more stable towards cyclometalation and remained unchanged upon heating in THF at 40 °C for 12 h. The presence of an electrophilic CuI center coordinated by the bridging Me group presumably offers some kinetic resistance to rollover cyclometalation.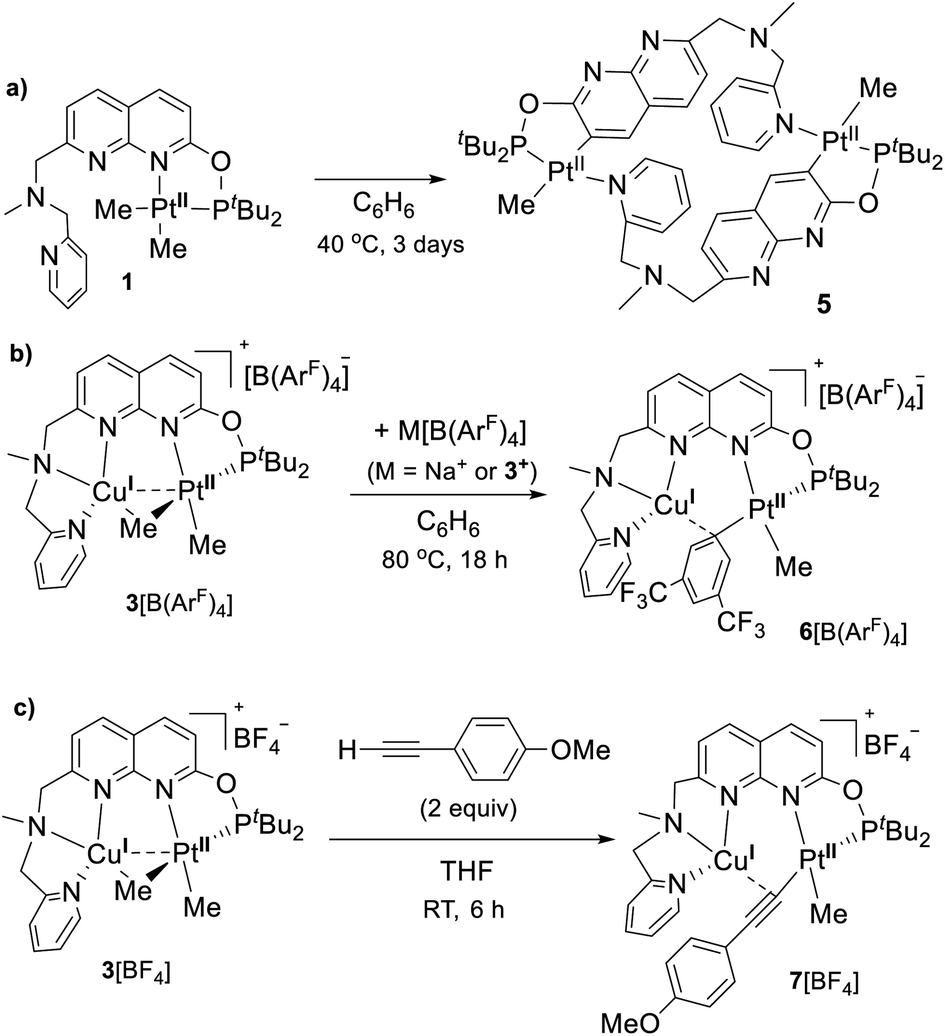 | ||
| Scheme 4 (a) Cyclometalation of 1; (b) aryl group transfer from the [B(ArF)4]− counterion to give 6; (c) terminal alkyne activation. | ||
Surprisingly, when complex 3[B(ArF)4] was heated in C6H6 at 80 °C for 18 h, a new complex 6[B(ArF)4] was obtained in 46% in situ yield resulting from an aryl group transfer from a [B(ArF)4]− counteranion to a Pt center (Scheme 4 and Fig. 4). Under the same conditions, complex 3[BF4] mostly decomposed (>80%) after heating in C6H6 at 80 °C for 18 h to form a mixture of unidentified products. Although such electron deficient aryl group transfer is known for some electrophilic monometallic complexes (Rh, Au, and Pt)25 and homobimetallic Cu2 and Fe2 complexes,6h,26 this is the first example of such transmetalation from a tetraarylborate anion by a heterobimetallic complex with a formally neutral Pt center. Aryl group transfer upon treatment with a Lewis-acidic BPh3 was also observed in Me-bridged homobimetallic Cu2 complexes.22 Although the fate of the Me group and B-containing product could not be determined, the less than 50% yield of complex 6[B(ArF)4] likely results from the necessity to sacrifice a [B(ArF)4]− counteranion for aryl group transfer.20 Indeed, when the reaction was performed in the presence of 4.5 equiv. of Na[B(ArF)4], the in situ yield of 6[B(ArF)4] increased to 80%.
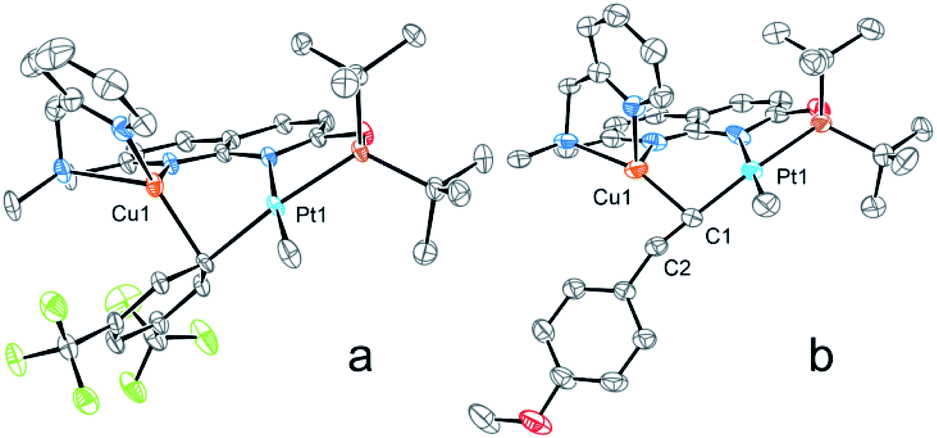 | ||
| Fig. 4 ORTEP of 6[B(ArF)4] (a) and 7[BF4] (b) at the 50% probability level. Hydrogen atoms, counterions, and solvent molecules together with the minor disorder component for 7[BF4] are omitted for clarity. | ||
The X-ray structure of 6[B(ArF)4] reveals close contacts of a CuI center with the ipso-carbon of an aryl group (2.098(3) Å) and an adjacent ortho-carbon (2.335(3) Å), while the distance between PtII and CuI atoms (2.7745(4) Å) is now longer than the sum of their covalent radii, indicating no metal–metal interactions when compared to 3[X] and also consistent with the lack of bcp according to AIM analysis (Fig. 3 and Table S4†).
NBO analysis for complex 6[B(ArF)4] reveals strong donation [E(2) = 62.36 kcal mol−1] from the Pt–Cipso fragment of Pt–Ar to an empty s-type orbital on Cu (Table S15 and Fig. S157†). Additionally, donation from the p-type orbitals of the π-bond (Cipso![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cortho) to an s-type orbital on Cu is also observed [E(2) = 27.35 kcal mol−1], along with the corresponding back-donation from the d-type orbital at Cu to an antibonding π*(CipsoCortho) orbital [E(2) = 15.66 kcal mol−1]. Compared to 3 and 4, only moderate donation from a d-type orbital at Pt to an s-orbital at Cu is observed, resulting in a lower NBI between Pt and Cu of 0.2281.
Cortho) to an s-type orbital on Cu is also observed [E(2) = 27.35 kcal mol−1], along with the corresponding back-donation from the d-type orbital at Cu to an antibonding π*(CipsoCortho) orbital [E(2) = 15.66 kcal mol−1]. Compared to 3 and 4, only moderate donation from a d-type orbital at Pt to an s-orbital at Cu is observed, resulting in a lower NBI between Pt and Cu of 0.2281.
We then examined the reactivity of 3 with a terminal alkyne as this substrate contains a reactive C–H bond and a π-system that can potentially interact with a cationic CuI center. The synergistic effect of CuI salts has been previously implicated in bimetallic alkyne activation.4a–c Importantly, monometallic complex 1 did not show any reaction with 2 equiv. of 4-ethynylanisole at RT for at least 24 h. On the other hand, when 3[BF4] was reacted with 2 equiv. of 4-ethynylanisole at RT, acetylide complex 7[BF4] was cleanly obtained (Scheme 4). The product was isolated in pure form in 59% yield and fully characterized.
A single crystal XRD study reveals a PtII center with a σ-bound acetylide ligand, which coordinates to a CuI center through the triple bond π-system (Cu1⋯C1 and Cu1⋯C2 distances of 1.982(5) Å and 2.141(4) Å). The distance between PtII and CuI is 3.0934(8) Å, indicative of a lack of interaction between two metals after coordination of the Cu center with the carbon atom of acetylide and consistent with AIM analysis (Fig. 3).
NBO analysis also shows strong donation from a Pt–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) fragment of Pt-acetylide to an empty s-orbital of Cu [E(2) = 56.69 kcal mol−1] (Table S17 and Fig. S159†). Donation from p-type orbitals of a CC fragment to Cu [E(2) = 56.69 kcal mol−1] and the corresponding back-donation from Cu to an antibonding π*(CC) orbital [E(2) = 16.14 kcal mol−1] is consistent with a π-coordination of Cu with a triple bond. Only weak donation is observed between a filled d-type orbital at Pt to the empty s-orbital on Cu [E(2) = 13.38 kcal mol−1] resulting in a low NBI between the Pt and Cu centers (0.1949).
fragment of Pt-acetylide to an empty s-orbital of Cu [E(2) = 56.69 kcal mol−1] (Table S17 and Fig. S159†). Donation from p-type orbitals of a CC fragment to Cu [E(2) = 56.69 kcal mol−1] and the corresponding back-donation from Cu to an antibonding π*(CC) orbital [E(2) = 16.14 kcal mol−1] is consistent with a π-coordination of Cu with a triple bond. Only weak donation is observed between a filled d-type orbital at Pt to the empty s-orbital on Cu [E(2) = 13.38 kcal mol−1] resulting in a low NBI between the Pt and Cu centers (0.1949).
The rate constants were measured in a reaction of 3[BF4] with an excess of phenylacetylene or phenylacetylene-d1 at −10 °C under pseudo-first order conditions to give the values of (3.9 ± 0.2) × 10−4 s−1 and (4.4 ± 0.9) × 10−5 s−1 for phenylacetylene and phenylacetylene-d1 labeled at the terminal CH group, respectively. The kinetic isotope effect (KIE) of 9 ± 2 (at −10 °C) suggests that the C–H bond cleavage likely happens at the rate determining step (vide infra).27 The relatively large value for the observed KIE, which is close to the theoretical maximum for the primary KIE, is not uncommon for C–H activation or protonolysis by Pt-methyl complexes28 and other transition metals.27 Unusually large values for the KIE (KIE ≥ 7) are often attributed to the tunnelling effect, or in some cases to the geometry of the transition state.27
The neutral complex 4 did not show clean reactivity with 2 equiv. of 4-ethynylanisole leading to its eventual decomposition to form multiple products. The lack of well-defined acetylide products is likely due to the significantly higher reactivity of 4 from the presence of a dearomatized ligand arm, which might lead to ligand-centered reactivity, and due to the lack of electrophilicity of the neutral, electron-rich Cu center stabilized by an amide donor.
A computational study of the role of Cu in alkyne activation
Based on the above reactivity, we hypothesize that a cationic CuI center plays a role in coordinating the alkyne, thus bringing the substrate in proximity to the Pt center, while increasing the acidity of the terminal C–H bond.4b We conducted preliminary DFT studies using a truncated system with propyne as a model substrate and a dimethylphosphinite-substituted ligand, and were able to find an energetically accessible pathway, which involves initial π-coordination of an alkyne with CuI of 3′ to initially give π-complex A (Fig. 5). NBO calculations reveal enhancement in the polarization of the terminal C–H bond in A (charges at C −0.369 and H +0.273) as compared to free propyne (C −0.265 and H +0.239). Oxidative addition to the PtII center leads to the PtIV hydride acetylide species B, where the triple bond of the alkyne is still π-coordinated to Cu. Due to the weak coordination of the naphthyridine N-donor,29 low barrier C–H (methane) reductive elimination results in the acetylide complex C with a net exergonicity of 28 kcal mol−1.30 The DFT-calculated KIE for this mechanism is 3.7 at −10 °C (for the truncated system), which is smaller than the observed KIE; however, it is consistent overall with C–H bond activation at the rate determining step.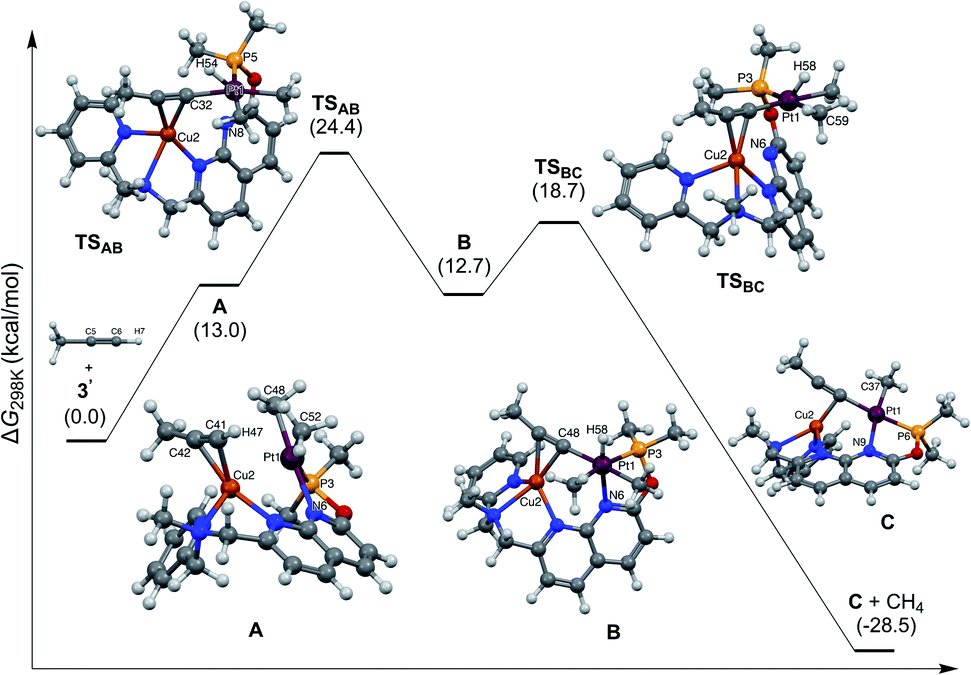 | ||
| Fig. 5 Calculated energy profile for alkyne activation and DFT-optimized structures for intermediates and transition states. | ||
The alternative concerted protonolysis pathway was also considered, but was found to have a significantly higher barrier of 38.7 kcal mol−1.30 Because of the use of a truncated system in the preliminary computational analysis, the absolute values should be considered only for qualitative evaluation, and contribution from other possible reaction pathways cannot be excluded.
Although the synergistic effect of CuI in terminal alkyne activation is also generally observed in the Sonogashira coupling,4a–c the nature of this effect in the case of the CuI/PtII complex described in this work is different and does not involve the transmetalation step, but rather oxidative addition of the C–H to the Pt center. We believe that this is facilitated due to the ability of Cu to act as a “docking site” for an alkyne bringing the C–H bond in proximity to a Pt center and its ability to polarize a C–H bond, making it more prone to further reactivity.
Conclusions
In summary, we designed and developed a reactive heterobimetallic Pt/Cu species that conclusively demonstrates that proximal interactions with a Cu center alters the reactivity of Pt. We found that a bridging alkyl group between the two metals prevents undesired rollover cyclometalation. The presence of a CuI center also induces facile transmetalation from an electron-deficient [B(ArF)4]− anion and enables facile C–H bond activation of a terminal alkyne. DFT studies elucidate the role of the copper center in coordination and activation of an alkyne substrate which otherwise remains unreactive in the presence of a Pt-only monometallic complex. Considering the increased interest towards the utilization of bimetallic catalysis, this study highlights the principles of ligand design for the study of the metal–metal cooperation effect in organometallic reactivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
O. R.-W. was a JSPS International Research Fellow. This work was supported by JSPS Kakenhi Grant Number 16F16038. The authors thank Mr A. Villar-Briones and Dr M. Roy for performing HR-MS analysis and acknowledge OIST for funding. We thank Dr K. Eguchi (JEOL RESONANCE Inc.) for advice regarding 195Pt NMR.Notes and references
-
(a) P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef PubMed
; (b) R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef
-
(a) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC
-
(a) N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 12264–12265 CrossRef PubMed
-
(a) R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC
-
(a) V. Farina, S. Kapadia, B. Krishnan, C. Wang and L. S. Liebeskind, J. Org. Chem., 1994, 59, 5905–5911 CrossRef
-
(a)
F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds between Metal Atoms, SpringerNew York, 2006 Search PubMed
- A. Nicolay and T. D. Tilley, Chem.–Eur. J., 2018, 24, 10329–10333 CrossRef CAS
-
(a) J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115–5127 RSC
-
(a) C. Dubs, A. Inagaki and M. Akita, Chem. Commun., 2004, 2760–2761 RSC
-
(a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS
- M.-E. Moret, D. Serra, A. Bach and P. Chen, Angew. Chem., Int. Ed., 2010, 49, 2873–2877 CrossRef CAS
- O. Rivada-Wheelaghan, S. L. Aristizábal, J. López-Serrano, R. R. Fayzullin and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2017, 56, 16267–16271 CrossRef CAS
- M.-E. Moret and P. Chen, J. Am. Chem. Soc., 2009, 131, 5675–5690 CrossRef CAS
-
(a) E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem.–Eur. J., 2014, 20, 15727–15731 CrossRef CAS PubMed
-
(a) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed
- Deposition numbers 1975187–1975194 contain the supplementary crystallographic data for this paper.†.
-
(a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC
- J. Campos, J. López-Serrano, R. Peloso and E. Carmona, Chem.–Eur. J., 2016, 22, 6432–6457 CrossRef CAS PubMed
-
(a) E. G. Hope, W. Levason and N. A. Powell, Inorg. Chim. Acta, 1986, 115, 187–192 CrossRef CAS
-
(a) C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Coord. Chem. Rev., 2017, 345, 150–181 CrossRef CAS
- M. S. Ziegler, N. A. Torquato, D. S. Levine, A. Nicolay, H. Celik and T. D. Tilley, Organometallics, 2018, 37, 2807–2823 CrossRef CAS
- R. Molteni, R. Bertermann, K. Edkins and A. Steffen, Chem. Commun., 2016, 52, 5019–5022 RSC
-
(a) M. L. Scheuermann, K. A. Grice, M. J. Ruppel, M. Rosello-Merino, W. Kaminsky and K. I. Goldberg, Dalton Trans., 2014, 43, 12018–12025 RSC
-
(a) H. Salem, L. J. W. Shimon, G. Leitus, L. Weiner and D. Milstein, Organometallics, 2008, 27, 2293–2299 CrossRef CAS
- S. L. Matthews and D. M. Heinekey, Inorg. Chem., 2011, 50, 7925–7927 CrossRef CAS PubMed
- M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed
-
(a) J. E. Bercaw, G. S. Chen, J. A. Labinger and B.-L. Lin, Organometallics, 2010, 29, 4354–4359 CrossRef CAS
-
(a) D. M. Crumpton and K. I. Goldberg, J. Am. Chem. Soc., 2000, 122, 962–963 CrossRef CAS
- See the ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1975187–1975194. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00646g |
| ‡ Current address: Orestes Rivada-Wheelaghan: Université de Paris, Laboratoire d'Electrochimie Moléculaire, UMR 7591 CNRS, 15 rue Jean-Antoine de Baïf, F-75205 Paris Cedex 13, France. |
| This journal is © The Royal Society of Chemistry 2020 |