
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn isocyanide ligand for the rapid quenching and efficient removal of copper residues after Cu/TEMPO-catalyzed aerobic alcohol oxidation and atom transfer radical polymerization†
Grzegorz
Szczepaniak
 *ab,
Jakub
Piątkowski‡
a,
Wojciech
Nogaś‡
a,
Francesca
Lorandi‡
b,
Saigopalakrishna S.
Yerneni
c,
Marco
Fantin
b,
Anna
Ruszczyńska
a,
Alan E.
Enciso
d,
Ewa
Bulska
a,
Karol
Grela
*a and
Krzysztof
Matyjaszewski
*b
*ab,
Jakub
Piątkowski‡
a,
Wojciech
Nogaś‡
a,
Francesca
Lorandi‡
b,
Saigopalakrishna S.
Yerneni
c,
Marco
Fantin
b,
Anna
Ruszczyńska
a,
Alan E.
Enciso
d,
Ewa
Bulska
a,
Karol
Grela
*a and
Krzysztof
Matyjaszewski
*b
aFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: szczepaniak.grzegorz@gmail.com; prof.grela@gmail.com
bDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, Pennsylvania 15213, USA. E-mail: km3b@andrew.cmu.edu
cDepartment of Biomedical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA
dDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
First published on 15th April 2020
Abstract
Transition metal catalysts play a prominent role in modern organic and polymer chemistry, enabling many transformations of academic and industrial significance. However, the use of organometallic catalysts often requires the removal of their residues from reaction products, which is particularly important in the pharmaceutical industry. Therefore, the development of efficient and economical methods for the removal of metal contamination is of critical importance. Herein, we demonstrate that commercially available 1,4-bis(3-isocyanopropyl)piperazine can be used as a highly efficient quenching agent (QA) and copper scavenger in Cu/TEMPO alcohol aerobic oxidation (Stahl oxidation) and atom transfer radical polymerization (ATRP). The addition of QA immediately terminates Cu-mediated reactions under various conditions, forming a copper complex that can be easily separated from both small molecules and macromolecules. The purification protocol for aldehydes is based on the addition of a small amount of silica gel followed by QA and filtration. The use of QA@SiO2 synthesized in situ results in products with Cu content usually below 5 ppm. Purification of polymers involves only the addition of QA in THF followed by filtration, leading to polymers with very low Cu content, even after ATRP with high catalyst loading. Furthermore, the addition of QA completely prevents oxidative alkyne–alkyne (Glaser) coupling. Although isocyanide QA shows moderate toxicity, it can be easily converted into a non-toxic compound by acid hydrolysis.
Introduction
The goal of synthetic chemistry is to efficiently produce diverse compounds of high purity. Since many important synthetic transformations require the use of transition metal catalysts,1–6 the problem of purifying the products from metal residues is of considerable practical importance. A metal impurity can promote the decomposition of the desired product or compromise the performance of advanced materials.7,8 Additionally, transition metal-catalyzed reactions are used in the large scale synthesis of modern active pharmaceutical ingredients (API).9–11 Therefore, this issue is crucial in the pharmaceutical industry, which is subject to stringent limits on the allowed concentration of heavy metals in the final API, typically less than 10 ppm. Traditional separation methods, such as chromatography or crystallization, often fail to give products with acceptably low metal content, especially when applied to compounds with functional groups that can bind to transition metals.12In recent years, more attention has been given to tackling the problem of metal contamination as early as possible in the synthetic pipeline.13 Possible approaches include heterogeneous catalysis, self-scavenging catalysts, and the use of metal scavengers.14,15 In 2015, inspired by the work of Diver and co-workers,16 we reported a new, stable, odorless, bidentate isocyanide scavenger and quenching agent QA (1,4-bis(3-isocyanopropyl)piperazine),17 which was used to remove ruthenium impurities from olefin metathesis products and palladium residues from cross-coupling products by straightforward purification protocols.17–19
Copper-based catalysts are used in many transformations of critical importance to both laboratory and industrial applications,20 including azide–alkyne Huisgen cycloaddition (CuAAC),21 atom transfer radical polymerization (ATRP),22 Ullmann coupling,23 and aerobic oxidation reactions.24 Since isocyanide ligands can coordinate to late 4d and 5d transition metals due to their σ-donor and π-acceptor properties,25 we hypothesized that QA could be used as a copper scavenger in the purification of Cu/TEMPO alcohol aerobic oxidation (Stahl oxidation) products and also for polymers made by Cu-catalyzed ATRP. The results presented in this study validate this hypothesis and show that QA could be used to purify both small molecules and macromolecules from copper impurities. In addition, QA immediately stops copper-mediated reactions, facilitating kinetic studies, and reaction design. Finally, we demonstrated that QA could be used to prevent side reactions, such as Glaser coupling, that can occur during polymers workup.
Alcohol oxidation is one of the most widely used oxidation reactions in the synthesis of complex organic molecules. In recent years, aerobic oxidation reactions have been the focus of considerable attention.26,27 Compared to traditional reagents and methods, these transformations are attractive due to the use of molecular oxygen as an inexpensive oxidant. Furthermore, aerobic oxidation is an environmentally benign method characterized by lower process mass intensity (PMI) compared to traditional alcohol oxidation protocols such us Swern,28 Parikh–Doering oxidation29 and TEMPO/NaOCl (TEMPO = 2,2,6,6-tetramethylpiperidine N-oxyl) based methods.30,31
The Stahl group developed a highly practical, selective, mild and efficient aerobic alcohol oxidation mediated by the Cu/TEMPO system,32–34 which exhibits high functional group tolerance and has found many applications in organic synthesis.26,35–37 However, to the best of our knowledge, there has been no study on the removal of copper residues for this type of transformation.
The catalytic cycle for Cu/TEMPO-catalyzed aerobic alcohol oxidation involves oxidation of CuI/L complex by molecular oxygen to a peroxo-bridged binuclear CuII intermediate (Scheme 1A). The [CuII/L]2O2 species oxidize the hydroxylamine (TEMPOH = 2,2,6,6-tetramethylpiperidin-1-ol) to TEMPO, leading to the formation of a HOO–CuII/L and a CuI/L complex. Then, the HOO–CuII/L intermediate reacts with water to afford a HO–CuII/L and hydrogen peroxide. Finally, the OH–CuII/L and TEMPO mediate the oxidation of alcohol via a six-membered transition state, resulting in aldehyde formation and regeneration of CuI/L complex.38
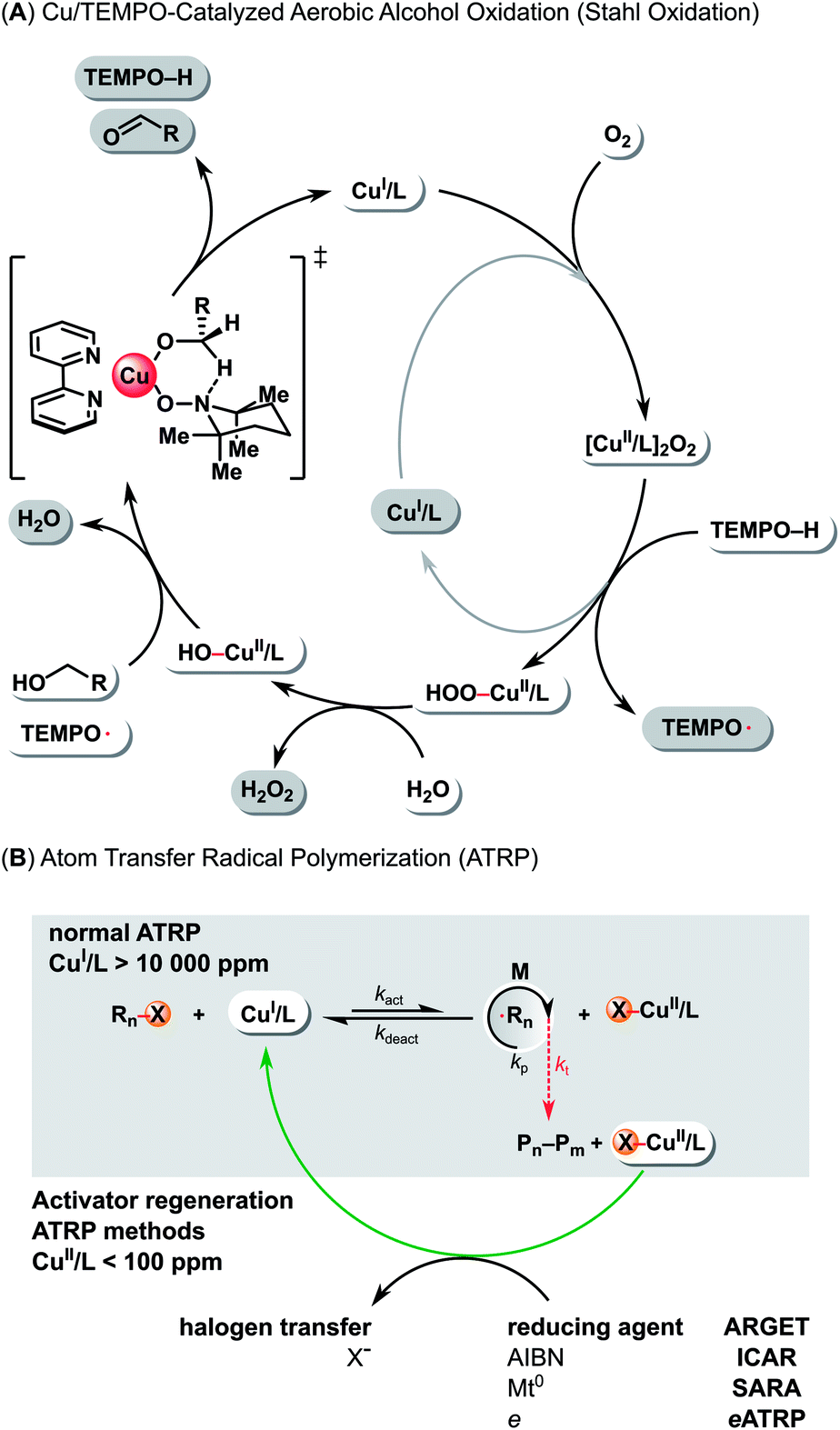 | ||
| Scheme 1 (A) Mechanism of Cu/TEMPO alcohol aerobic oxidation (Stahl oxidation). (B) Mechanism of Cu-catalyzed normal ATRP and low-ppm ATRP in the presence of excess reducing agents. | ||
Atom transfer radical polymerization (ATRP) is now considered one of the stock transformations in the toolbox of modern polymer chemistry.22,39–41 Due to its compatibility with a wide variety of functional groups, easy setup, and mild conditions, it is used in a wide range of applications. ATRP is based on a reversible redox reaction catalyzed by transition metal complexes in their lower oxidation state. The most common ATRP catalysts are Cu complexes.42
The control over radical propagation in ATRP is provided via an atom transfer equilibrium between a CuI/L and a X–CuII/L complex, where L represents a multidentate nitrogen-based ligand and X is a halogen (Scheme 1B). The CuI/L complex activates the C(sp3)–X polymer chain end, resulting in the formation of a propagating carbon-based radical and the deactivator form (X–CuII/L) of the catalyst. Then, the X–CuII/L reacts with the propagating radical in a reverse reaction, regenerating the active catalyst CuI/L and the dormant polymer chain end C(sp3)–X.22
Standard ATRP requires a large amount of an oxygen sensitive CuI/L catalyst (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm with respect to the monomer),43,44 the residues of which are difficult to remove from the polymer (Scheme 1B). Recently, several new ATRP techniques were developed to overcome this (Scheme 1B). They are based on the regeneration of the active form of the catalyst using various external stimuli, which allows reducing the catalyst loading to less than 100 ppm. These methods include: activators generated by electron transfer (AGET),45 activators regenerated by electron transfer (ARGET),46 initiators for continuous activator regeneration (ICAR),47 supplemental activators and reducing agent (SARA),48 electrochemically mediated ATRP (eATRP),49 photoinduced ATRP (pATRP),50 and mechanically/ultrasound induced ATRP (mechano/sonoATRP).51,52
000 ppm with respect to the monomer),43,44 the residues of which are difficult to remove from the polymer (Scheme 1B). Recently, several new ATRP techniques were developed to overcome this (Scheme 1B). They are based on the regeneration of the active form of the catalyst using various external stimuli, which allows reducing the catalyst loading to less than 100 ppm. These methods include: activators generated by electron transfer (AGET),45 activators regenerated by electron transfer (ARGET),46 initiators for continuous activator regeneration (ICAR),47 supplemental activators and reducing agent (SARA),48 electrochemically mediated ATRP (eATRP),49 photoinduced ATRP (pATRP),50 and mechanically/ultrasound induced ATRP (mechano/sonoATRP).51,52
The ideal approach to overcoming copper contamination in traditional ATRP systems is the recently developed metal-free ATRP, catalyzed by an organic photoredox catalyst.53–56 However, the narrow monomer scope is a major limitation of this technique. Moreover, it creates a new challenge – the product can be contaminated with the organocatalyst.
Despite significant progress in the development of ATRP techniques, many modern applications that give access to complex macromolecular architectures still require the use of high loadings of copper catalysts.57 Moreover, in biomedical applications, copper residues in polymers can induce cellular oxidative damage.58 For these reasons, the development of economical and practical methods for the removal of copper impurities after ATRP is highly desirable.
The problem of copper contamination in polymers can be solved by precipitation, dialysis, extraction, or filtration through a silica gel or aluminum oxide. Unfortunately, standard polymer purification protocols are often insufficient to bring the copper content below 10 ppm, in particular when polymers exhibit high affinity towards copper.59 To address this issue, many advanced methods of purification have been developed,60,61 including heterogeneous catalysis,62–64 self-scavenging catalysts,65–67 metal scavengers,68 switchable systems,69 ion-exchange resins,70 electrolysis of copper residues59,71 and other related electrochemical methods to reduce copper.72 However, most of these purification protocols suffer from at least one critical flaw, such as complexity and limited availability, the need of specialized equipment, long purification time, issues with end-group fidelity or inability to decrease the copper content below 10 ppm.
Herein, we demonstrate that the introduction of the quenching agent QA rapidly stops ATRP under various conditions, forming a copper complex that precipitates, allowing for facile removal by filtration. After this treatment, the residual copper contamination of the polymers was <2 ppm, even when a high initial catalyst loading was used. Similarly, the addition of QA to Stahl oxidation products decreased the copper content below 5 ppm.
Results and discussion
Quenching of Stahl oxidation by isocyanide QA
We started our investigation by examining QA as a quenching agent of Stahl oxidation (Fig. 1A). Our hypothesis assumed that QA might form a stable complex with copper(I) that does not oxidize further. Aerobic oxidation of nerol (1a) was performed in MeCN at room temperature, in a reaction vessel open to the air, with CuI/bpy/TEMPO/DMAP (bpy = 2,2′-bipyridine, DMAP = 4-(dimethylamino)pyridine) as the catalytic system. After 24 h, in the presence of QA, the conversion of alcohol 1a was 0% (Fig. 1A), while in the control experiment without the addition of QA, it was >99%. These experiments showed that isocyanide QA is binding to copper, inhibiting the oxidation reaction.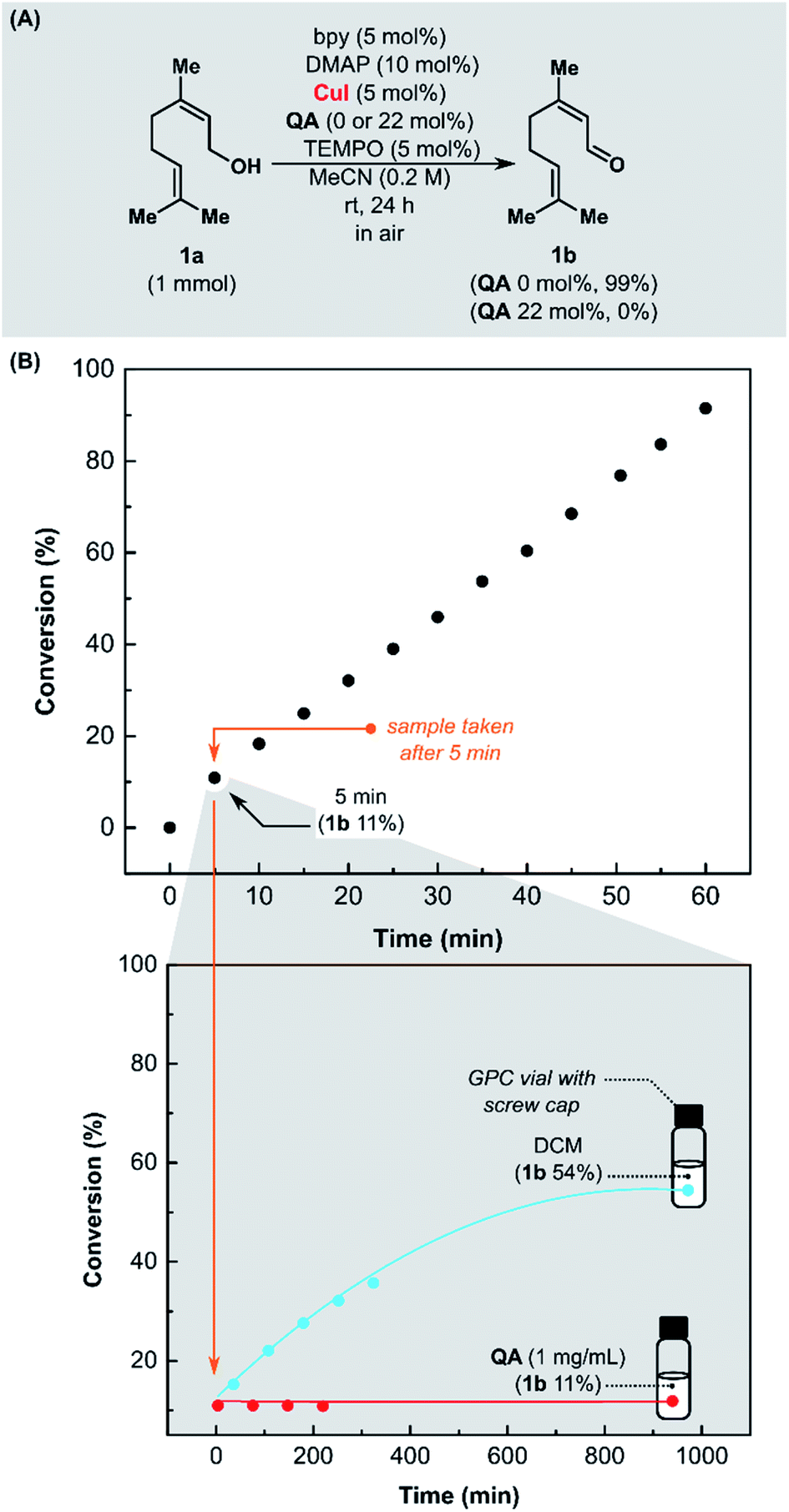 | ||
| Fig. 1 (A) Quenching of Stahl oxidation by QA. (B) Evaluation of the performance of QA as a quenching agent in Stahl oxidation of nerol (1a), reaction conditions: 1a (1 mmol), bpy (5 mol%), DMAP (10 mol%), CuI (5 mol%), TEMPO (5 mol%). | ||
Next, we evaluated the performance of QA as a quenching agent in the Cu-catalyzed oxidation of 1a. The kinetic study was performed at room temperature in a reaction vessel open to the air, using CuI/bpy/TEMPO/DMAP catalytic system (Fig. 1B). Aliquots of the reaction mixture (0.2 mL) were added to a solution of QA in DCM (1.0 mL, 1 mg mL−1) at selected time and then analyzed with gas chromatography (GC).
After 5 min, a sample was taken from the reaction mixture. Half of it was added to a solution of scavenger QA in DCM, and the remainder was added to pure DCM (Fig. 1B). Even though the reaction mixture was significantly diluted and the access to air was limited (vial with screw cap), the oxidation of alcohol 1a was still proceeding (blue curve). In the vial containing QA, the conversion did not change over time (red curve).
Then, we examined the activity of various copper catalytic systems utilizing QA (Fig. 1S†). This study demonstrated that QA could be used as a fast-acting quenching agent to facilitate the kinetic studies of Cu-mediated oxidation of alcohols by halting the reaction progress at a specific point in time and using an offline analytic method, such as GC, to analyze the samples.73 To the best of our knowledge, no quenching agent for this transformation has yet been described in the literature. To determine the conversion as a function of time by GC, it was necessary to conduct an independent experiment for every time interval and terminate it by filtration through silica gel.74 The proposed quenching method is fast and easy to carry out. Encouraged by these promising results, we evaluated QA as a metal scavenger in the removal of copper residues.
Removal of copper residues after Stahl Oxidation
Recently, we reported a semi-heterogeneous purification protocol for the removal of ruthenium impurities after olefin metathesis reaction.19 It is based on the noncovalent immobilization of QA on unmodified silica gel during the purification process. Herein, we applied this protocol to the removal of copper residues after Cu-mediated oxidation of alcohols.For the evaluation of QA performance in the removal of copper impurities after Stahl oxidation, we selected aliphatic and aromatic alcohols with different functional groups (Table 1). All reactions used 1.0 mmol of the alcohol and were performed in MeCN at room temperature with ambient air as the source of oxidant, using a CuI/bpy/TEMPO/DMAP catalytic system. After 2 h, the conversions for all tested substrates were >99%.
| a Conversions were determined by GC analysis and are based on the ratio of product/(product + starting material). Cu content was determined by ICP-MS. |
|---|
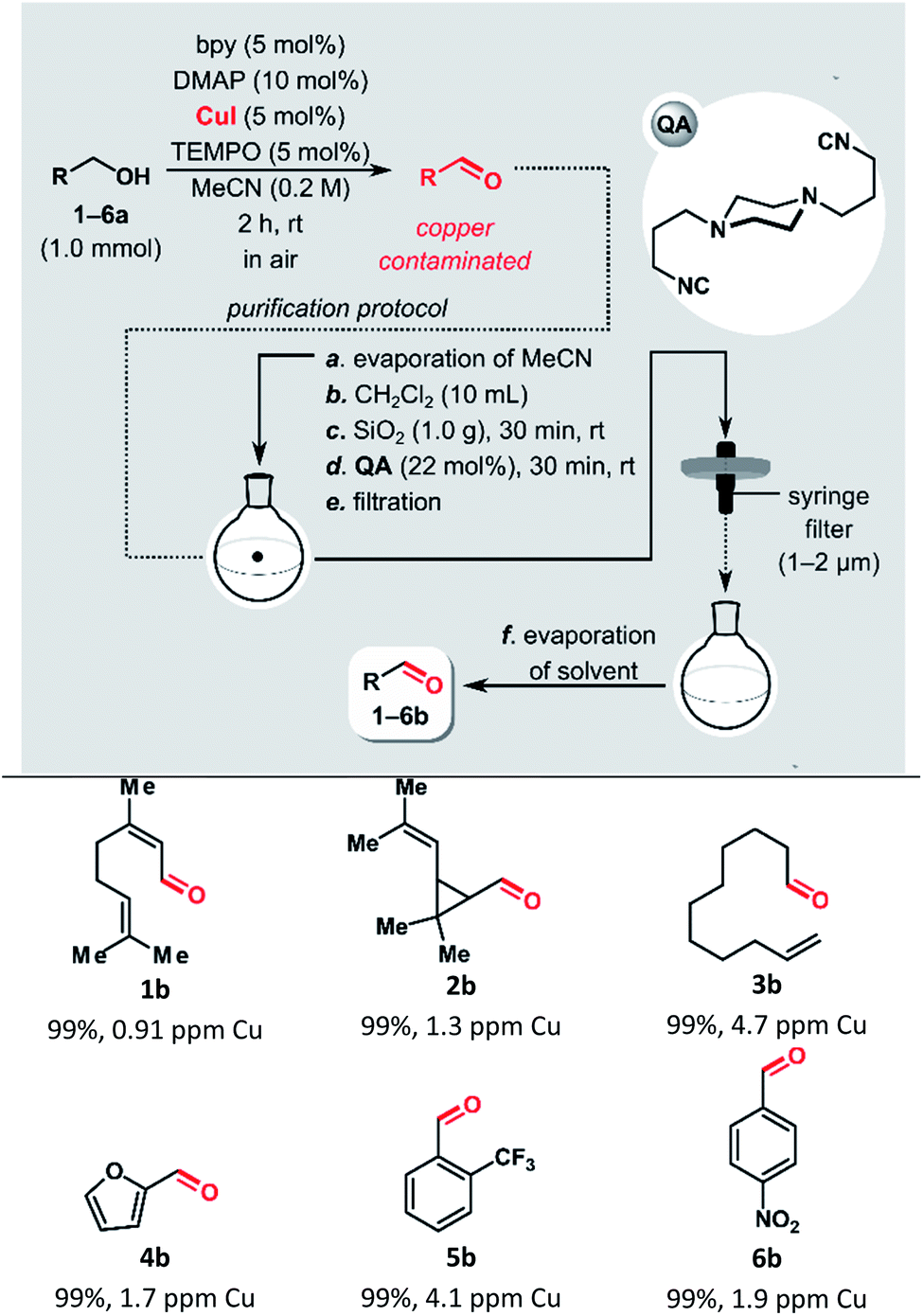
|
To minimize the risk of leaching the copper-containing contaminants from the silica gel, MeCN was evaporated before removing the copper, and then DCM was added. The solvent switch can be avoided. However, the best results were obtained when MeCN was removed before the purification (ESI Table S1†). The addition of silica gel, followed by the isocyanide treatment, allowed for isolating aldehydes with very low copper content, below 5 ppm. Meanwhile, a control experiment with furfural (4b), in which no QA was added, yielded a product that remained contaminated with copper at a very high level, 800 ppm. In addition, since most of the catalytic system components bind strongly to silica gel after simple filtration through a syringe filter, followed by evaporation of the solvent, the isolated products were pure, based on 1H NMR spectroscopic analysis.
Cu/TEMPO catalyst systems show reduced reactivity with aliphatic and sterically hindered alcohols. Stahl and co-workers demonstrated that replacement of TEMPO with the less sterically hindered ABNO (ABNO = 9-azabicyclo[3.3.1]nonane N-oxyl) diminishes the steric and electronic constraints of the Cu/TEMPO.75 The aerobic oxidation of the sterically hindered alcohol 7a in a gram scale (4 mmol, 1.41 g) was performed using the highly active CuI/bpy/ABNO/DMAP catalytic system (Fig. 2). The new purification protocol proved to be scalable. After purification, the copper content in the isolated aldehyde 7b, a key intermediate in the synthesis of cardiovascular disease drug rosuvastatin,76 was only 3.6 ppm.
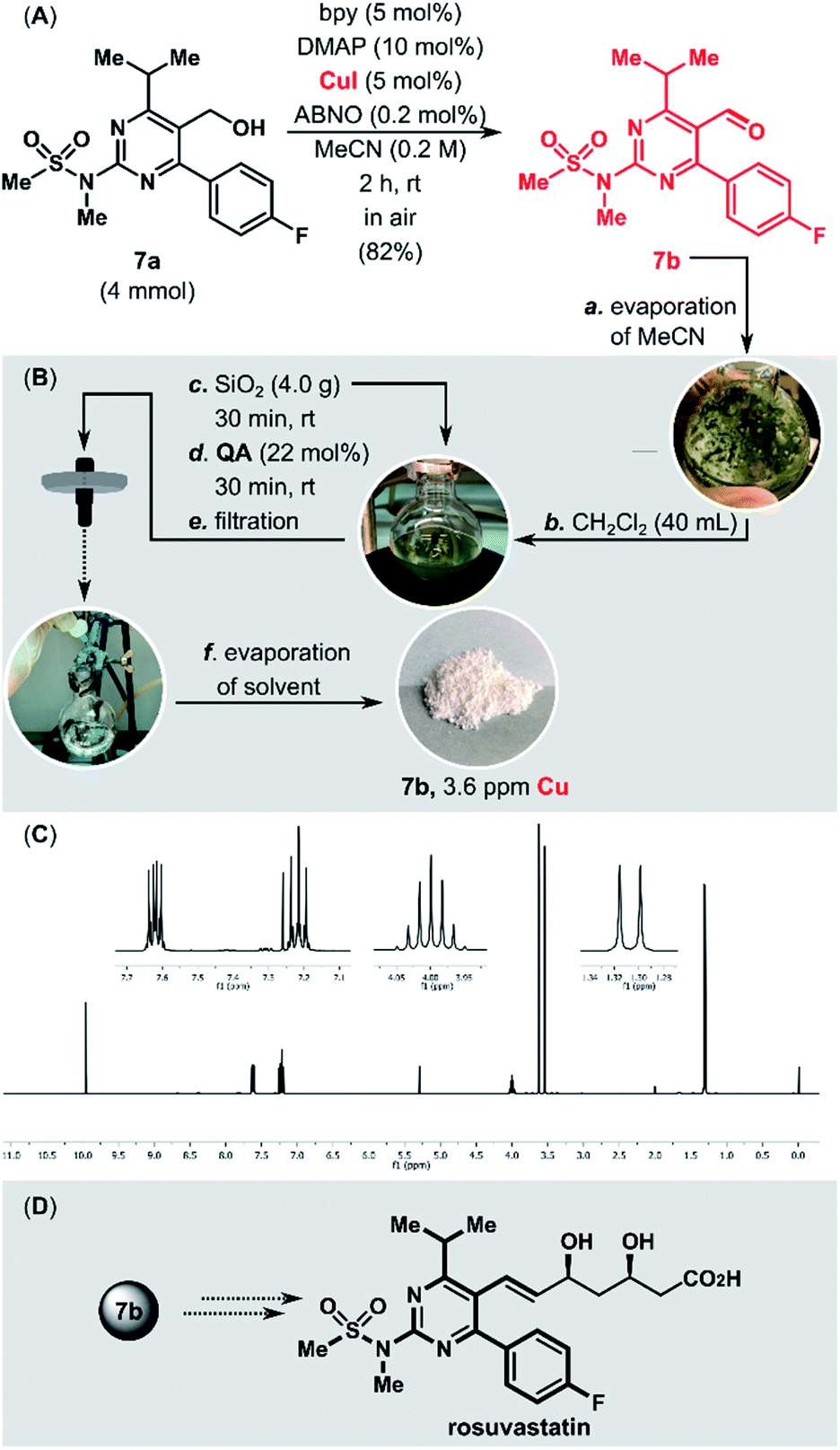 | ||
| Fig. 2 (A) Stahl oxidation of the rosuvastatin precursor (7a) on a gram scale. (B) Removal of Cu residues from 7b. (C) 1H NMR spectrum of 7b after purification. (D) Rosuvastatin structure. | ||
Reactivity of QA toward ATRP catalysts
The promising results on the removal of copper residues after Stahl oxidation prompted us to utilize QA in ATRP. First, we examined QA reactivity towards benchmark ATRP CuII-catalysts. Nitrogen-based polydentate ligands commonly used in Cu-mediated ATRP have strong σ-donor properties,77 which ensure effective coordination of the ligand to copper in both oxidation states. However, for ATRP ligands, the stability constant βII (binding constant for CuII) is much higher than βI (binding constant for CuI), providing a thermodynamic driving force for the activation of C(sp3)–X bonds. βII values ranging from 1018 to 1027 M−1 and corresponding βI between 107 and 1013 M−1 have been reported for conventional ATRP catalysts in organic solvents.78 The first question we had to answer was whether QA could form a complex with CuII in the presence of strong σ-donor ligands.To test the performance of QA, its reaction with [CuII(TPMA)Br]+ (TPMA = tris(2-pyridylmethyl)amine) was performed in DMSO under ambient atmosphere (Fig. 3). The progress of the reaction was monitored by UV-vis spectroscopy. After the addition of QA (4 equiv.) to [CuII(TPMA)Br]+, no ligand exchange occurred in the coordination sphere of copper (Fig. 3B). For Me6TREN (Me6TREN = tris[2-(dimethylamino)ethyl]amine) and PMDETA (PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) ligand, the results were similar (ESI Fig. S2 and S3†). However, the addition of ascorbic acid (AA) in excess (10 equiv.) resulted in the immediate replacement of TPMA with QA. The absorption band of [CuII(TPMA)Br]+ immediately disappeared in the UV-vis spectrum (Fig. 3C). Conversely, the addition of AA alone did not cause any changes in the spectrum (ESI Fig. S4B†).
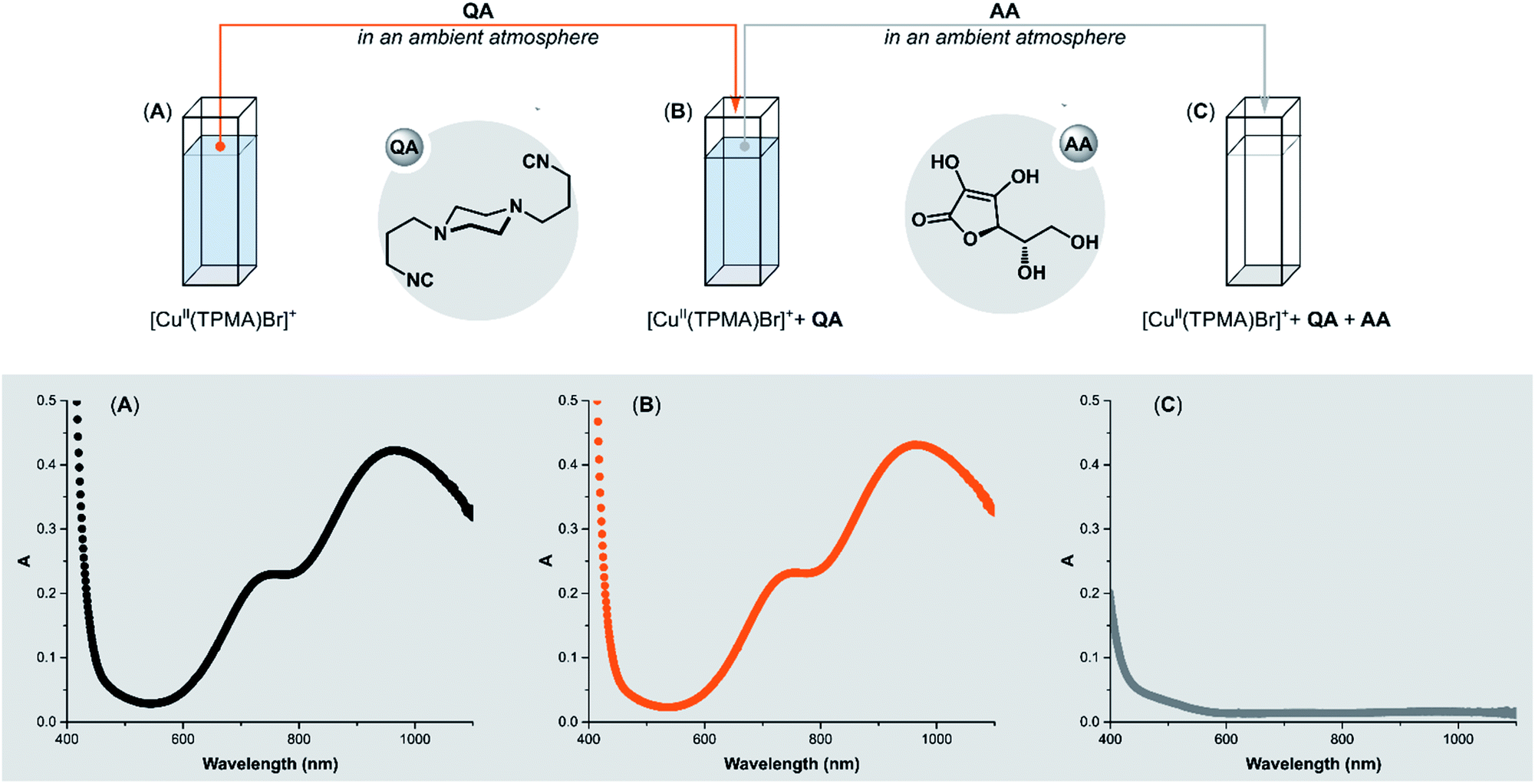 | ||
| Fig. 3 QA reactivity towards the [CuII(TPMA)Br]+. (A) UV-vis spectra of [CuIIBr2] = 2.5 mM; [TPMA] = 2.5 mM in DMSO at 22 °C in air. (B) UV-vis spectra of copper complex after addition of QA (4 equiv.). (C) UV-vis spectra of copper complex after addition of QA (4 equiv.) and AA (10 equiv.). | ||
Ascorbic acid reduces CuII to CuI, which results in a swift reaction between the [CuI(TPMA)]+ complex and the isocyanide QA, even though atmospheric oxygen is present in the system (Fig. 4C). In the absence of QA, the atmospheric oxygen immediately oxidizes [CuI(TPMA)]+. Therefore, the UV-vis spectrum does not show any changes after the addition of AA (ESI Fig. S4B†). Due to their strong σ-donor properties and their weak π-acidity, isocyanides preferentially bind to transition metals in a low oxidation state.25 As expected, the compound QA has very high affinity to CuI/L and does not react with CuII/L with electron-rich ligands that strongly bind to CuII.
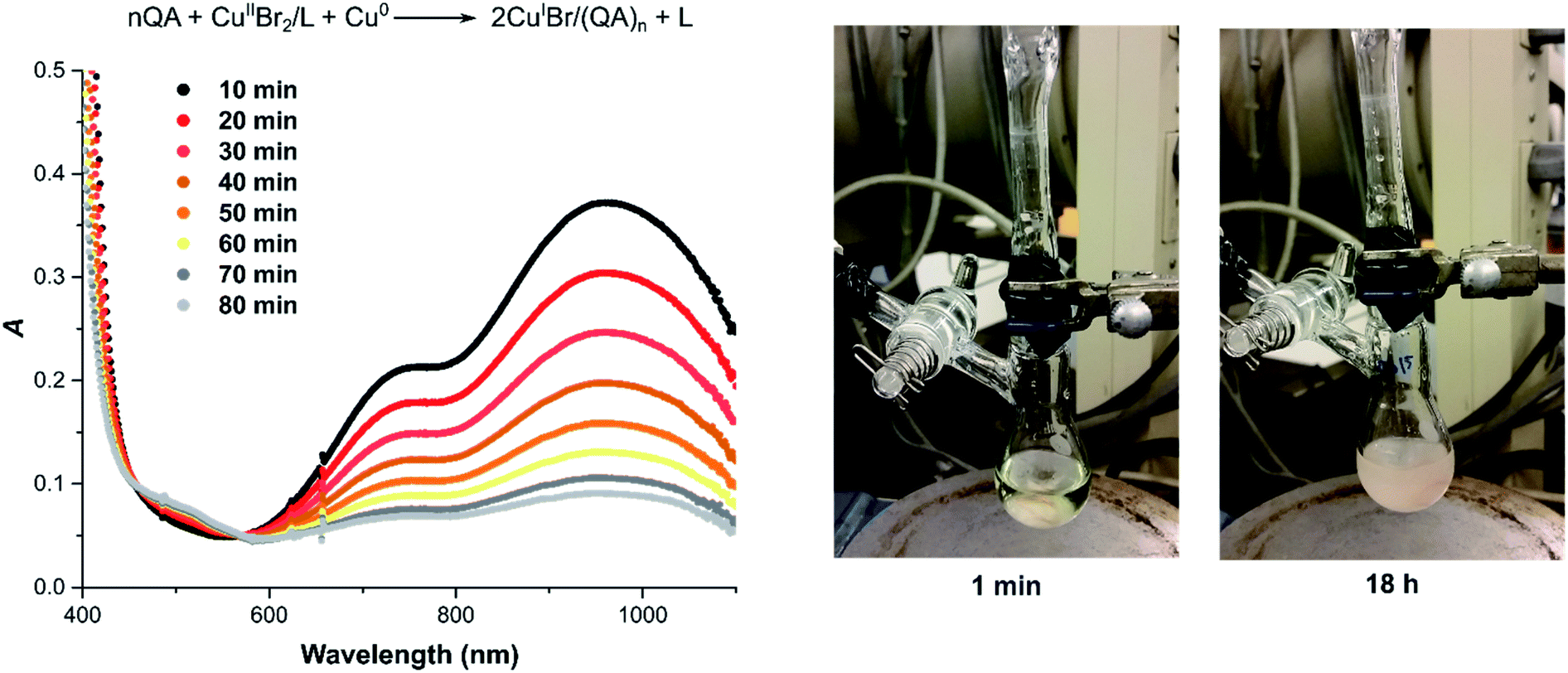 | ||
| Fig. 4 Comproportionation of [CuIIBr2] = 2.5 mM with [TPMA] = 5.25 mM in the presence of [QA] = 10.0 mM and 4 cm Cu(0) wire (d = 1 mm, S = 1.27 cm2); in DMSO at 22 °C. | ||
Similar conclusions can be drawn from the experiment where the complex [CuII(TPMA)Br]+ was reduced by comproportionation with Cu(0) in the presence of excess QA under anaerobic conditions in DMSO (Fig. 4). In the presence of QA, a white solid precipitated as the copper comproportionation proceeded. After 18 hours, the reaction mixture was exposed to air and filtered through a syringe filter. The UV-vis spectrum showed the complete disappearance of the CuII/L complex (ESI Fig. S5A†). The same result was recorded after 19 hours of air exposure, indicating that the CuI bound to QA ligand was not re-oxidized to CuII (ESI Fig. S5B†). In the control experiment in which no QA was added, [CuI(TPMA)]+ was oxidized (ESI Fig. S6†). These experiments have shown that QA can quantitatively bind to the CuI generated by the action of a reducing agent, even in the presence of strong σ-donor ligands, to form air-stable, polar copper(I) complexes with QA.
These results were confirmed by electrochemical analysis. Cyclic voltammetry (CV) is commonly used to investigate the reactivity of ATRP catalysts,79 which typically exhibit a quasi-reversible voltammetric behavior (e.g., Fig. 5, black line). Therefore CV was used to analyze the effect of QA on [CuII(TPMA)Br]+ in DMSO. When a relatively small amount of QA was present (<0.5 equiv.), a new cathodic peak appeared, shifted at more positive potentials compared to the cathodic peak of [CuII(TPMA)Br]+. While the latter gradually decreased in intensity, a more pronounced decrease was observed in the intensity of the anodic peak, corresponding to the re-oxidation of [CuI(TPMA)Br] to [CuII(TPMA)Br]+. A new broad oxidation peak appeared at more positive potentials. When the concentration of QA was increased, only the cathodic peak at more positive potentials was detected. Its intensity slightly decreased when the amount of QA was increased. No oxidation peak could be recorded when ≥1 equiv. of QA was present. The described behavior is consistent with the occurrence of a fast and irreversible chemical reaction following the electron transfer process,80 which results in the shift of the voltammetric signal to more positive potential values. In our experiment, QA reacted with the CuI species that was generated in the vicinity of the electrode during the potential scan. This reaction rendered the reduction of [CuII(TPMA)Br]+ to [CuI(TPMA)Br] more favorable, thus shifting its potential to more positive values. Moreover, QA formed a stable species with the copper(I) complex (at least in the timescale of CV), and therefore the cathodic peak corresponding to the re-oxidation of [CuI(TPMA)Br] decreased in intensity and then disappeared for relatively high amounts of QA. The fact that no other peaks ware detected in the explored potential range supports the formation of a non-electroactive complex between Cu(I) and QA (i.e. that cannot be oxidized), which is therefore unable to catalyze Stahl oxidation or ATRP reactions.
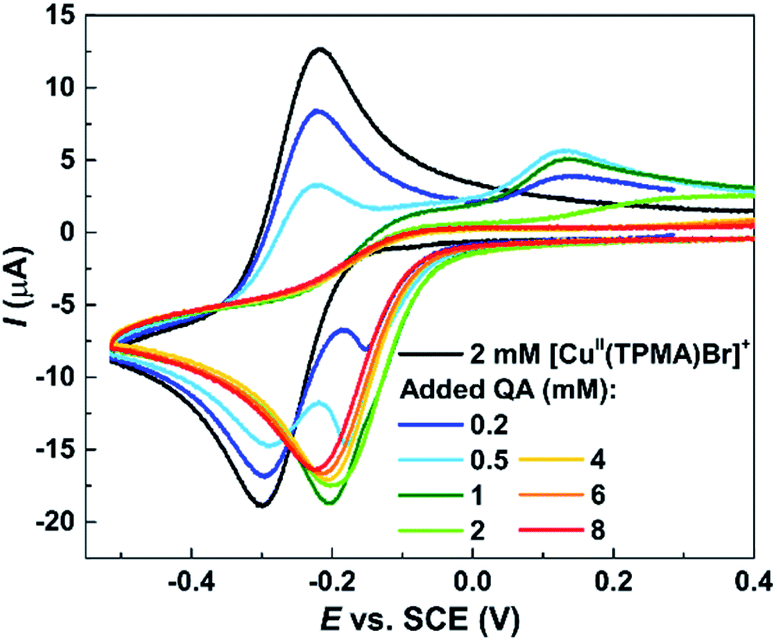 | ||
| Fig. 5 Cyclic voltammetry of 2 mM [CuII(TPMA)Br]+ in DMSO + 0.1 M Et4NBF4, in the absence and in the presence of QA. Scan rate = 0.2 V s−1, T = 25 °C. | ||
The CV experiment supported the ability of QA to strongly bind to CuI species. However, one can expect a similar voltammetric response both when QA binds to CuI/L and when it displaces the ligand and binds to “naked” CuI. One possibility to discriminate between these two scenarios is to look at a more positive potential range in a similar CV experiment. When the amount of QA increased, several anodic signals appeared or increased in intensity at E > 0.8 V vs. SCE, which could be ascribed to the oxidation of free Br− anions and TPMA (ESI Fig. S7†). This suggested that TPMA was at least partially displaced by QA.
A rotating disk electrode (RDE) was used to directly observe the effect of QA on [CuI(TPMA)]+ in DMSO. Chronoamperometry under hydrodynamic conditions was used to monitor the concentration of [CuI(TPMA)]+ by applying a potential of 0.29 V vs. SCE (Fig. 6). Initially, only [CuI(TPMA)]+ was present in the solution and a slowly decreasing limiting current was recorded due to the slow disproportionation of the complex. When 4 equiv. of QA were added, the current immediately dropped to zero, indicating that no [CuI(TPMA)]+ remained in the solution. If only a stoichiometric amount of QA was added, the current did not drop to zero but to a small value (ESI Fig. S8A†),81 indicating that an excess of QA was needed to effectively bind to all CuI species. Nevertheless, the current drop was instantaneous, both for [CuI(TPMA)]+ and for a CuI complex with 3 orders of magnitude higher ATRP activity (ESI Fig. S8B†), suggesting that QA can be used for the removal of Cu even when ATRP is performed with highly active catalysts.
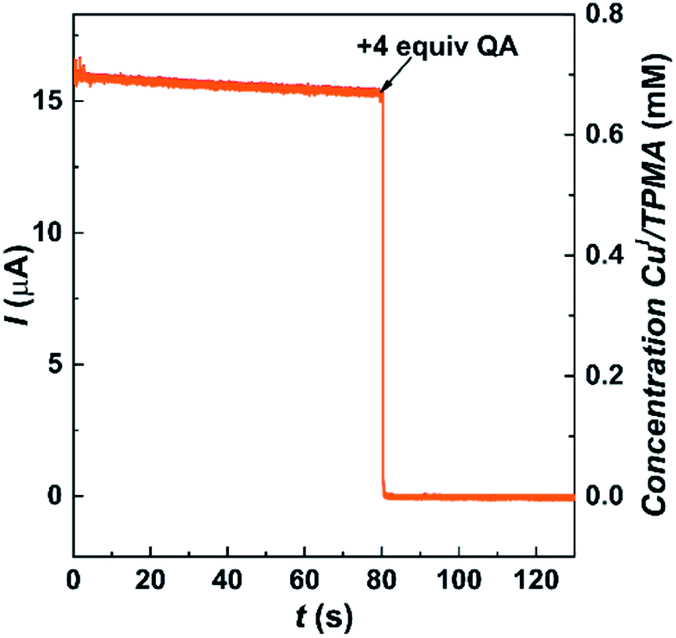 | ||
| Fig. 6 Decay of limiting current vs. time for the reaction between 0.7 mM [CuI(TPMA)]+ and 2.8 mM QA in DMSO + 0.1 M Et4NBF4, recorded at Eapp = 0.29 V vs. SCE on a RDE, l = 4000 rpm, T = 25 °C. | ||
Quenching of ATRP by QA
On a lab scale, typical ATRP quenching involves opening the post-reaction mixture to air, which provides oxidative conditions to form the deactivator CuII species. However, the amount of oxygen needed depends on the reaction volume, and thus, it cannot be scaled to industrial settings. Moreover, exposing a reaction mixture containing catalyst residues to oxidative conditions may cause undesired side products to form.82,83 Therefore, anaerobic methods of ATRP quenching are strongly desirable.The performance of QA as a quenching agent was evaluated in SARA ATRP. Methyl 2-bromopropionate (MBP) was used as an initiator with CuBr2/TPMA/Cu(0) as the catalytic system. The reaction was carried out in DMSO with [MA]/[MBP]/[CuIIBr2]/[TPMA]/[QA] molar ratios of 200/1/0.02/0.12/0.08 in the presence of copper wire, under anaerobic conditions (Fig. 7). After 90 min at room temperature, the conversion of methyl acrylate (MA) measured by 1H NMR was 0%, while in the corresponding polymerization without the addition of QA it was 81% (Fig. 7). Furthermore, the addition of QA to the reaction mixture after 40 min of polymerization resulted in an immediate quenching of ATRP (Fig. 7).
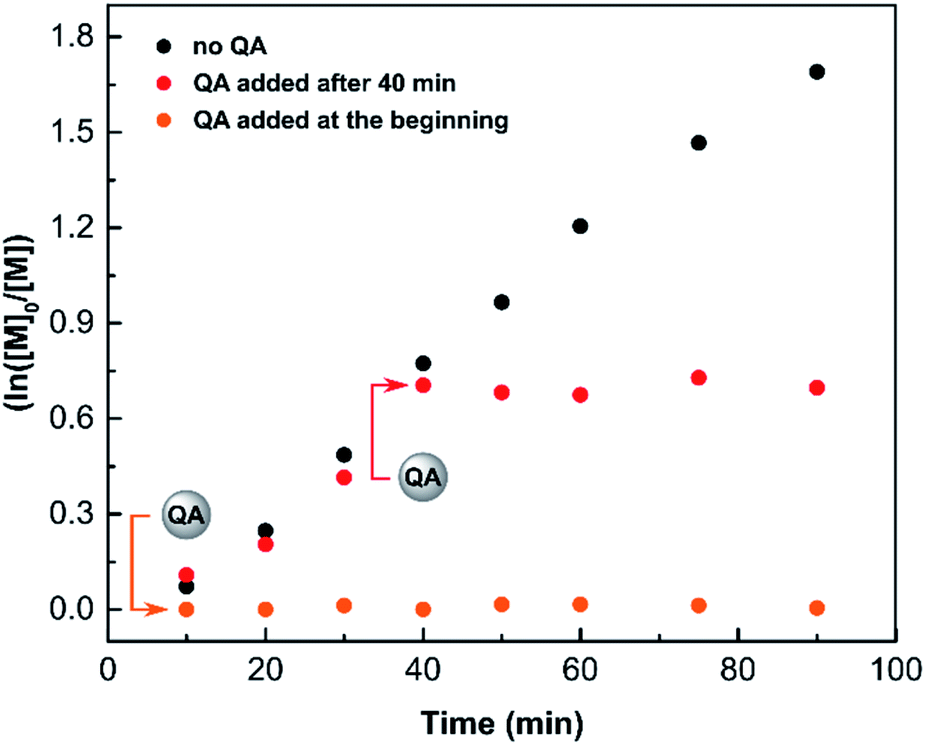 | ||
| Fig. 7 Quenching of SARA ATRP by QA, reaction conditions: [MA]/[MBP]/[CuIIBr2]/[TPMA]/[QA] = 200/1/0.02/0.12/0.08, Cu0 wire (l = 4 cm, S = 1.27 cm2); in DMSO ([MA] = 7.4 M) at 22 °C, under anaerobic conditions. | ||
Temporal control was successfully demonstrated in ARGET ATRP of MA using QA. Ethyl α-bromoisobutyrate (EBiB) was used as an initiator with CuBr2/Me6TREN/AA as the catalytic system. The polymerization was performed in DMSO at 22 °C with [MA]/[EBiB]/[CuIIBr2]/[Me6TREN]/[AA] molar ratios of 200/1/0.05/0.15/0.25 (Fig. 8A). After 10 min, the conversion of MA was 48%. Size exclusion chromatography (SEC) analysis showed that the polymer had a low dispersity (Đ = 1.10), and a molecular weight (MW) matching theoretical value (Mn,th = 8300, Mn = 9100) (Fig. 8B). The addition of QA (2 equiv. with respect to CuIIBr2) immediately stopped the ATRP. No increase in monomer conversion was observed during the 20 min period after adding QA. Then the polymerization was switched on by adding a second batch of [CuIIBr2]/[Me6TREN] (with a molar ratio of 0.05/0.05 respect to [EBiB]). After a 20 min period, the monomer conversion reached 68%, confirming the retention of chain-end functionality in the presence of QA. SEC traces showed a clear shift toward higher MWs (Mn = 12000, Đ = 1.10) after restarting the ATRP, verifying a well-controlled polymerization (Fig. 8B).
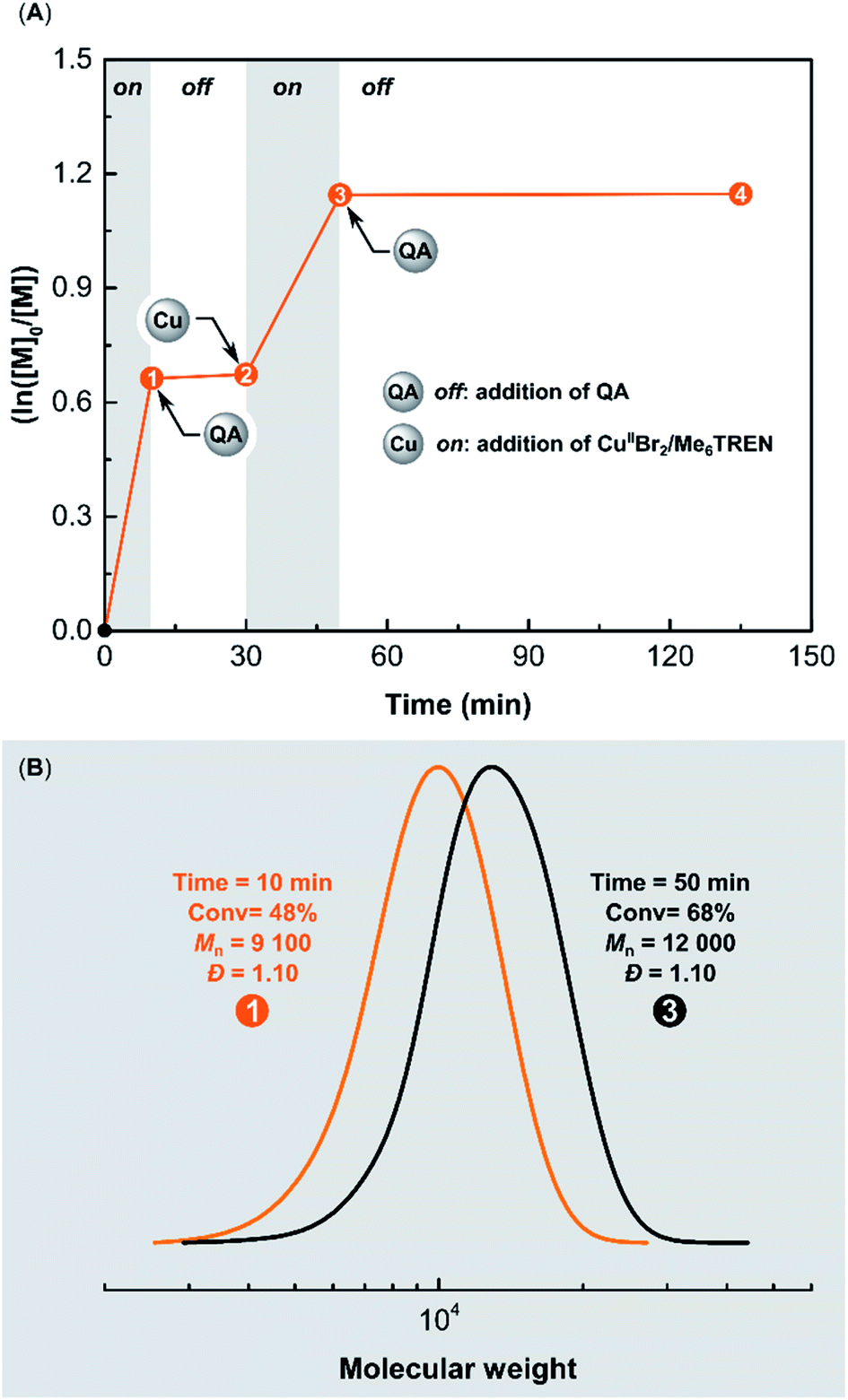 | ||
| Fig. 8 (A) Kinetics of temporal control in ARGET ATRP using QA, reaction conditions: [MA]/[EBiB]/[CuIIBr2]/[Me6TREN]/[AA] = 200/1/0.05/0.15/0.25 in DMSO ([MA] = 7.4 M) at 22 °C, in the presence of limited amounts of air (without degassing of DMSO and MA). (B) SEC traces after addition of QA (2 equiv. with respect to CuIIBr2) and [CuIIBr2]/[Me6TREN] (with a molar ratio of 0.05/0.05 respect to EBiB). | ||
Removal of copper residues after ATRP
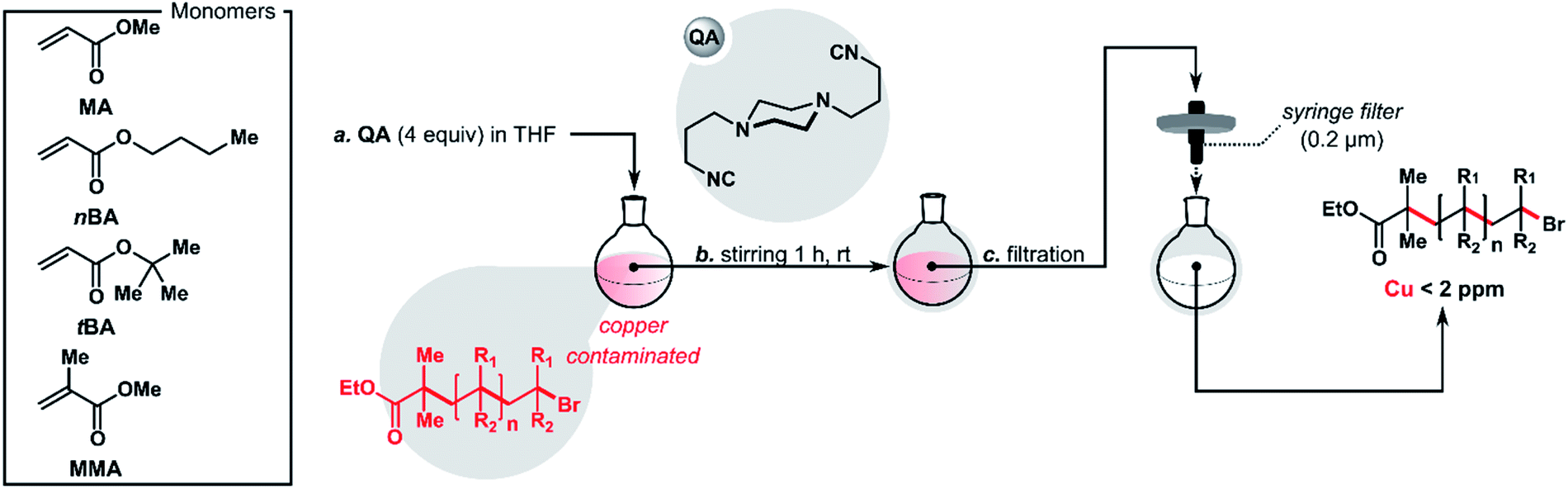
The purification of the polymers consisted of the addition of a solution of QA (4 equiv.) in THF once the reaction was complete, followed by filtration using a syringe filter (0.2 μm). QA proved to be highly effective at removing metal residues after ATRP, usually reducing the copper content to a level below 2 ppm (Table 2). In control experiments without QA, metal contamination remained above 10 ppm. In extreme cases, it was over 50 times higher, up to 525 ppm (Table 2, entry 3). Additionally, the new purification protocol proved to be compatible with various monomers and ATRP techniques, including ARGET, SARA, and pATRP. The use of QA did not affect the molecular weight or dispersity of the purified polymers.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Monomer | ATRPa | Time (h) | α (%) | M n,th | M n,GPC | Đ | QA (equiv.) | Cuf (ppm) |
| a Reaction conditions: [monomer]/[EBiB]/[CuIIBr2]/[Me6TREN] = 200/1/0.05/0.25 in MeCN, ([MA] = 7.4 M, [nBA] = 4.7 M, [tBA] = 4.6 M, [MMA] = 6.2 M) at 22 °C. b Monomer conversion was determined by using 1H NMR spectroscopy. c Theoretical molecular weight was calculated using the equation Mn,th = [M] × MWM × α + MWEBiB, where [M], MWM, α, and MWEBiB correspond to initial monomer concentration, molar mass of monomer, conversion, and molar mass of EBiB initiator, respectively. d Molecular weight and dispersity (Đ) were determined by GPC analysis (THF as eluent) calibrated to poly(methyl methacrylate) standards. e Equivalents of QA with respect to CuIIBr2. f Copper content was determined by ICP-MS. g ARGET ATRP: ascorbic acid (5 equiv. with respect to CuIIBr2), under anaerobic conditions. h SARA ATRP: 4 cm Cu(0) wire (d = 1 mm, S = 1.27 cm2), under anaerobic conditions. i pATRP: UV irradiation (λ = 365 nm), under anaerobic conditions. j pATRP: UV irradiation (λ = 365 nm), in the presence of limited amounts of air (without degassing of MeCN and monomer). | |||||||||
| 1g | MA | ARGET | 3 | 68 | 11900 |
12400 |
1.05 | — | 26.7 |
| 2g | MA | ARGET | 3 | 68 | 11900 |
13800 |
1.03 | 4 | 1.38 |
| 3h | MA | SARA | 3 | 90 | 15700 |
15400 |
1.05 | — | 525 |
| 4h | MA | SARA | 3 | 85 | 14800 |
14400 |
1.07 | 4 | 0.97 |
| 5i | MA | pATRP | 3 | 80 | 14000 |
13100 |
1.05 | — | 51.6 |
| 6i | MA | pATRP | 3 | 79 | 13800 |
13600 |
1.04 | 4 | 0.91 |
| 7j | MA | pATRP | 5 | 77 | 13500 |
15400 |
1.08 | 4 | 0.80 |
| 8g | nBA | ARGET | 24 | 86 | 22200 |
21600 |
1.05 | 4 | 2.20 |
| 9j | nBA | pATRP | 5 | 65 | 16900 |
14200 |
1.04 | 4 | 1.41 |
| 10j | tBA | pATRP | 5 | 81 | 21000 |
18500 |
1.18 | 4 | 0.71 |
| 11g | MMA | ARGET | 10 | 90 | 18100 |
17100 |
1.24 | 4 | 1.07 |
Preventing post-ATRP glaser coupling using QA
Azide–alkyne Huisgen cycloaddition (CuAAC) in combination with ATRP allows the synthesis of strictly defined, tailored polymers with various architectures,84–88 especially macrocyclic polymers that are difficult to prepare by other methods.89 This synthetic route involves the preparation of a linear precursor with a terminal alkyne group by ATRP, followed by the nucleophilic displacement of the bromide chain end with azide and finally CuAAC-mediated macrocyclization reaction at high dilution. However, during these transformations, side reactions can occur that compromise the fidelity of the chain end, such as (i) radical termination at high monomer conversions and (ii) Glaser coupling of the alkyne moiety under oxidative conditions.90 Both side reactions jeopardize the subsequent CuAAC-mediated macrocyclization.While the conventional radical termination can be minimized by decreasing the rate of polymerization and by terminating the reaction at lower monomer conversion, suppression of the Glaser coupling remains a challenge.82,91,92 One of the methods of preventing the Glaser coupling is the protection of the alkyne group with a trimethylsilyl or triisopropylsilyl group.93 However, this approach requires an additional protection/deprotection sequence, which has a low atom and time economy. In addition, the deprotection step requires the use of reagents that can cause the degradation of the synthesized polymer.
Recently, in an inspiring work, Koberstein and co-workers showed two methods for preventing post-ATRP alkyne–alkyne coupling.82 First, Glaser coupling can be completely prevented by cooling the post-reaction mixture below −28 °C before exposing it to air and then maintaining a low temperature during the polymer workup. Second, the coupling is suppressed by the use of an excess of reducing agents, such as tin(II) 2-ethylhexanoate or (+)-sodium L-ascorbate. Moreover, it was found that high-denticity ligands can reduce Glaser coupling rates. However, the proposed approaches are difficult to scale up because of the necessity of using aluminum oxide during the purification process. For this reason, the development of economical and simple methods of preventing oxidative alkyne–alkyne coupling is desirable.
The linear polystyrene (α-alkyne,ω-Br-PS) was prepared using standard ATRP. 3-Butynyl 2-bromoisobutyrate (PgBiB) was used as initiator with CuBr/dNbpy (dNbpy = 4,4′-dinonyl-2,2′-bipyridine) as the catalyst system. The reaction was carried out in bulk at 110 °C with [styrene]/[PgBiB]/[CuIBr]/[dNbpy] molar ratios of 230/1/1/2. After 1.5 h, two samples of 4.5 mL each were taken from the reaction mixture and purified using method A or B (Fig. 9A).
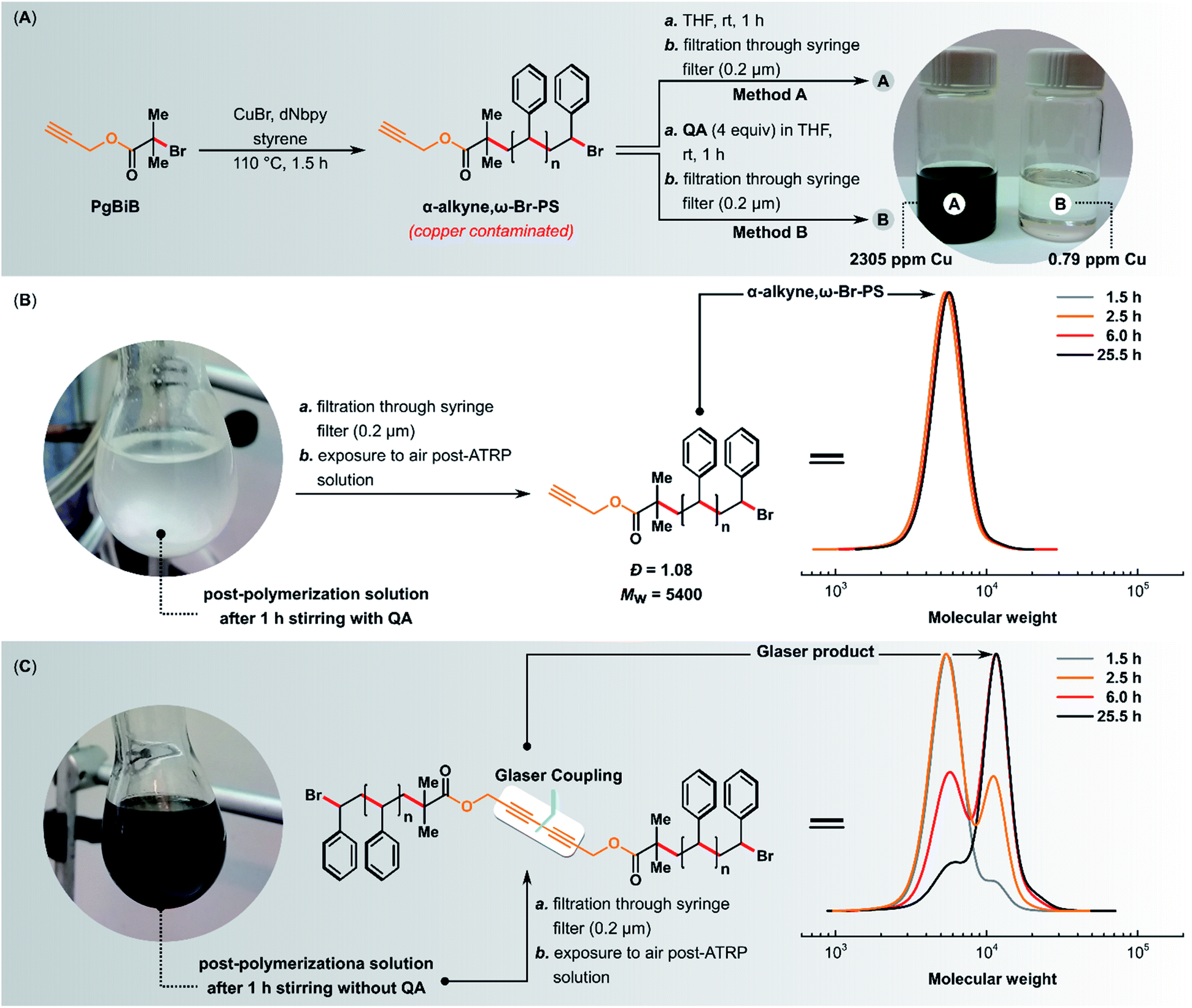 | ||
| Fig. 9 (A) Preventing post-ATRP Glaser coupling using QA. Normal ATRP of styrene, reaction conditions: [styrene]/[PgBiB]/[CuIBr]/[dNbpy] = 1/1/2/230 in bulk at 110 °C. (B) Exposure to air post-ATRP mixture after the addition of QA (4 equiv. with respect to CuIBr). (C) Exposure to air post-ATRP mixture without the addition of QA. | ||
The addition of QA (4 equiv. with respect to CuIBr) to the post-ATRP reaction mixture not only completely prevented the Glaser coupling (i.e., the average molecular weight of the polymer did not change over time when the mixture was exposed to air) but also caused the quantitative precipitation of the copper(I) complex with the QA ligand (Fig. 9B). The complex was easily removed by simple filtration, giving a colorless polymer with copper content below 1 ppm and low Đ (1.08). In the control experiment in which QA was not added, the average molecular weight and Đ of the resulting polymer increased over time (Fig. 9C), and the obtained polymer had an intense color and a very high copper content of 2305 ppm.
Cytotoxicity study of QA
Finally, we investigated the cytotoxicity of QA and its acidic hydrolysis product HQA (N,N′-(piperazine-1,4-diylbis(propane-3,1-diyl))diformamide). HQA was obtained by adding 4 equiv. of HCl to QA dissolved in water and stirring for 2 hours. The resulting solution was diluted in buffer to the desired concentration without isolating HQA (Fig. 10). To ensure that hydrolysis was quantitative, the same procedure was performed in D2O and monitored by 1H NMR spectroscopy. After 2 hours, no peaks from QA were visible in the spectrum.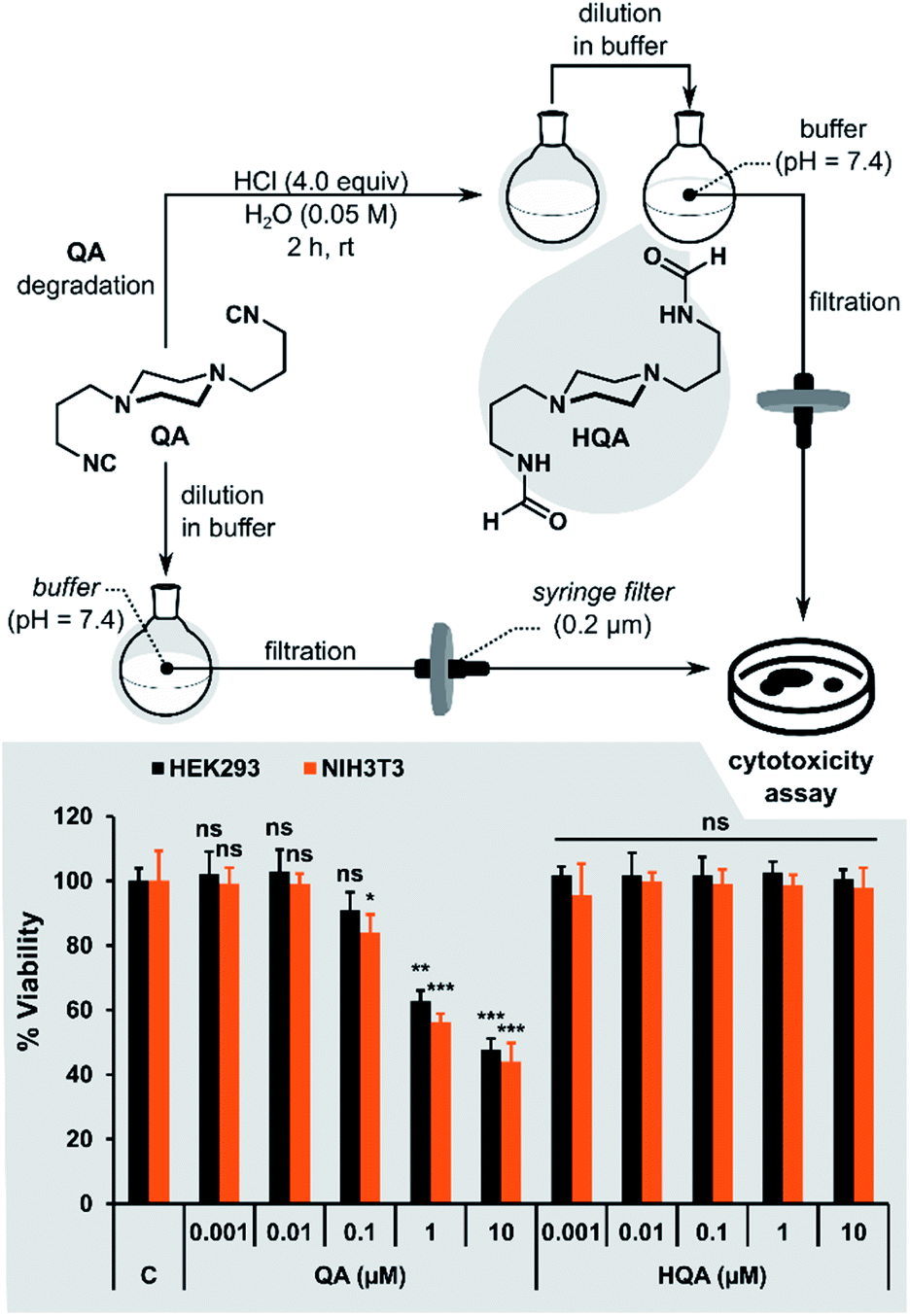 | ||
| Fig. 10 The cytotoxicity assay performed using HEK293 (black) and NIH3T3 cells (orange). Cells were treated with five concentrations of QA or HQA for 72 hours and compared to control group (no treatment). Bars indicate the mean percent viability ± SEM (n = 3), ns: no significant difference, *p < 0.5, **p < 0.01, ***p < 0.001 (vs. control group). | ||
The toxicity study was performed using two different cell lines: HEK293 (human embryonic kidney cells) and NIH3T3 (mouse fibroblast cells). QA and HQA were tested at five different concentrations (Fig. 10). The cells were tolerant to 0.01 μM of either of the compounds up to 72 hours. However, at concentrations above 0.01 μM, QA started exhibiting cytotoxicity in a time-dependent manner (ESI Fig. S9†) in both cell lines. The highest level of cytotoxicity of QA was observed at 72 hours, with approximately 50% of cells remaining viable after treatment with 10 μM QA compared to the control (no treatment) group. On the other hand, HQA was not cytotoxic even at 10 μM, suggesting that acidic hydrolysis of QA is an effective way to neutralize its toxicity.
Conclusions
In this study, we have shown that QA is a rapid quenching agent for Stahl oxidation and ATRP, as well as a highly efficient Cu scavenger. The use of a small amount of silica gel in combination with a small excess of QA reduced the Cu content in aldehydes below 5 ppm. This purification protocol was effective on a gram scale in the synthesis of the cardiovascular disease drug precursor (rosuvastatin), leading to a product containing only 3.6 ppm Cu.Spectroscopic and electrochemical studies provided insights into the reactivity of QA toward Cu complexes used as ATRP catalysts and showed that QA is highly reactive against CuI/L, even for ligands with very high copper binding constants (1013 M−1). At the same time, QA proved to be inert towards CuII/L complexes with strong σ-donor ligands. However, the use of a reducing agent allowed QA to bind copper quantitatively.
QA very efficiently removed Cu after ATRP without compromising the molecular weight and dispersity of the purified polymers. The addition of QA followed by simple filtration using a syringe filter proved to be highly effective in removing Cu contamination, affording polymers with Cu content below 2 ppm. This protocol was of great significance for normal ATRP, leading to polystyrene with less than 1 ppm Cu. Treatment with QA not only facilitated the removal of Cu, but also completely prevented the Glaser coupling under oxidative conditions.
Finally, we examined the cytotoxicity of QA. The isocyanide showed moderate cytotoxicity, with approximately 50% of cells remaining viable after 72 hours of treatment with 10 μM of the compound. However, it could be easily hydrolyzed by adding a small excess of HCl. The hydrolysis product showed no cytotoxicity even at relatively high concentrations.
In summary, we presented the applicability of QA in two fundamental reactions catalyzed by copper complexes. We expect this straightforward approach to find application in other significant transformations catalyzed by transition metal complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. S. wishes to thank the National Science Centre (“Etiuda” fellowship) and the Foundation for Polish Science (“Ventures” Program) for financial support. The project “Ventures/2013-11/8” is funded by the “Ventures” program of the Foundation for Polish Science, co-financed from the European Union Regional Development Fund. J. P., W. N., and K. G. are grateful to the Catalysis for the Twenty-First Century Chemical Industry Project carried out within the TEAM-TECH Programme of the Foundation for Polish Science co-financed by the European Union from the European Regional Development under the Operational Programme Smart Growth. The study was partially carried out at the Biological and Chemical Research Centre, University of Warsaw, established within a project co-financed by the European Union from the European Regional Development Fund under the Operational Programme “Innovative Economy”, 2007–2013. The support from the National Science Foundation (CHE 1707490) is also acknowledged. The authors thank Krzysztof Kosiński and Emilee Tkacik for fruitful discussions and proofreading.References
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- P. O'Brien, Angew. Chem., Int. Ed., 1999, 38, 326–329 CrossRef.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160–17161 CrossRef CAS PubMed.
- Ö. Usluer, M. Abbas, G. Wantz, L. Vignau, L. Hirsch, E. Grana, C. Brochon, E. Cloutet and G. Hadziioannou, ACS Macro Lett., 2014, 3, 1134–1138 CrossRef.
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552–3565 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS.
- C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS.
- E. J. Flahive, B. L. Ewanicki, N. W. Sach, S. A. O'Neill-Slawecki, N. S. Stankovic, S. Yu, S. M. Guinness and J. Dunn, Org. Process Res. Dev., 2008, 12, 637–645 CrossRef CAS.
- P. Wheeler, J. H. Phillips and R. L. Pederson, Org. Process Res. Dev., 2016, 20, 1182–1190 CrossRef CAS.
- G. Szczepaniak, K. Kosiński and K. Grela, Green Chem., 2014, 16, 4474–4492 RSC.
- G. C. Vougioukalakis, Chem.–Eur. J., 2012, 18, 8868–8880 CrossRef CAS PubMed.
- B. R. Galan, K. P. Kalbarczyk, S. Szczepankiewicz, J. B. Keister and S. T. Diver, Org. Lett., 2007, 9, 1203–1206 CrossRef CAS PubMed.
- G. Szczepaniak, K. Urbaniak, C. Wierzbicka, K. Kosiński, K. Skowerski and K. Grela, ChemSusChem, 2015, 8, 4139–4148 CrossRef CAS PubMed.
- G. Szczepaniak, A. Ruszczyńska, K. Kosiński, E. Bulska and K. Grela, Green Chem., 2018, 20, 1280–1289 RSC.
- G. Szczepaniak, W. Nogaś, J. Piątkowski, A. Ruszczyńska, E. Bulska and K. Grela, Org. Process Res. Dev., 2019, 23, 836–844 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC.
- A. J. Mancuso and D. Swern, Synthesis, 1981, 1981, 165–185 CrossRef.
- J. R. Parikh and W. v. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS.
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116 RSC.
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC.
- J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161–1166 CrossRef CAS PubMed.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- R. C. Walroth, K. C. Miles, J. T. Lukens, S. N. MacMillan, S. S. Stahl and K. M. Lancaster, J. Am. Chem. Soc., 2017, 139, 13507–13517 CrossRef CAS PubMed.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247–1251 CrossRef CAS.
- A. Ochen, R. Whitten, H. E. Aylott, K. Ruffell, G. D. Williams, F. Slater, A. Roberts, P. Evans, J. E. Steves and M. J. Sanganee, Organometallics, 2019, 38, 176–184 CrossRef CAS.
- B. L. Ryland, S. D. McCann, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 12166–12173 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS.
- T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, Science, 1996, 272, 866–868 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS.
- E. H. Discekici, A. Anastasaki, J. Read de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- G. Xie, M. R. Martinez, M. Olszewski, S. S. Sheiko and K. Matyjaszewski, Biomacromolecules, 2019, 20, 27–54 CrossRef CAS PubMed.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- N. Jasinski, A. Lauer, P. J. M. Stals, S. Behrens, S. Essig, A. Walther, A. S. Goldmann and C. Barner-Kowollik, ACS Macro Lett., 2015, 4, 298–301 CrossRef CAS.
- M. Ding, X. Jiang, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2015, 36, 1702–1721 CrossRef CAS PubMed.
- Y. Shen, H. Tang and S. Ding, Prog. Polym. Sci., 2004, 29, 1053–1078 CrossRef CAS.
- H.-C. Lee, M. Fantin, M. Antonietti, K. Matyjaszewski and B. V. K. J. Schmidt, Chem. Mater., 2017, 29, 9445–9455 CrossRef CAS.
- H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2016, 7, 7199–7203 RSC.
- G. Kickelbick, H. Paik and K. Matyjaszewski, Macromolecules, 1999, 32, 2941–2947 CrossRef CAS.
- Y. Wang, F. Lorandi, M. Fantin, P. Chmielarz, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2017, 50, 8417–8425 CrossRef CAS PubMed.
- B. Zhang, L. Yao, X. Liu, L. Zhang, Z. Cheng and X. Zhu, ACS Sustainable Chem. Eng., 2016, 4, 7066–7073 CrossRef CAS.
- M. E. Honigfort, W. J. Brittain, T. Bosanac and C. S. Wilcox, Macromolecules, 2002, 35, 4849–4851 CrossRef CAS.
- M. F. Ebbesen, D. Itskalov, M. Baier and L. Hartmann, ACS Macro Lett., 2017, 6, 399–403 CrossRef CAS.
- X. Su, P. G. Jessop and M. F. Cunningham, Macromolecules, 2018, 51, 8156–8164 CrossRef CAS.
- K. Matyjaszewski, T. Pintauer and S. Gaynor, Macromolecules, 2000, 33, 1476–1478 CrossRef CAS.
- A. J. D. Magenau, N. Bortolamei, E. Frick, S. Park, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 4346–4353 CrossRef CAS.
- F. Canturk, B. Karagoz and N. Bicak, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3536–3542 CrossRef CAS.
- J. M. Blacquiere, T. Jurca, J. Weiss and D. E. Fogg, Adv. Synth. Catal., 2008, 350, 2849–2855 CrossRef CAS.
- J. E. Steves and S. S. Stahl, J. Org. Chem., 2015, 80, 11184–11188 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- J. E. Steves, Y. Preger, J. R. Martinelli, C. J. Welch, T. W. Root, J. M. Hawkins and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1548–1553 CrossRef CAS.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- N. Bortolamei, A. A. Isse, V. B. Di Marco, A. Gennaro and K. Matyjaszewski, Macromolecules, 2010, 43, 9257–9267 CrossRef CAS.
- A. A. Isse, F. Lorandi and A. Gennaro, Curr. Opin. Electrochem., 2019, 15, 50–57 CrossRef CAS.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M'Halla and J. M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1980, 113, 19–40 CrossRef CAS.
- K. Schröder, R. T. Mathers, J. Buback, D. Konkolewicz, A. J. D. Magenau and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1037–1040 CrossRef.
- P. Leophairatana, S. Samanta, C. C. De Silva and J. T. Koberstein, J. Am. Chem. Soc., 2017, 139, 3756–3766 CrossRef CAS PubMed.
- W. P. Gallagher and A. Vo, Org. Process Res. Dev., 2015, 19, 1369–1373 CrossRef CAS.
- S. Zhang, T. Vi, K. Luo and J. T. Koberstein, Macromolecules, 2016, 49, 5461–5474 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- J. A. Johnson, D. R. Lewis, D. D. Díaz, M. G. Finn, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 6564–6565 CrossRef CAS PubMed.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS PubMed.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS PubMed.
- P. Leophairatana, C. C. De Silva and J. T. Koberstein, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 75–84 CrossRef CAS.
- C. J. Duxbury, D. Cummins and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3795–3802 CrossRef CAS.
- W. K. Storms-Miller and C. Pugh, Macromolecules, 2015, 48, 3803–3810 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00623h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |