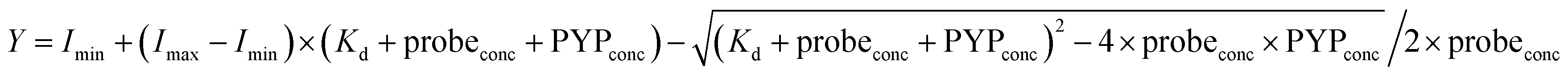DOI:
10.1039/D0SC00499E
(Edge Article)
Chem. Sci., 2020,
11, 3694-3701
Rapid no-wash labeling of PYP-tag proteins with reactive fluorogenic ligands affords stable fluorescent protein conjugates for long-term cell imaging studies†
Received
28th January 2020
, Accepted 7th March 2020
First published on 9th March 2020
Abstract
Covalent labeling systems that employ protein-tags or chemical probes to convert proteins into fluorescent conjugates are powerful tools for carrying out real time imaging and pulse-chase tracking studies that enable the spatiotemporal role of proteins in complex biological systems to be investigated. In this study, we have covalently modified a specific nucleophilic cysteine residue of the PYP-tag protein with weakly fluorescent α,β-unsaturated ketone (conjugate addition) and α-halomethyl ketone (SN2 reaction) acceptors to afford highly fluorescent PYP-tag-dimethylaminocoumarin (DMAC) conjugates, whose ligands are covalently bound to the PYP-protein through stable thioether linkers. A chloromethylketone derived DMAC-CMK reagent was found to afford the best kinetic and stability profile for labeling the PYP-tag in cellular systems, with in vitro studies demonstrating that PYP-DMAC-CMK conjugates exhibit excellent photostability and cellular stability profiles which enables them to be used for long-term protein imaging studies in cellular systems. The potential of using this no wash fluorescent labeling PYP-tag-DMAC system to visualise dividing cells undergoing mitosis and for imaging a PYP-tag fused telomere binding protein bound to chromatin in cell nuclei has been demonstrated.
Introduction
The visualization of the cellular localization and dynamics of an individual protein is important not only to elucidate its function, but also to gain insight into its spatiotemporal role in biological processes.1–5 Covalent labeling of a protein with a protein-tag that is capable of binding to a fluorescent chemical probe has been shown to be a very powerful tool to enable intracellular protein visualisation.6–8 The stable nature of these covalent constructs is particularly beneficial if pulse-chase analysis or protein trafficking studies are required to investigate cellular processes over time.9,10 One labeling approach is to employ a tagged fusion protein that can bind to a weakly fluorescent probe to afford a highly fluorescent conjugate.11–19 This requires that the probe is photostable, exhibits fast labeling kinetics, demonstrates a good specificity profile for the target protein and produces a stable fluorescent protein–probe conjugate. Notable examples of this type of approach include Halo-tag,12,13 SNAP/CLIP-tag,12,13 TMP-tag,14 or dC10α-tag,15 all of which have been used to create fusion proteins containing reactive groups that react selectively with weakly fluorescent probes to afford highly fluorescent tagged proteins. These ‘no-wash’ labeling systems are convenient, because they employ probes that only fluoresce strongly when bound to their target protein, thus avoiding the need to wash out excess probe before carrying out cell visualisation studies.
A non-enzymatic photoactive yellow protein (PYP) tag has previously been used as a small protein (14 kDa) no-wash tag to produce fluorescently labeled fusion proteins with favourable imaging properties,18–22 For example, the FAST-tag is a PYP mutant that binds to hydroxybenzylidene rhodanine derivatives through a series of non-covalent binding interactions.18 Alternatively, fluorogenic probes for covalent labeling of the PYP-tag have been developed, with a thioester bond used to link the acid group of a 7-dimethylaminocoumarin (DMAC) ligand to a Cys69 residue in the binding pocket of PYP.20,21 However, the thioester bonds of these DMAC probes were found to be labile to nucleophilic cleavage by endogenous thiols [e.g. glutathione (GSH)], which resulted in the cleavage of the DMAC ligand and a corresponding decrease in protein fluorescence over time.23 Attempts to address this problem using PYP-tag mutants that could bind DMAC ligands more effectively were only partially successful,23 resulting in a gradual decrease in fluorescence in living cells over time (ca. 10% per h). In order to address these instability issues, we now report a new PYP-tag-ligand system that employs a reactive DMAC α-chloromethyl ketone based ligand that can react with the Cys-69 residue of the PYP-tag to form a stable thioether bond that is resistant to cleavage in cellular environments. The increased stability of this new fluorescent PYP-tag-DMAC conjugate system enables its use for long-term cellular imaging studies, with its potential for real time monitoring of dynamic cellular processes demonstrated through visualisation of dividing cells undergoing mitosis and imaging of a PYP-tag fused telomere binding protein bound to chromatin in cell nuclei.
Results and discussion
Design of new DMAC ketone ligands for PYP-tag labeling
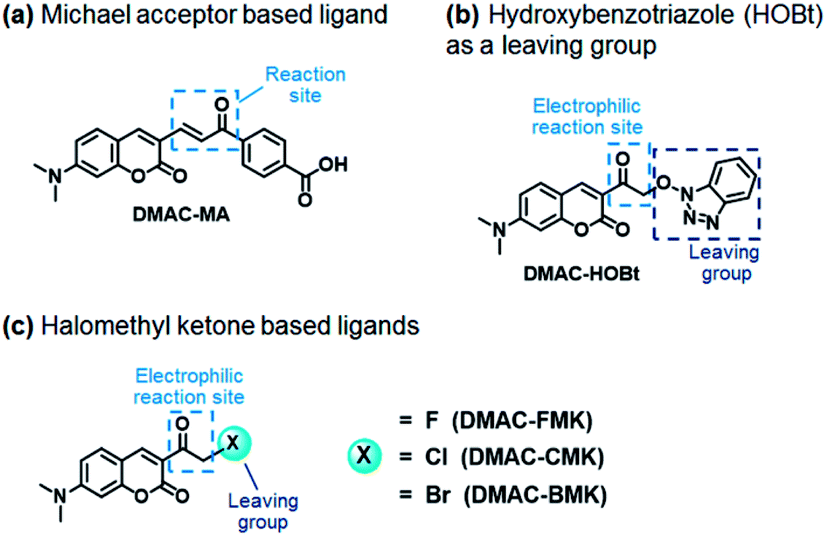
Our first attempt to design a PYP-tag-ligand with improved cellular stability was based on functionalising the PYP-tag with a DMAC ligand (DMAC-MA) that contained an α,β-unsaturated ketone group (Fig. 1a, see the ESI for synthesis†). It was anticipated that the nucleophilic thiol group of the Cys69 residue of PYP would undergo irreversible 1,4-conjugate addition to DMAC-MA to afford a stable conjugate through the formation of a stable thioether bond. A second approach was also envisaged based on the use of DMAC ketone ligands containing reactive α-halo leaving groups that could undergo nucleophilic SN2 displacement by the same Cys69 residue of the PYP-tag (Fig. 1b and c, see the ESI for syntheses of ligands†). DMAC-HOBt was designed to contain a hydroxybenzotriazole (HOBt) residue, whose use as a leaving group for protein labeling studies was unprecedented (Fig. 1b), whilst DMAC-FMK, DMAC-CMK and DMAC-BMK contained more conventional fluoride, chloride, and bromide leaving groups, respectively (Fig. 1c). These types of polarity sensitive DMAC based ligands are only weakly fluorescent in polar aqueous solution; however it was anticipated that their introduction into the hydrophobic binding pocket of the PYP-tag would result in a strong enhancement in their fluorescence intensity. This increase in fluorescence would not only enable the rate of covalent attachment of the DMAC ligands to the PYP-tag to be easily monitored but would also enable cell labeling studies to be carried out in vitro, with no need to wash out excess DMAC ligand. Overall, the use of these new DMAC ketone labeling agents would result in PYP-tag conjugates whose DMAC-ligands were attached to the PYP-tag through stable thioether bonds, which we anticipated would allow PYP-tag conjugate derived fusion proteins to be used for long-term cell imaging studies.
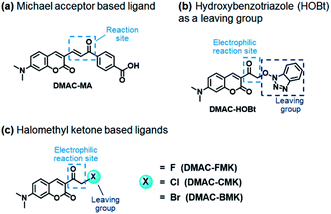 |
| | Fig. 1 Electrophilic DMAC derivatives react selectively with the nucleophilic thiol of Cys69 of the PYP-tag to afford PYP-tag-DMAC conjugates whose DMAC ligands are attached to the protein through stable thioether bonds. (a) DMAC-MA contains a reactive α,β-unsaturated ketone group that can act as a Michael acceptor for sulfur nucleophiles; (b) ketone ligand DMAC-HOBt contains a reactive α-HOBt leaving group that can react with sulfur nucleophiles; (c) ketone ligands DMAK-FMK, DMAK-CMK and DMAK-BMK contain reactive α-halomethyl ketone groups that can react with sulfur nucleophiles at their α-positions. | |
Synthesis and analysis of fluorescent PYP-tag-DMAC conjugates
Protein labeling reactions were carried out by incubating the PYP-tag with DMAC-MA in HEPES buffer (pH 7.4) at 37 °C for 1 h and the resulting products were analysed using SDS-PAGE (see the ESI for experimental details†). We were initially disappointed to find that SDS-PAGE analysis did not reveal the presence of a fluorescence band expected for a labeled PYP-tag (Fig. S1†). However, when the PYP-tag was incubated with DMAC-HOBt, DMAC-FMK, DMAC-CMK, and DMAC-BMK in HEPES buffer (pH 7.4) at 37 °C for 1 h (see the ESI for experimental details†), then fluorescence bands for PYP-tag conjugates were detected in the gel (Fig. S2†). The heat shock conditions and denaturing reagents used to carry out SDS analysis of the DMAC ketone labeled PYP-tag products indicated that their DMAC fragments were likely to be covalently bound to the binding pocket of the PYP-tag protein (Fig. S3†). To confirm this, the reaction product of DMAC-CMK was digested with trypsin and the resultant cleavage products were analysed by SDS-PAGE, which revealed a fluorescence band at a lower gel position than for the intact DMAC-CMK-labeled PYP-tag (Fig. S4,† lane 4). This band corresponded to a peptide fragment conjugated to DMAC-CMK, with reverse phase HPLC analysis (ODS-3 column, linear gradient of acetonitrile in 0.1% formic acid aqueous solution from 10 to 90% in 30 min) revealing a retention time of 10.2 min (Fig. S5c†) – as compared to free DMAC-CMK that had a retention time of 23.8 min (Fig. S5a†). ESI-MS analysis of the fluorescent fraction eluting at 10.2 min revealed that the DMAC-CMK ligand was bound to a 14-mer peptide fragment, “DVAPCTDSPEFYGK”, whose cysteine residue corresponded to the Cys69 residue of the PYP-tag (Fig. S6†). The digestion of an unlabeled PYP-tag with trypsin afforded the corresponding unlabeled peptide fragment with a HPLC retention time of 8.7 min (Fig. S5b and S7†). Finally, MS/MS analyses of the ligand-bound 14-mer peptide fragment clearly revealed molecular ion fragments that were consistent with the DMAC-CMK ligand being covalently attached to the peptide cysteine residue via a thioether bond (Fig. S8†). MacroModel was then used to model the structure of the PYP-tag covalently bound to DMAC-CMK through a thioether bond to its Cys-69 residue, which clearly revealed that the binding pocket of the PYP-tag could sterically accommodate the DMAC ligand (Fig. S9†).
Photophysical characterization and cellular stability studies of PYP-tag-DMAC conjugates
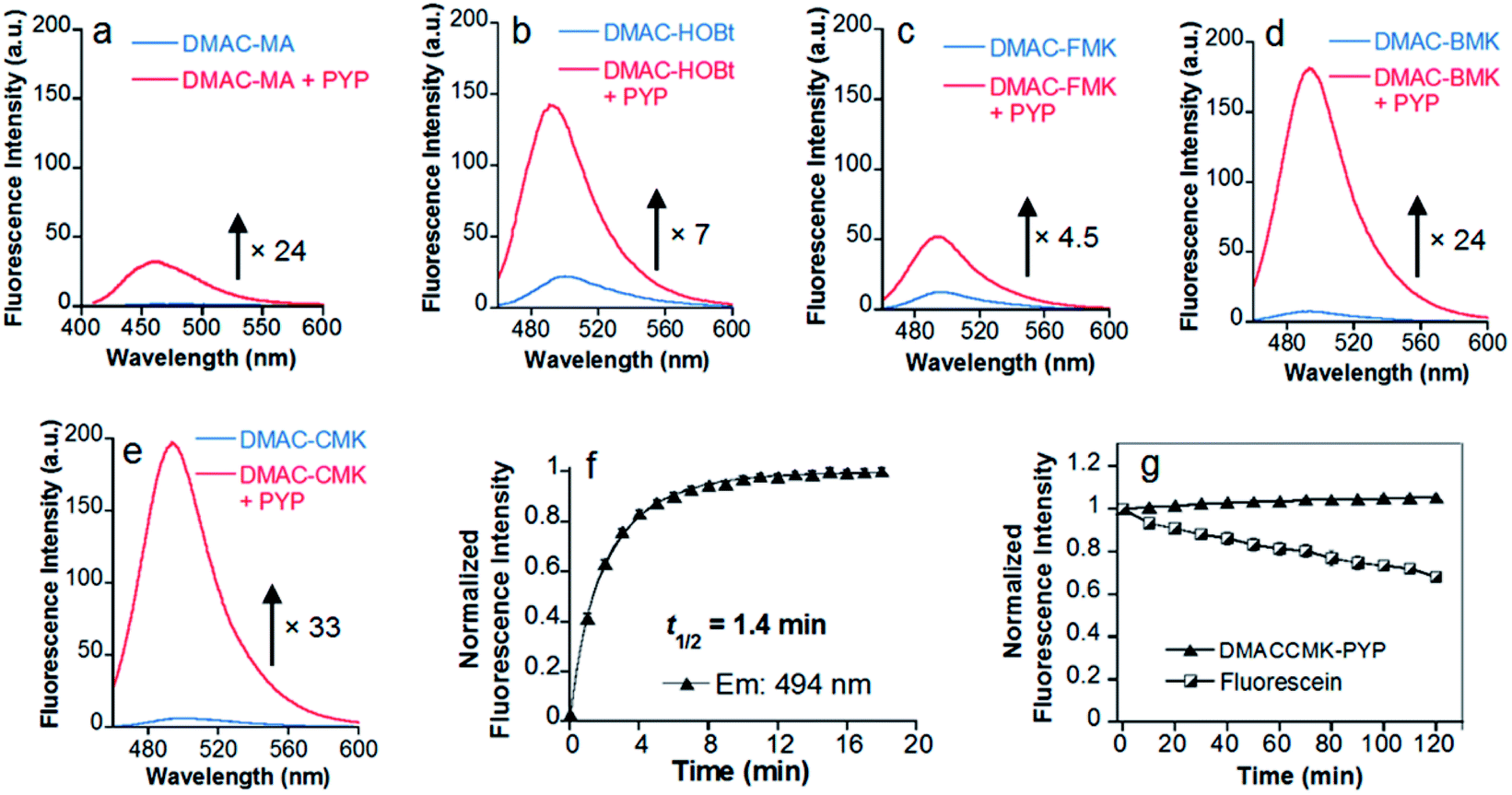
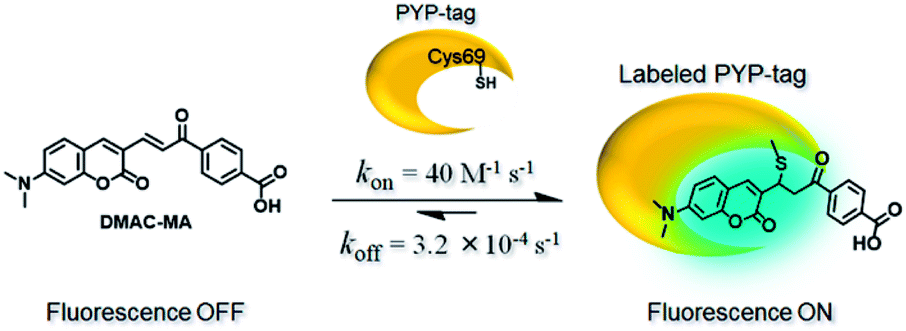
Twisted intramolecular charge transfer (TICT) fluorescence quenching meant that DMAC ligands only exhibit weak fluorescence emission in polar aqueous buffer (Fig. 2a–e).24,25 In contrast, covalent attachment of these DMAC ligands to the PYP-tag induced a significant increase in fluorescence emission, which was consistent with the DMAC ligands occupying the hydrophobic binding pocket of the PYP-tag (Fig. 2a–e). The labeling of PYP-tag with DMAC-MA resulted in a new absorption peak at 398 nm with simultaneous reduction in absorbance levels at 470 nm (Fig. S10a†). Although SDS-PAGE analysis did not show a fluorescence band corresponding to a DMAC-MA-labeled PYP-tag (Fig. S1†), changes to the fluorescence and UV/vis spectra of this ligand indicated that it had successfully labeled the PYP-tag (Fig. 2a and S10a†). We reasoned that this inconsistency might be due to the DMAC-MA fragment being cleaved from the PYP-tag complex under the conditions used for SDS-PAGE analysis. This hypothesis was validated by carrying out UV/vis analysis of a heated sample of PYP-tag-DMAC-MA, which gave spectra consistent with the formation of a free DMAC-MA ligand (Fig. S10b†). A purified sample of PYP-tag-DMAC-MA was subsequently isolated by gel filtration chromatography and shown to exhibit a time-dependent decrease in absorbance at 398 and an increase in absorbance at 470 nm, respectively (see pages S12 and S13†). This is consistent with DMAC-MA being slowly released from the PYP-tag over time, which presumably occurs through a relatively slow retro-Michael pathway (Fig. 3). Therefore, these studies confirm that the attachment of DMAC-MA to the PYP-tag occurs via reversible conjugate addition of the Cys69 residue of the PYP-tag to the DMAC-MA ligand. The thermodynamic and kinetic dissociation constants for this reversible Michael addition reaction were determined to be 7.4 μM and 8.0 μM, respectively (Fig. S12†), with the rate of forward (kon) and backward (koff) reactions found to be 40 M−1 s−1 and 3.2 × 10−4 s−1, respectively.
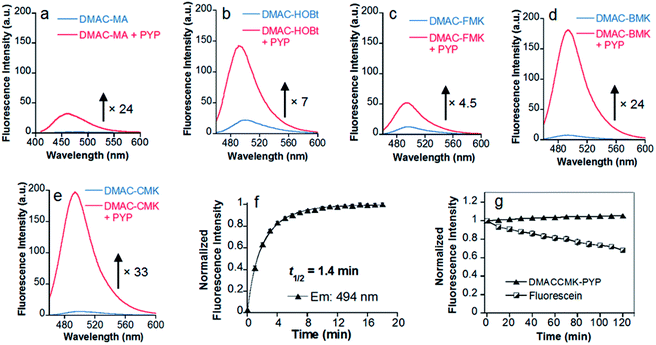 |
| | Fig. 2 Fluorescence spectra of DMAC ketone ligands in the absence/presence of the PYP-tag. Spectra for (a) DMAC-MA; (b) DMAC-HOBt; (c) DMAC-FMK; (d) DMAC-BMK; (e) DMAC-CMK were obtained after the incubation of these ligands (5 μM) with the PYP-tag (10 μM) for 68 min, 38 min, 48 h, 6 min, and 12 min, respectively, during which time their fluorescence intensities became saturated. The excitation wavelengths for DMAC-MA and the DMAC-ketone ligands were 398 and 450 nm, respectively. (f) Time course of the normalized fluorescence intensity of DMAC-CMK (5 μM) reacted with the PYP-tag (7 μM) at 494 nm (λex 450 nm). (g) Photostability of PYP-tag-DMAC-CMK and fluorescein (5 μM) during repeated light irradiation. DMAC-CMK (5 μM) and PYP-tag (10 μM) were incubated for 1 h before being irradiated, with fluorescence intensity levels were recorded every 10 min for 120 min. Excitation/emission wavelengths of 450/494 nm and 490/514 nm were used for PYP-tag-bound DMAC-CMK and fluorescein, respectively. All analyses were carried out at 37 °C in 20 mM HEPES buffer (pH 7.4) containing 150 mM NaCl and 1.5% DMSO, except DMAC-HOBt which included 5% DMSO. The error bars in (f) and (g) denote standard deviations (n = 3). | |
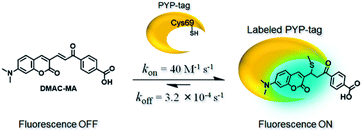 |
| | Fig. 3 Reversible Michael addition of the thiol group of the Cys-69 residue of the PYP-tag to the α,β-unsaturated fragment of DMAC-MA. | |
The addition of the DMAC-based methyl ketone ligands to the PYP-tag also resulted in changes to their absorption and emission profiles, with all of the bound ligands producing a significant increase in their fluorescence response (Fig. 2 and S13†). The incubation of DMAC-HOBt with the PYP-tag resulted in a 7-fold increase in fluorescence after 38 min (Fig. 2b), with 7.0 min (t1/2) required for 50% of the ligand to react with the PYP-tag (Fig. S14†). HPLC analysis of free DMAC-HOBt that had been incubated in aqueous buffer (Fig. S15†) revealed that it was unstable towards hydrolysis over time. However, both SDS-PAGE and fluorescence analyses indicated that the Cys69 residue of the PYP-tag could react effectively with DMAC-HOBt under aqueous conditions (Fig. S3†). The addition of DMAC-BMK and DMAC-CMK to the PYP-tag resulted in a 24 and 33-fold increase in fluorescence intensity after 6 and 12 min, respectively (Fig. 2d and e). However, DMAC-FMK only showed a slow increase in fluorescence indicating that it exhibits relatively slow labeling kinetics towards the PYP-tag (Fig. 2c). Although DMAC-BMK showed rapid fluorescence intensity saturation, it was found to be unstable to storage in DMSO solution.
The superior labeling properties, hydrolytic stability and fluorescence properties of DMAC-CMK in these PYP-tag labeling studies (Fig. S16†) led to it being chosen as the best candidate for further characterisation (Fig. 2e). The fluorescence quantum yields of DMAC-CMK in its free and PYP-tag bound states were determined to be 0.008 and 0.18, respectively. The higher quantum yield observed for bound DMAC is consistent with the hydrophobic binding pocket of the PYP-tag enhancing the fluorescence output of the bound ligand. The t1/2 value for the reaction of DMAC-CMK with the PYP-tag was found to be 1.4 min for PYP-tag concentrations of 5.0 and 7.0 μM, respectively (Fig. 2f), which corresponds to a second order rate constant (k2) of 1.2 × 103 M−1 s−1 (Fig. S17†). This rate value is similar to those of many other fluorogenic probes used for covalent protein labeling, although much higher k2 values have been reported for some HaloTag and SNAP-tag probes12 (Table S1†). The photostability of the DMAC-CMK ligand bound to the PYP-tag remained constant under continuous light irradiation (450 ± 5 nm; 4 mW cm−2) for 2 h (Fig. 2g). In contrast, the irradiation of fluorescein under these conditions resulted in a steady decrease in fluorescence (Fig. 2g), thus demonstrating the superior photostability of the DMAC-CMK ligand. The overall specificity of DMAC-CMK for the PYP-tag was confirmed by carrying out labeling studies of cell lysates (Fig. S18†), which showed a strongly fluorescence band for PYP-tag/DMAC-CMK, with bands for all other cellular proteins only demonstrating weak fluorescence. We also confirmed the stability of the PYP-tag/DMAC-CMK complex in the presence of competing biological thiols (Fig. S19†), with no cleavage reaction observed upon treatment with excess glutathione (10 mM). Consequently, given their favourable stability profiles and good labeling kinetics, DMAC-CMK and DMAC-HOBt were chosen as probes to carry out long-time PYP-tag fluorescence imaging of fusion proteins in live cells.
Live-cell imaging studies using fluorescent PYP-tag-DMAC fusion proteins
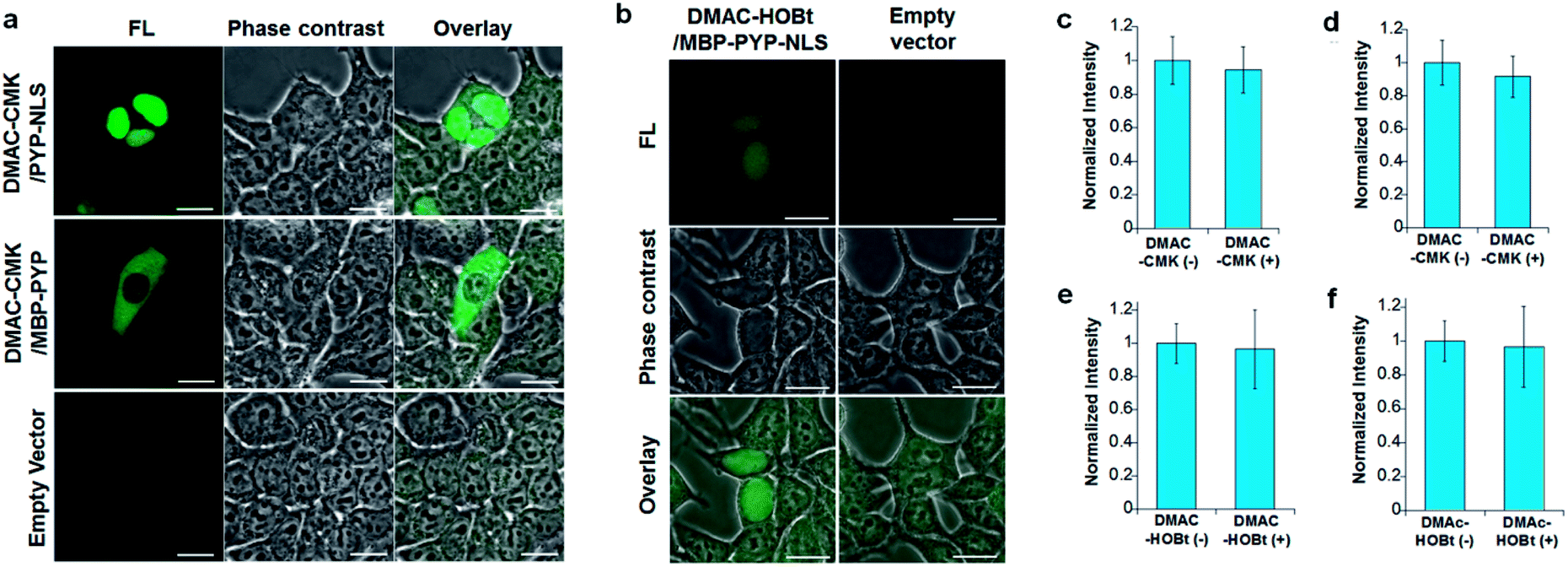
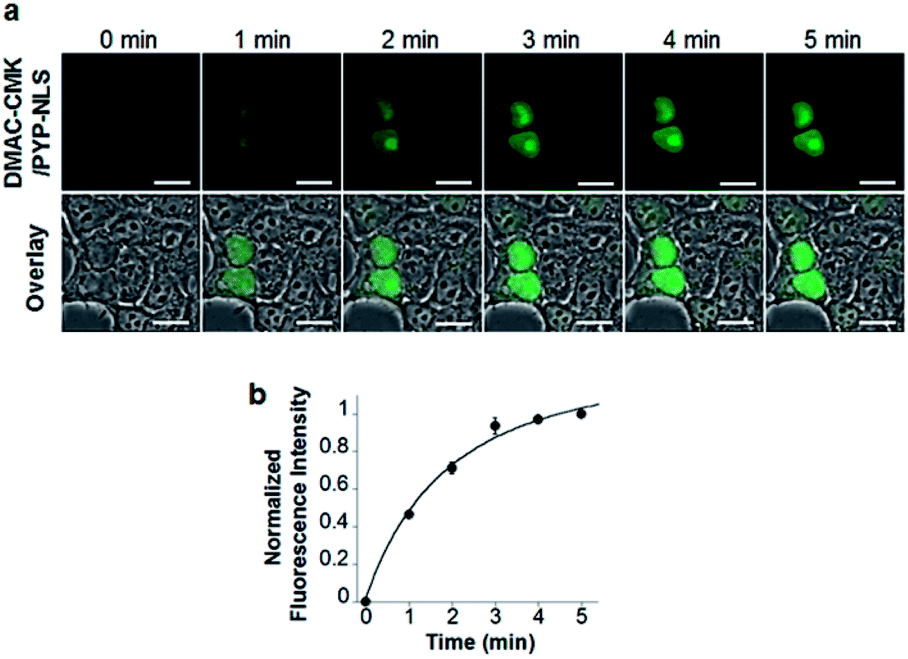
DMAC-CMK (or DMAC-HOBt) was incubated with HEK293T cells transfected with genes encoding PYP fused to nuclear localization signals (PYP-NLS or MBP-PYP-NLS) and maltose binding proteins (MBP-PYP). Fluorescence images of these cells were obtained using a confocal laser-scanning microscope, with fluorescence being observed in the nuclei of cells expressing PYP-NLS, whilst cells expressing MBP-PYP showed fluorescence in their cytoplasm (Fig. 4a). Untransfected cells, or cells transfected with an empty vector, did not exhibit any fluorescence (Fig. 4a, lower row). DMAC-HOBt could also be used for specific fluorescence labeling of MBP-PYP-NLS in the nuclei (Fig. 4b). These images clearly demonstrate that DMAC-CMK and DMAC-HOBt can freely cross the cell membrane to produce highly selective no-wash images of intracellular PYP-tag proteins with good fluorescence contrast. WST-assays of HEK293T cells that had been incubated with DMAC-CMK (2 μM) and DMAC-HOBt (2 μM) for 1 or 12 h revealed that they were not cytotoxic, with no difference in cell viability observed when compared to untreated cells (Fig. 4c–f). Time-lapse imaging of PYP-NLS was used to evaluate the rate of intracellular labeling of the PYP-tag after the addition of DMAC-CMK to cells in vitro (Fig. 5), with the nuclei becoming fluorescent after 1 min and a maximal fluorescence response achieved after 5 min. We noticed that DMAC-CMK tends to rapidly pass through the cell membrane and accumulate inside cells (Fig. S20†). The reaction kinetics and the intracellular accumulation of DMAC-CMK enable rapid labeling of PYP-fused proteins with DMAC-CMK with no need to carry out cell washing to remove excess probe, thus allowing any cell imaging studies to be commenced immediately after probe addition. The intracellular kinetics of the PYP labeling process were more rapid than many other fluorogenic probes used as covalent protein tags and comparable to that of the widely used HaloTag probe26 (Table S1†). The excellent in vitro photostability of the DMAC-CMK ligand bound to the PYP-tag in cellular systems was demonstrated by carrying out multiple irradiations of PYP-NLS-DMAC-CMK in living U2OS cells using a confocal laser scanning microscope (473 nm; 12.5 mW cm−2, measured using a Hioki 3664 optical power meter and Hioki 9742-10 optical sensor; sampling speed: 2.0 μs per pixel) (Fig. S21†). Repeated imaging of these cells (1000 times) resulted in only a 15% decrease in the nuclear fluorescence intensity of the bound DMAC fluorophore over the course of 1000 irradiations. This compares favourably with the irradiation of U2OS cells containing AcFCANB-bound PYP-NLS (a fluorescein PYP-tag-labeled probe), whose nuclear fluorescence was decreased by 39% under the same multiple irradiation conditions. We also conducted similar photostability experiments using EGFP. In comparison with EGFP, the PYP-DMAC-CMK complex also showed higher photostability (Fig. S22†). 24 hour cell imaging experiments of actively dividing DMAC-CMK/PYP-NLS cells (Fig. S23†) were then carried out, which revealed that DMAC-PYP-NLS remained intact throughout the mitosis process, as shown by the presence of fluorescence signals in the nuclei of daughter cells after cell division. These results clearly demonstrate that DMAC-CMK can be used to rapidly label PYP-tag proteins in living cells to afford stable fluorescent protein–ligand conjugates whose cellular locations can be monitored over long periods of time.
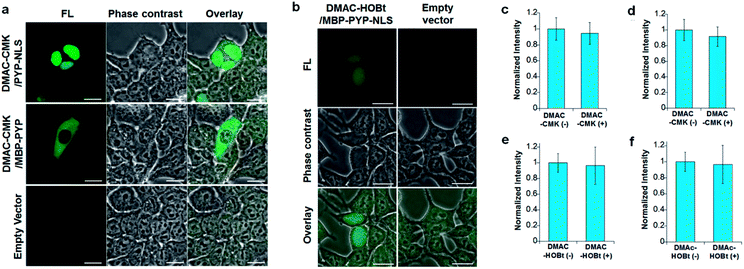 |
| | Fig. 4 (a) Images of HEK293T cells transfected with plasmids encoding PYP-NLS (top row), MBP-PYP (middle row) and an empty vector (lower row) after incubation with DMAC-CMK (200 nM) for 8 min. (b) Images of HEK293T cells transfected with plasmids encoding MBP-PYP-NLS (left column) and an empty vector (right column) after incubation with DMAC-HOBt (5 μM) for 60 min. λex/em = 473/490–590 nm. Scale bar: 20 μm. (c–f). Cell viabilities of DMAC-CMK (top) and DMAC-HOBt (below) determined using HEK293T cells in a 96 well plate (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) that were incubated with a 2 μM probe for 1 h (c and e) or 12 h (d and f) before WST-assay analysis was carried out. Data acquired using three independent experiments with values reported as mean values ±SD. 000 cells per well) that were incubated with a 2 μM probe for 1 h (c and e) or 12 h (d and f) before WST-assay analysis was carried out. Data acquired using three independent experiments with values reported as mean values ±SD. | |
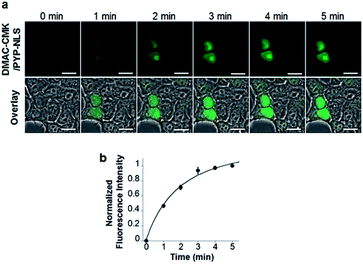 |
| | Fig. 5 (a) Time-lapse live-cell imaging of PYP-NLS using DMAC-CMK in HEK293T cells. Images were recorded at 1 min intervals after the addition of DMAC-CMK (200 nM). (b) Plot of the average fluorescence intensities of DMAC-CMK against incubation time for the images shown in (a) (N = 4). λex/em = 473/490–590 nm. Scale bar: 20 μm. | |
Live-cell imaging of the telomere binding protein TRF2
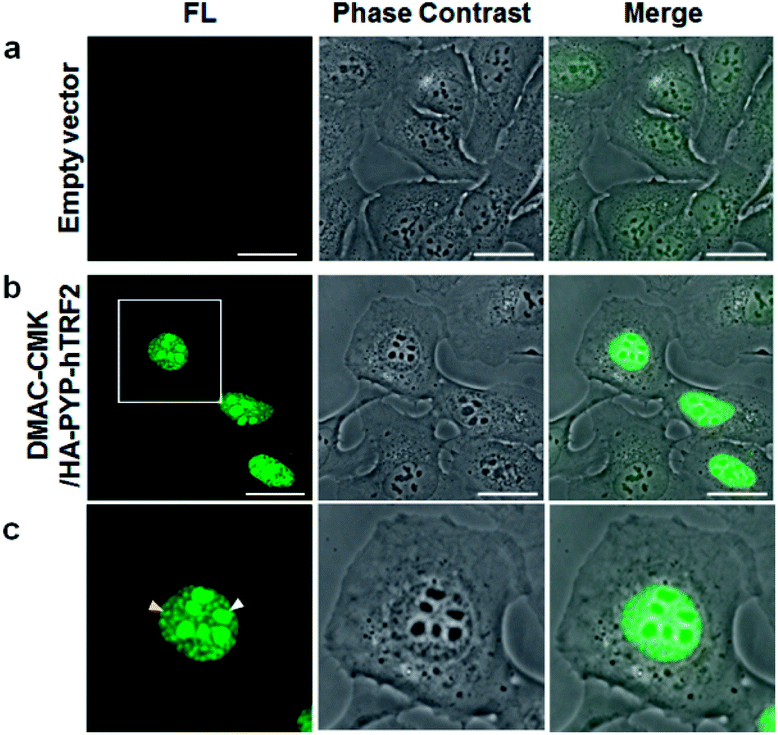
We then demonstrated that the DMAC-CMK/PYP-tag construct could be used for intracellular imaging of TRF2 (Telomeric repeat binding factor 2), which is a key telomere binding protein involved in maintaining telomere integrity and repairing DNA.27–30 A plasmid encoding a HA-PYP-hTRF2 fusion protein was first constructed and its expression in U2OS cells was confirmed by western blot analysis (Fig. S24†). Live-cell imaging of U2OS cells was carried out by transiently expressing HA-PYP-hTRF2 in the presence of DMAC-CMK (Fig. 6 and S25†), which revealed the presence of fluorescent proteins in two distinct cellular locations – the nucleoli (Fig. 6c, white triangles) and the nucleoplasm (Fig. 6c, yellow triangles). The appearance of fluorescence in the nucleoli is consistent with the presence of non-telomeric TRF2 in the nucleolus,31–33 whilst the discrete fluorescent dots in the nucleoplasm correspond to telomeric TRF2.31,32 These imaging results suggest that photostable DMAC-CMK may be a useful ligand for visualising/tracking a wide range of different PYP-tag fused proteins in cellular systems, thus providing a fluorescence reporting mechanism to probe their spatiotemporal roles in different cellular pathways.
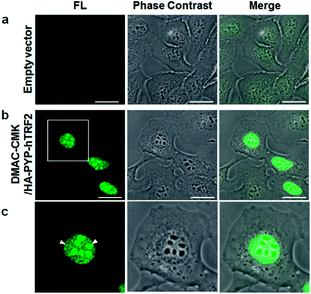 |
| | Fig. 6 Live-cell imaging studies: (a) U2OS cells expressing an empty vector; (b) U2OS cells expressing HA-PYP-hTRF2; (c) expansion of the square region of (b). Images recorded after the addition of DMAC-CMK (2 μM; 30 min incubation). λex/em = 473/490–590 nm. Scale bar: 20 μm. | |
Conclusions
New photostable DMAC ketone ligands have been developed that enable PYP-tag proteins to be selectively functionalised through the formation of stable covalent thioether bonds. The labeling of the PYP-tag with these weakly fluorescent DMAC ligands affords highly stable fluorescent PYP-tag-DMAC conjugates that can be used for long term cellular visualisation studies. The best DMAC-CMK probe enabled rapid no wash labeling of the PYP-tag, with the remarkable stability of its PYP-tag conjugates due to the formation of a strong thioether bond between the Cys69 residue of the PYP-tag and the DMAC ligand. The potential of using this no wash fluorescent labeling PYP-tag-DMAC system to visualise dividing cells undergoing mitosis and for imaging a PYP-tag fused telomere binding protein bound to chromatin in cell nuclei has been demonstrated. We believe that the cellular stability and photostability of these fluorescent DMAC-CMK/PYP-tag fusion proteins provide an excellent tool to enable their spatiotemporal location in complex biological systems to be tracked.
Experimental procedures
Materials and instruments
High purity chemicals were purchased from Tokyo Chemical Industries, Sigma-Aldrich Co., Kishida Chemical Co. and Wako Pure Chemical Industries and used without further purification. Enzymes used to carry out genetic engineering studies were purchased from Takara Bio and New England Biolabs. Synthetic oligonucleotides were purchased from Invitrogen or Greiner Bio-One. ESI-MS spectra were recorded using a WATERS 2695 mass spectrometer. High-resolution mass spectra (HRMS) were recorded using a JEOL JMS-700. NMR spectra (Bruker Ascend 500 (500 MHz for 1H and 125 MHz for 13C NMR)) were recorded using tetramethylsilane as an internal standard. Silica gel, BW-300 (Fuji Silysia Chemical Ltd.), was used for normal phase column chromatography. Reverse-phase high-performance liquid chromatography (RP-HPLC) purification was conducted using an Inertsil ODS-3 column (4.6 or 10.0 mm × 250 mm, GL-Science, Inc.). The PYP-tag was purified by size exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare Life Sciences) connected to an ÄKTA explorer system (GE Healthcare Life Sciences) or an NGC Chromatography System (Bio-Rad). Fluorescence spectra were recorded using a Hitachi F7000 spectrometer with a photomultiplier voltage of 700 V. Absorption spectra were recorded using a JASCO V-650 spectrometer. Ligands were dissolved in DMSO (biochemical grade, Wako) and then diluted to the desired concentration using buffer prior to use. Fluorescence bands in SDS PAGE gels were visualised using an AE 6935B VISIRAYS-B (ATTO Co) illuminator, with gels stained using Coomassie Brilliant Blue (CBB). Fluorescent live-cell images were obtained using a confocal laser-scanning microscope (Olympus FLUOVIEW FV10i, 60× lens) or a fluorescence microscope (KEYENCE, BZ-X700, 40× lens).
Plasmid construction
pcDNA-PYP-NLS, pcDNA-MBP-PYP-NLS and pcDNA-MBP-PYP plasmids were constructed as described previously.33 The primers 5′-TAGGCGTGTACGGTGGGA-3′/5′-CTTGCCAAA CCTACAGGT-3′ were used to transform pLPC-NMYC-TRF2 (gift from Titia de Lange (Addgene plasmid # 16066)) into pcDNA-HA-PYP-hTRF2 and hTRF2 (1-1548) genes using standard PCR techniques. This amplified gene was then cleaved with ClaI and EcoRI and the resultant sequence attached to a similarly cleaved plasmid, pcDNA-HA-PYP-mCherry, to give the gene pcDNA-HA-PYP-hTRF2. DNA sequencing was then carried out to confirm that all the genes had been correctly inserted into the plasmid.
Preparation of the protein (PYP-tag)
The PYP-tag was expressed in BL21 (DE3) and purified as reported previously,16 with its size and purity evaluated by SDS-PAGE analysis. The purified protein was dissolved in 20 mM HEPES buffer (pH 7.4) containing 150 mM NaCl before being flash-frozen in liquid N2 and stored at −80 °C.
Fluorescence spectroscopy
An excitation wavelength of 398 nm was used to record the fluorescence spectra of DMAC-MA, with the fluorescence spectra of DMAC ketone derivatives recorded at 450 nm using an ex/em slit width of 2.5/2.5 nm. The relative fluorescence quantum yields of DMAC-CMK were determined using a 0.1 N NaOH solution of fluorescein.34
Kinetic analyses of protein labeling reactions
The second-order rate constant for the reaction of DMAC-CMK and the PYP-tag was obtained using the following method. The probe was first reacted with an excess PYP-tag and the fluorescence intensity of the reaction was monitored (excitation at 450 nm, emission at 494 nm, and slit width of 5 nm) over time. Fluorescence intensity values were then transformed into labeled fractions using the following equation:
| [Labeled fraction] = (Ft − F0)/(Fmax − F0) |
where Ft, Fmax, and F0 represent the observed, maximum and initial fluorescence intensities, respectively.
The pseudo-first-order rate constant, kobs, was obtained by fitting the fluorescence data to the following equation:
| [Labeled fraction] = 1 − exp(−kobst) |
The pseudo-first-order rate constant was then plotted against the protein concentration and the plot was then fitted using the following equation to obtain the second-order-rate constant, k2:
Measurement of the equilibrium constant of DMAC-MA for the labeling of the PYP-tag using a fluorescence titration experiment
The fluorescence intensities of DMAC-MA at 456 nm (λex 398 nm) were plotted against different concentrations of the PYP-tag. The thermodynamic equilibrium constant was then obtained by fitting the fluorescence data to the following equation:
where Imin and Imax = the minimum and maximum fluorescence intensities, respectively; probeconc = the concentration of the probe used; PYPconc = concentration of PYP used.
Time course fluorescence intensities at 456 nm (λex 398 nm) for the labeling reaction of DMAC-MA with different concentrations of the PYP-tag were monitored to obtain an observed rate constant (kobs). These kobs values were then plotted as a function of PYP-tag concentration, which enabled the kinetic dissociation constant (Kd) to be calculated using the following equations:
| kobs = koff + kon × PYPconc |
and,
Cell viability
A WST-assay was used to evaluate cell viability in the presence of DMAC-CMK and DMAC-HOBt. HEK293T cells were incubated with DMAC-CMK or DMAC-HOBt (2 μM in each case) at 37 °C for 1 h (or 12 h) in a 96 well plate (40000 cells per well). These cells were then washed three times with HBSS followed by addition of DMEM (90 μL) and WST (10 μL) before being further incubated at 37 °C for 1 h. The treated cells were then analysed using a plate reader to monitor their absorption at 450 nm.
Fluorescence imaging of proteins in living cells
The transfection of HEK293T or U2OS cells with pcDNA-PYP-NLS, pcDNA-MBP-PYP, pcDNA-MBP-PYP-NLS, or pcDNA-HA-PYP-hTRF2 was conducted using Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer's manual. The resultant cells were incubated at 37 °C for 24 h and then washed twice with HBSS. The cells were then incubated with DMAC-CMK or DMAC-HOBt in DMEM for an appropriate time, followed by imaging using a confocal laser-scanning microscope employing an excitation frequency of 473 nm. Time-lapse imaging experiments were carried out by acquiring fluorescence images of PYP-NLS-expressing cells treated with DMAC-CMK derivatives over a specified period of time.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by the JSPS KAKENHI (Grant Numbers: JP17H06409 “Frontier Research on Chemical Communications”, JP18H03935 and JP25220207 to K. K.; JP16F16331 to K. K. and N. K.; and JP17H02210, JP18K19402, JP17H06005, and JP18H04735 “Resonance Bio” to Y. H.), JSPS A3 Foresight Program, JSPS Asian CORE Program, “Asian Chemical Biology Initiative”, Japan (JSPS)-UK (RSC) Research Cooperative Program (JPJSBP120195705 to K. K.), AMED-CREST, SICORP from JST, and Asahi Glass Foundation. We would like to thank Dr Masafumi Minoshima for fruitful discussions.
Notes and references
- K. M. Marks and G. P. Nolan, Nat. Methods, 2006, 3, 591 CrossRef CAS PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512 CrossRef CAS PubMed.
- L. Xue, I. A. Karpenko, J. Hiblot and K. Johnsson, Nat. Chem. Biol., 2015, 11, 917 CrossRef CAS PubMed.
- L. Leder, Methods Mol. Biol., 2015, 1266, 7 CrossRef CAS PubMed.
- M. Ferreboeuf, V. Mariot, D. Furling, G. Butler-Browne, V. Mouly and J. Dumonceaux, Hum. Mol. Genet., 2014, 23, 4125 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784 CrossRef CAS PubMed.
- S. Mizukami, Y. Hori and K. Kikuchi, Acc. Chem. Res., 2014, 47, 247 CrossRef CAS PubMed.
- H. M. O'Hare and K. Johnsson, Curr. Opin. Struct. Biol., 2007, 17, 488 CrossRef PubMed.
- D. L. Bodor, M. G. Rodriguez, N. Moreno and L. E. Jansen, Curr. Protoc. Cell Biol., 2012, 55, 8.8.1 Search PubMed.
- J. Lotze, P. Wolf, U. Reinhardt, O. Seitz, K. Mörl and A. G. Beck-Sickinger, ACS Chem. Biol., 2018, 13, 618 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269 CrossRef CAS PubMed.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132 CrossRef PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, ACS Chem. Biol., 2013, 8, 1704 CrossRef CAS PubMed.
- Y. Chen, C. M. Clouthier, K. Tsao, M. Strmiskova, H. Lachance and J. W. Keillor, Angew. Chem., Int. Ed., 2014, 53, 13785 CrossRef CAS PubMed.
- C. Szent-Gyorgyi, B. A. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235 CrossRef CAS PubMed.
- S. Mizukami, S. Watanabe, Y. Akimoto and K. Kikuchi, J. Am. Chem. Soc., 2012, 134, 1623 CrossRef CAS PubMed.
- M.-A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Querard, B. Pan, J. Rossignol, K. Moncoq, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. Le Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497 CrossRef CAS PubMed.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610 CrossRef CAS PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360 CrossRef CAS PubMed.
- Y. Hori, S. Hirayama, M. Sato and K. Kikuchi, Angew. Chem., Int. Ed., 2015, 54, 14368 CrossRef CAS PubMed.
- S. Hirayama, Y. Hori, Z. Benedek, T. Suzuki and K. Kikuchi, Nat. Chem. Biol., 2016, 12, 853 CrossRef CAS PubMed.
- J. Gao, Y. Hori, O. Takeuchi and K. Kikuchi, Bioconjugate Chem., 2019 DOI:10.1021/acs.bioconjchem.9b00696.
- S. Nad, M. Kumbhakar and H. Pal, J. Phys. Chem. A, 2003, 107, 4808 CrossRef CAS.
- G. Jones, W. R. Jackson, S. Kanoktanaporn and A. M. Halpern, Opt. Commun., 1980, 33, 315 CrossRef CAS.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165 CrossRef CAS PubMed.
- R. J. O'Sullivan and J. Karlseder, Nat. Rev. Mol. Cell Biol., 2010, 11, 171 CrossRef PubMed.
- B. van Steensel, V. Smogorzewska and T. de Lange, Cell, 1998, 92, 401 CrossRef CAS PubMed.
- K. Okamoto, C. Bartocci, I. Ouzounov, J. K. Diedrich, J. R. Yates III and E. L. Denchi, Nature, 2013, 494, 502 CrossRef CAS PubMed.
- Z. Mao, A. Seluanov, Y. Jiang and V. Gorbunova, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13068 CrossRef CAS PubMed.
- S. Zhang, P. Hemmerich and F. Grosse, J. Cell Sci., 2004, 117, 3935 CrossRef CAS PubMed.
- F. Yuan, G. Li and T. Tong, Cell Death Discovery, 2017, 3, 17043 CrossRef CAS PubMed.
- F. Yuan, C. Xu, G. Li and T. Tong, Cell Death Dis., 2018, 9, 518 CrossRef PubMed.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic schemes and details, figures and characterization data. See DOI: 10.1039/d0sc00499e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc