
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic basis for tuning iridium hydride photochemistry from H2 evolution to hydride transfer hydrodechlorination†
Seth M.
Barrett
 ab,
Bethany M.
Stratakes
a,
Matthew B.
Chambers
ac,
Daniel A.
Kurtz
a,
Catherine L.
Pitman
a,
Jillian L.
Dempsey
a and
Alexander J. M.
Miller
*a
ab,
Bethany M.
Stratakes
a,
Matthew B.
Chambers
ac,
Daniel A.
Kurtz
a,
Catherine L.
Pitman
a,
Jillian L.
Dempsey
a and
Alexander J. M.
Miller
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA. E-mail: ajmm@email.unc.edu
bDepartment of Chemistry, Muskingum University, New Concord, OH 43762-1118, USA
cDepartment of Chemistry, Louisiana State University, Baton Rouge, LA 70803-1804, USA
First published on 6th March 2020
Abstract
The photochemistry of metal hydride complexes is dominated by H2 evolution, limiting access to reductive transformations based on photochemical hydride transfer. In this article, the innate H2 evolution photochemistry of the iridium hydride complexes [Cp*Ir(bpy-OMe)H]+ (1, bpy-OMe = 4,4ʹ-dimethoxy-2,2′-bipyridine) and [Cp*Ir(bpy)H]+ (2, bpy = 2,2ʹ-bipyridine) is diverted towards photochemical hydrodechlorination. Net hydride transfer from 1 and 2 to dichloromethane produces chloromethane with high selectivity and exceptional photochemical quantum yield (Φ ≤ 1.3). Thermodynamic and kinetic mechanistic studies are consistent with a non-radical-chain reaction sequence initiated by “self-quenching” electron transfer between excited state and ground state hydride complexes, followed by proton-coupled electron transfer (PCET) hydrodechlorination that outcompetes H–H coupling. This unique photochemical mechanism provides a new hope for the development of light-driven hydride transfer reactions.
Introduction
The photochemistry of transition metal complexes underpins a range of applications, including organic light-emitting diode (OLED) development,1,2 solar energy conversion,3–6 and photoredox catalysis.7–11 The photocatalysts in these systems usually operate via pathways that involve excited state single-electron transfer (SET), and careful photophysical and photochemical studies have established factors that give rise to photocatalysts with long excited-state lifetimes and good long-term stability.12–15 In terms of electronic structure, a large ligand field splitting (or a d10 configuration) is often critical. In terms of chemical structure, complexes that successfully engage in outer-sphere SET are typically supported by rigid chelating ligands that are completely inert towards chemical reactivity.New opportunities can be imagined for photocatalysts that go beyond SET reactivity to tightly couple light absorption with bond-forming and bond-breaking chemical reactions. Photochemical ligand dissociation from transition metal complexes, such as CO release from carbonyl complexes, has been the subject of intense study.16,17 Photodissociation can initiate thermal catalytic reactions via the generation of highly reactive coordinatively unsaturated intermediates.3,18–21 Other systems capable of photochemical bond-forming reactions include metal–halide complexes (e.g. dihalogen generation);3,4,22 metal–amide complexes (e.g. C–N cross-coupling reactions);23–29 and metal–aryl complexes (e.g. dual photoredox cross-coupling catalysis).10,30–32
Complexes with metal–hydride bonds would appear to be ideal candidates for photocatalytic reductions involving net hydride transfer to an organic substrate, but examples of photochemical organic transformations utilizing metal hydride complexes are scarce.33 The lack of development is likely due to a challenge that is unique to metal–hydride photocatalysts: there is an inherent competition between hydride transfer and H2 evolution. Indeed, metal hydride complexes are quintessential H2 evolution photocatalysts,33 with particular application in the synthesis of solar fuels.3 Understanding how to tune the photochemical reactivity of metal hydride complexes between H2 evolution and hydride transfer represents a fundamental challenge.
Iridium hydride complexes of the type [Cp*Ir(bpy-R)H]+ (bpy-R is 4,4′-disubstituted-2,2′-bipyridine) are promising candidates for exploring the photochemical activation of metal–hydride bonds.34 Photoexcitation generates a weakly emissive metal–ligand charge transfer (MLCT) excited state.35 The high degree of MLCT character derives from the strong σ-donor ability of the hydride ligand.36 The photochemistry of these Ir hydride complexes is dominated by H2 evolution (Scheme 1). Photocatalytic H2 evolution was first reported in aqueous conditions,37–41 with subsequent research demonstrating photoelectrochemical H2 production and photocatalytic formic acid dehydrogenation.42,43 In acetonitrile solvent, photolysis of [Cp*Ir(bpy)H]+ (either alone or with added acids) also produces H2 in high yield.44
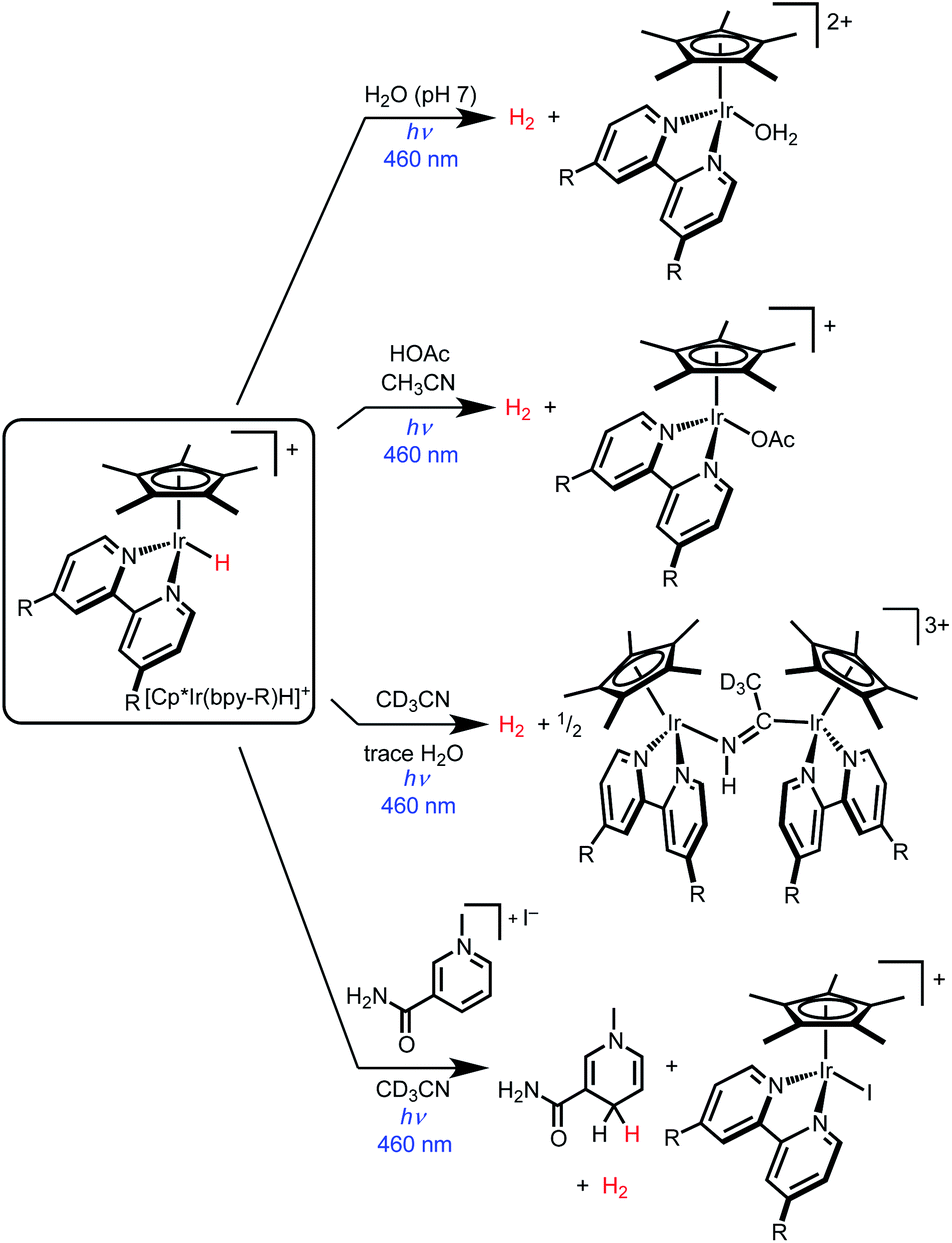 | ||
| Scheme 1 Photochemical reactivity of iridium hydride complexes [Cp*Ir(bpy-R)H]+ (bpy-R is 4,4ʹ-disubstituted-2,2ʹ-bipyridine). | ||
Complex [Cp*Ir(bpy)H]+ undergoes H2 evolution via a mechanism that is thus far unique amongst molecular hydrides. The MLCT excited state undergoes “self-quenching” electron transfer with a ground state hydride complex, followed by rapid bimolecular H–H coupling.45 As such, the photochemical quantum yield for H2 production approaches unity as the initial concentration of iridium complex is increased above 20 mM. Due to the bimolecular mechanism, H2 is produced in good yield even in the absence of external proton sources.
While prior studies of [Cp*Ir(bpy-R)H]+ complexes have focused on photochemical H2 evolution, we were surprised to observe net photochemical hydride transfer to a nicotinamide derivative in acetonitrile solvent (Scheme 1).44 While promising as the first example of even a stoichiometric light-driven hydride transfer, this reaction suffered from low yields and competing H2 evolution (even though no proton source was added). The reaction mechanism has not yet been explored. Nonetheless, the precedent for both H2 evolution and hydride transfer in the same iridium hydride complex led us to embark on further studies to elucidate the factors that control reactivity between H2 evolution and hydride transfer.
Here we report near-complete suppression of H2 evolution and a switch to net hydride transfer in the presence of dichloromethane or other alkyl chlorides. Mechanistic studies of dichloromethane photoreduction elucidate a self-quenching electron transfer pathway that generates reactive organometallic intermediates capable of hydride transfer and hydrogen atom transfer reduction of CH2Cl2. Because a single photon absorption triggers the reactivity of two different metal centers, the maximum theoretical quantum yield is 2; experimental conditions were found that achieve Φ = 1.3. The mechanistic insight into the factors that can suppress H2 evolution and enable hydride transfer provides general guidance for future catalyst structures with photocontrolled reactivity.
Results and discussion
Photochemical hydrodechlorination of alkyl chlorides
Hydride complexes [Cp*Ir(bpy-OMe)H][OTf] (1) and [Cp*Ir(bpy)H][OTf] (2) were prepared following previously developed approaches.44,46 Hypothesizing that high concentrations of a hydride acceptor substrate could promote hydrodechlorination, dichloromethane was explored as both substrate and solvent.Hydride 1 was dissolved in dichloromethane-d2 (CD2Cl2) and monitored by NMR spectroscopy while protected from light, with only 3% conversion to [Cp*Ir(bpy-OMe)Cl]+ (3) observed after 2 h at 25 °C. Illumination of 1 in CD2Cl2 with a 460 nm LED lamp for 10 min produced chloride complex 3 quantitatively (Scheme 2). A diagnostic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:2:1 pentet for dissolved chloromethane-d2 gas (CHD2Cl δ 2.99, JHD = 1.6 Hz) was also observed (49% yield relative to 1 by 1H NMR). The excitation wavelength was chosen to match the MLCT transition (λmaxca. 420 nm, Fig. S23†).43 When the same experiment was run in CH2Cl2, chloromethane (CH3Cl) was produced in 69% yield relative to 1 (1H NMR). Some of the volatile chloromethane product is lost to the headspace over time, so the solution yields are considered lower limits. Soluble H2 was not detectable by 1H NMR spectroscopy. The gas phase yield of H2 was less than 4% relative to 1, according to headspace analysis by gas chromatography (GC).
:2:3:2:1 pentet for dissolved chloromethane-d2 gas (CHD2Cl δ 2.99, JHD = 1.6 Hz) was also observed (49% yield relative to 1 by 1H NMR). The excitation wavelength was chosen to match the MLCT transition (λmaxca. 420 nm, Fig. S23†).43 When the same experiment was run in CH2Cl2, chloromethane (CH3Cl) was produced in 69% yield relative to 1 (1H NMR). Some of the volatile chloromethane product is lost to the headspace over time, so the solution yields are considered lower limits. Soluble H2 was not detectable by 1H NMR spectroscopy. The gas phase yield of H2 was less than 4% relative to 1, according to headspace analysis by gas chromatography (GC).
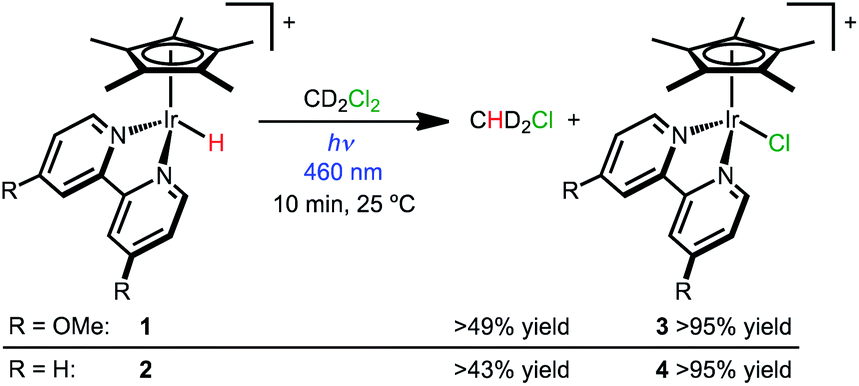 | ||
| Scheme 2 Visible light photochemical hydrodechlorination of CD2Cl2. | ||
Hydride 2 exhibits similar reactivity, producing chloride complex [Cp*Ir(bpy)Cl]+ (4; >95% yield) and CHD2Cl (43% yield relative to 2) after 10 min of 460 nm illumination in CD2Cl2 (Scheme 2). When the same experiment was run in CH2Cl2, chloromethane (CH3Cl) was produced in 50% yield. When protected from light, only 6% conversion of 2 was observed after 1 h.
To probe the effect of substrate concentration on the selectivity between H2 formation and hydrodechlorination systematically, photolysis was carried out in various dichloromethane/acetonitrile mixtures. Fig. 1 shows the yields of dissolved CH3Cl and H2 determined by 1H NMR spectroscopy after illumination of hydride 1 in CH2Cl2/CD3CN mixtures for 10 min. Even when CD3CN makes up the majority of the solvent, the yield of dissolved CH3Cl is higher than that of H2.
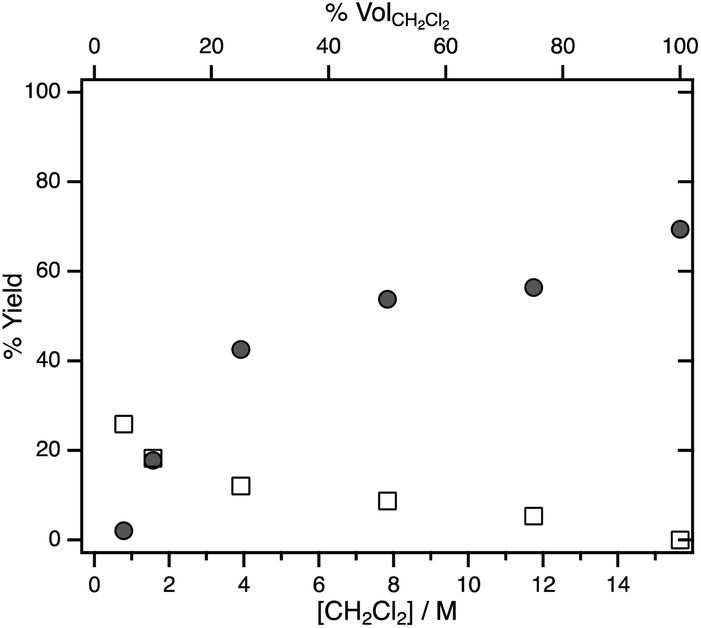 | ||
| Fig. 1 Yield of dissolved CH3Cl (filled circles) and H2 (empty squares) measured by 1H NMR spectroscopy after 10 min photolysis of 1 in CH2Cl2/CD3CN mixtures of varying solvent ratios (where % volCH2Cl2 is the volume percent of CH2Cl2 and [CH2Cl2] is the concentration of CH2Cl2). | ||
To further probe the competition between H2 evolution and hydride transfer, reactions were carried out in the presence of acid. Illumination of hydride complex 1 in CD2Cl2 solutions containing 1 equiv. acetic acid generated the chloride complex 3—and not the acetate complex expected upon H2 release.44 Even in the presence of acid, selective hydrodechlorination of dichloromethane is observed.
The tolerance of proton donors suggested that an aqueous formate solution could close a catalytic cycle by regenerating the iridium hydride after initial hydride transfer to the substrate. Illumination of biphasic mixtures of chloride complex 3 in CD2Cl2 and 1 M formate in H2O indeed produced chloromethane (TON > 2, see Section III in the ESI† for details).
The reactivity can also be extended to other simple chlorinated organic substrates dissolved in acetonitrile. Three model substrates were selected, 2-chloroethylbenzene, 3-chloropropiophenone, and 2-chloroethoxybenzene. As shown in Scheme 3, illumination of CD3CN solutions of 1 and the primary alkyl chloride substrate produced the dechlorinated products in approximately 20% yield relative to 1 by 1H NMR spectroscopy (with the balance presumably producing H2 gas, detected in solution in up to 66% yield; no other organic products were detected).
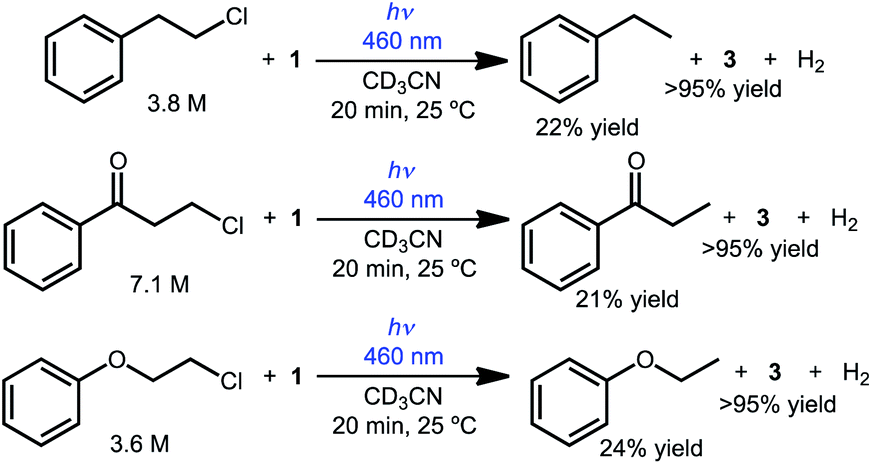 | ||
| Scheme 3 Photochemical hydrodechlorination of primary alkyl chlorides. | ||
The selective hydride transfer reactivity of 1 and 2 in dichloromethane solution draws a sharp contrast to the efficient H2 evolution reactivity of these same hydride complexes in acetonitrile or water solutions.41–44 In pure acetonitrile solutions without any acid present, some H2 evolution is observed, followed by Ir nucleophilic attack on the solvent to form an iminoacyl complex.44 In the presence of acetic acid, triethylammonium, or pyridinium, quantitative H2 evolution is observed.
In this context, we also examined chlorobenzene as a chlorinated substrate that would be unlikely to undergo reduction via hydride transfer.47 When chlorobenzene-d5 (C6D5Cl) solutions of 1 or 2 were illuminated with a 443 nm lamp, a signal for H2 was detected by 1H NMR spectroscopy without evidence for the hydrodechlorination product benzene. Photolysis of a 2 mM solution of 2 in C6H5Cl for 35 min produced H2 in 52% yield (GC). An unidentified iridium-containing yellow precipitate formed in these reactions. The results in chlorobenzene underscore the innate H2 evolution reactivity of 1 and 2 in the absence of a suitable hydride acceptor, even when no proton donor is added.
To understand the origin of the selective photochemical hydride transfer reactivity, we carried out a series of thermodynamic and kinetic studies to examine the detailed mechanism of photochemical hydrodechlorination by iridium hydride complexes, focusing on the highest yielding substrate, dichloromethane.
Overview of mechanistic pathways considered
Scheme 4 shows several possible reaction pathways for photochemical hydrodechlorination. The pathways can be divided into two categories: in monometallic pathways, the excited state of 1 (1*) interacts with CH2Cl2 directly; in bimetallic pathways, the excited state undergoes a “self-quenching” process with an equivalent of 1 in the ground state before interacting with CH2Cl2.31,45,48–50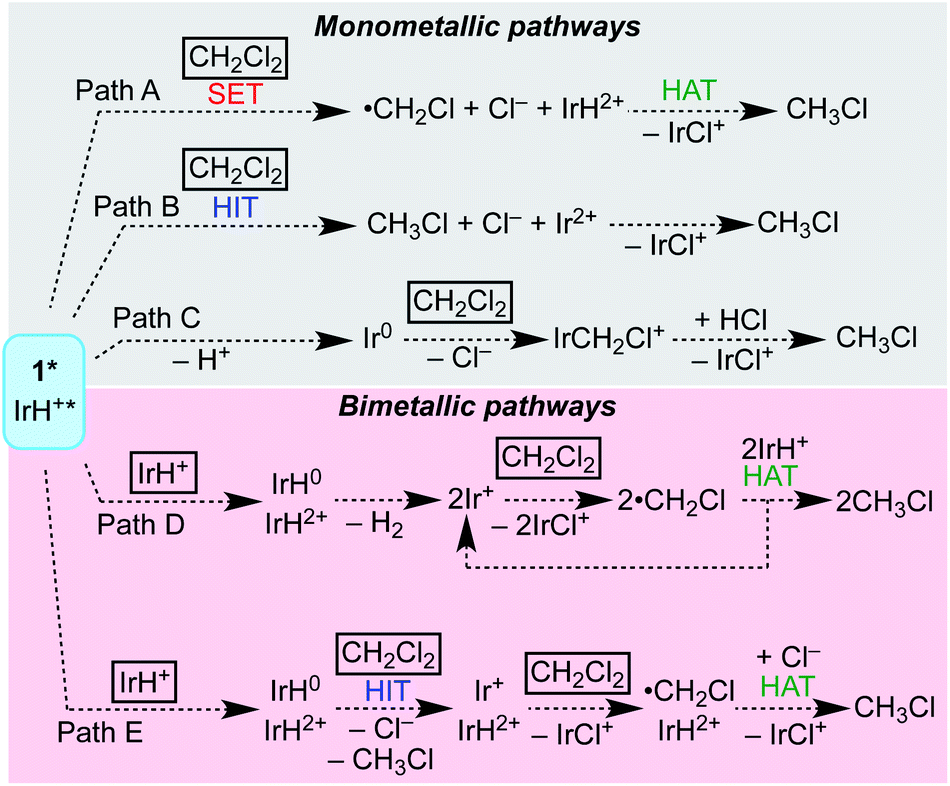 | ||
| Scheme 4 Photochemical hydrodechlorination pathways considered. | ||
Monometallic pathways could proceed via Path A, single electron transfer (SET) followed by hydrogen atom transfer (HAT); via Path B, hydride ion transfer (HIT); or via Path C, photoacid generation of H+ and a nucleophilic Ir(I) center.51 Complex 2 catalyzes thermal hydrodehalogenation of alkyl bromides (but not alkyl chlorides) via a HIT mechanism.52
Bimetallic pathways could proceed via initial self-quenching electron transfer to form a reactive pair of Ir(II) and Ir(IV) hydrides,45 with Path D proceeding by a radical chain reaction; or the PCET Path E proceeding by stoichiometric HIT and HAT steps. Radical chain hydrodehalogenation is observed for reagents such as Bu3SnH, which requires a radical initiator to generate ·CH2Cl.53 In the present case, a small amount of Ir hydride could undergo photochemical H2 release, generating an Ir(II) species that could undergo Cl-atom abstraction from CH2Cl2 to generate ·CH2Cl, which in turn could react with an Ir hydride to furnish CH3Cl and initiate a new chain (Path D). The alternative bimetallic pathway would involve hydride transfer to CH2Cl2 from the neutral hydride complex Cp*Ir(bpy-R)H, followed by Cl˙ abstraction from a second CH2Cl2 and finally HAT from the oxidized hydride [Cp*Ir(bpy-R)H]2+ (Path E).
Thermochemical analyses of the pathways shown in Scheme 4 were carried out, as detailed in Section V in the ESI.† Each pathway appears to be thermodynamically viable. Some steps are close to ergoneutral, however, and other steps are likely to face significant kinetic challenges. To distinguish between these viable pathways, a kinetic analysis of the photochemical hydrodechlorination reaction was undertaken.
Photoluminescence quenching kinetic studies
The kinetics of excited state quenching, probed by time-resolved photoluminescence measurements, can differentiate amongst the pathways considered in Scheme 4. In monometallic pathways, the luminescence lifetime should shorten with increasing concentration of CH2Cl2. In bimetallic pathways, the luminescence lifetime should shorten with increasing concentration of iridium hydride.The role of CH2Cl2 was probed by analyzing the photoluminescence lifetime of 1* in solutions of CH3CN with varying concentration of CH2Cl2 (and a constant 1 mM concentration of 1). As shown in Fig. 2, the lifetime of 1* is essentially invariant up to 8 M CH2Cl2, suggesting that monometallic pathways A and B that involve CH2Cl2 quenching are not operative. While the lifetime decreases above 8 M (50 vol%) CH2Cl2, the lack of a linear correlation in the Stern–Volmer analysis (Fig. 2) shows that the change in lifetime is not related to a diffusional quenching process. The change in lifetime is instead attributed to solvation effects changing with solvent composition, as has been observed for other charge-transfer-based excited states.49,54–57
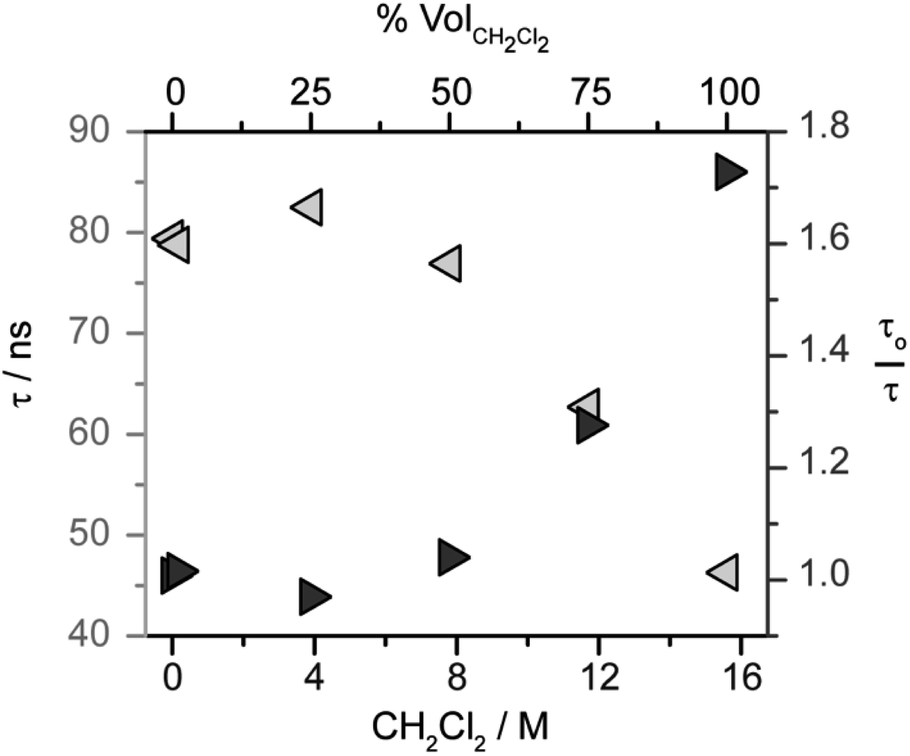 | ||
| Fig. 2 Evaluation of excited state quenching of [Cp*Ir(bpy-OMe)H][PF6] (1) by CH2Cl2 by time-resolved photoluminescence spectroscopy. Observed lifetimes (τ, light gray left arrows) and Stern–Volmer analysis (τo = 80 ns, dark gray right arrows) in CH2Cl2/CH3CN mixtures at a constant 1 mM concentration of 1. Excitation at 445 nm, luminescence decay monitored at 650 nm. In the axes, % volCH2Cl2 is the volume percent of CH2Cl2 and [CH2Cl2] is the concentration of CH2Cl2. | ||
The other monometallic pathway, Path C, involves initial photochemical H+ release to form Cp*Ir(bpy) (5) followed by Ir nucleophilic attack on CH2Cl2 to form a chloromethyliridium intermediate [Cp*Ir(bpy)(CH2Cl)]+ (6) that could undergo protonolysis to release CH3Cl. This pathway could not be ruled out by the Stern–Volmer quenching experiment, but was discounted based on independent reactivity of complexes 5 and 6. Dissolving 5 in CH2Cl2 leads to formation of chloromethyliridium complex 6 within 20 min at room temperature (Scheme 5). Chloromethyl complex 6 is quite stable, however. Treatment with various acids does not generate significant amounts of CH3Cl, even under illumination (Scheme 5). The chloromethyl complex 6 is therefore not kinetically competent to carry out the observed reactivity.
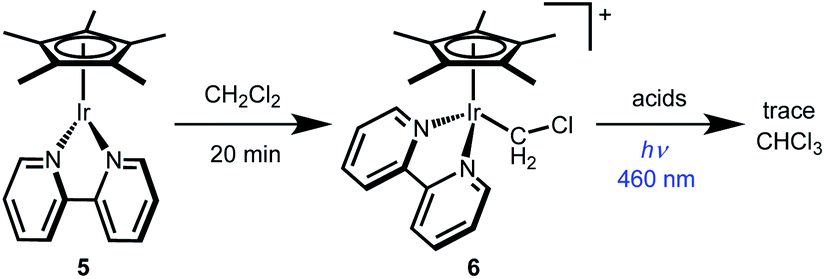 | ||
| Scheme 5 Chloromethyl complex 6 was synthesized and shown to be kinetically incompetent for CH2Cl2 reduction. | ||
To assess the possibility of a bimetallic self-quenching pathway in which excited state 1* is quenched by hydride 1, the luminescence lifetime of 1* in CH2Cl2 was evaluated as the concentration of 1 was increased from 0.094 mM to 31 mM (Fig. 3). The luminescence lifetime decreases with increasing concentration of 1, from which the intrinsic lifetime, τo = 50 ns, can be obtained by extrapolation to infinite dilution (Fig. 3). The intrinsic lifetime of 1 in CH2Cl2 is shorter than the intrinsic lifetime of 2 in CH3CN with 0.1 M [nBu4][PF6] (τo = 98 ns),45 consistent with the radiative decay rates being affected by the solvent composition.
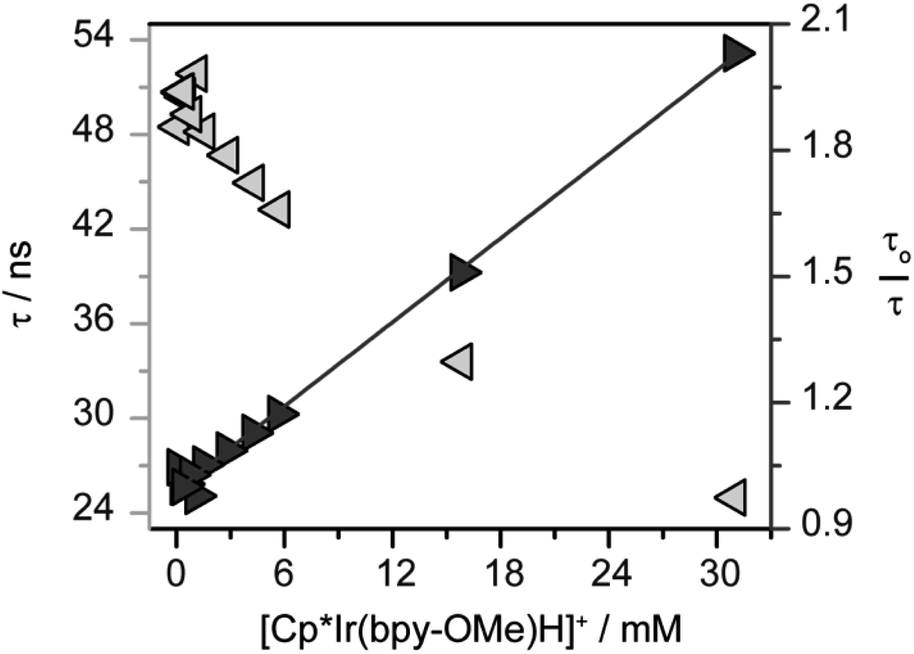 | ||
| Fig. 3 Evaluation of excited state self-quenching in [Cp*Ir(bpy-OMe)H][PF6] (1) by time-resolved photoluminescence spectroscopy. Observed lifetimes (τ, light gray left arrows) and Stern–Volmer analysis (τo = 54 ns, dark gray right arrows) in CH2Cl2. Excitation at 445 nm, luminescence decay monitored at 650 nm. The linear fit corresponds to a kq = 6.6 × 108 M−1 s−1. | ||
A linear correlation in the Stern–Volmer analysis varying the concentration of 1 (Fig. 3) is consistent with a bimetallic self-quenching pathway, with kq = 6.6 × 108 M−1 s−1. The rate of self-quenching in CH2Cl2 is notably slower than in CH3CN (kq = 3.8 × 109 M−1 s−1),45 perhaps due to the relatively low polarity of CH2Cl2 disfavoring the reaction of two cationic metal complexes.
Although the net photochemistry is starkly different in CH3CN (H2 evolution) and CH2Cl2 (hydrodechlorination), the Ir hydrides undergo the same initial self-quenching reaction in each solvent. The self-quenching step is estimated to be slightly thermodynamically unfavorable (by about 5 kcal mol−1 on the basis of excited state redox potentials, Fig. S19†). Other examples of excited state self-quenching are also close to ergoneutral, including a recent nickel example that is substantially endergonic.31,45,48–50
A self-quenching mechanism should give rise to strongly concentration-dependent photochemistry. Fig. 4 presents the quantum yields for consumption of 1 (based on UV-vis spectroscopic monitoring of the initial rate, see ESI† for details) upon photolysis with 443 nm light in CH2Cl2, revealing a dramatic dependence on iridium concentration that reaches a maximum exceeding unity, Φ = 1.3. The linear increase in quantum yield with increasing iridium concentration up to 8 mM 1 is further evidence of a bimetallic mechanism. The maximum quantum yield exceeding unity suggests either a photochemically triggered radical chain reaction (Path D) or a bimetallic self-quenching pathway that generates two equivalents of chloromethane per photon absorbed (Path E).
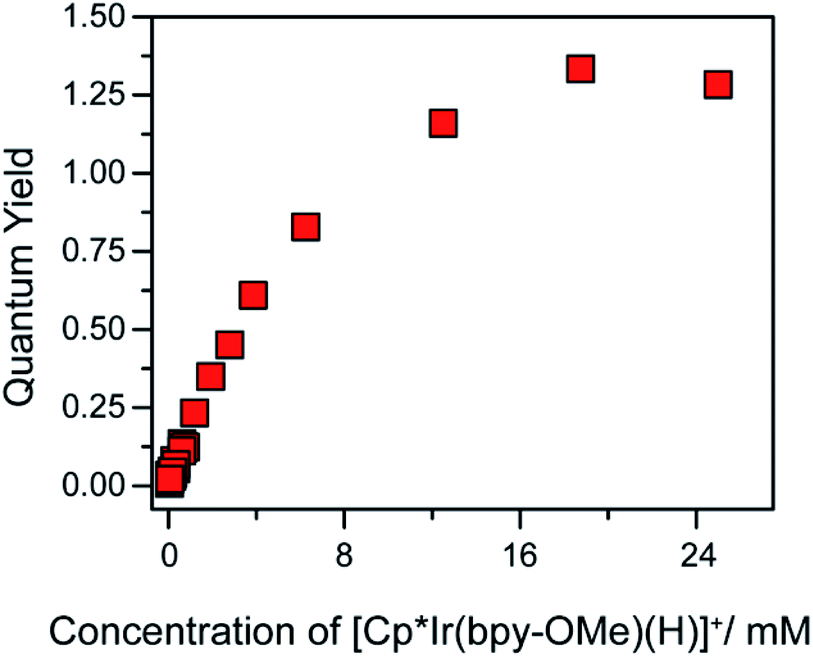 | ||
| Fig. 4 Quantum yield of photochemical CH2Cl2 hydrodechlorination (443 nm illumination) by [Cp*Ir(bpy-OMe)H][PF6] (1) as a function of concentration of 1. | ||
Distinguishing between bimetallic pathways
Of the mechanisms considered in Scheme 4, only Paths D and E can explain the bimetallic reactivity with a quantum yield greater than 1. In each pathway, photoexcitation of 1 is followed by self-quenching electron transfer to generate a pair of Ir(IV) and Ir(II) hydrides. In the radical chain mechanism of Path D, photochemical H2 release would generate an Ir(II) intermediate capable of abstracting a chlorine atom from CH2Cl2, which would initiate radical hydrodehalogenation reminiscent of Bu3SnH reactivity.47,53 The quantum yield for this pathway could be much higher than unity,58 depending on the rates of chain propagation and termination. In the stoichiometric mechanism of Path E, the Ir(II) hydride formed after self-quenching would hydrodechlorinate CH2Cl2 by hydride transfer. The resulting Ir(II) intermediate would abstract a chlorine atom from CH2Cl2, followed by H atom transfer from the Ir(IV) hydride. This mechanism would have a theoretical quantum yield of 2.To probe the viability of chlorine atom abstraction from CH2Cl2 by an Ir(II) intermediate, cyclic voltammograms of [Cp*Ir(bpy)(NCCH3)][PF6]2 were obtained in CH2Cl2 (0.25 M [nBu4N][PF6]). At scan rates above 1 V s−1, a single irreversible reduction feature was observed. At slower scan rates, however, a feature attributed to the reduction of chloride complex 4 was observed (Fig. S24†), consistent with formation of 4 upon reduction of [Cp*Ir(bpy)(NCCH3)][PF6]2. The product was confirmed as 4 based on 1H NMR spectroscopic analysis of the products produced during controlled potential electrolysis of [Cp*Ir(bpy)(NCCH3)][PF6]2 in CH2Cl2 (Fig. S25†). These results indicate that the reduced Ir species (Ir+ in Scheme 4, above) can abstract a chlorine atom from CH2Cl2. A mechanism for this reaction is proposed in Scheme 6. The formally Ir(II) intermediate in Scheme 6 has been proposed previously in CH3CN solution, where further reduction or disproportionation steps produce the formally Ir(I) complex 5.59,60
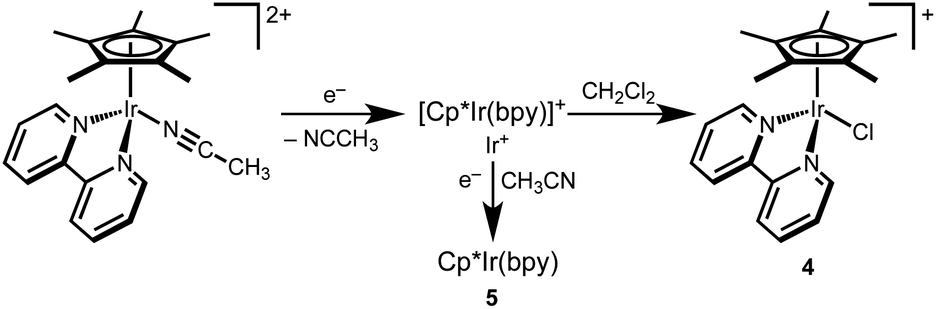 | ||
| Scheme 6 Proposed mechanism of electrochemically triggered formation of 4. | ||
A key difference between Paths D and E is that the radical chain pathway (D) requires a freely diffusing ·CH2Cl radical, whereas the HIT/HAT pathway (E) does not. Two key pieces of evidence are inconsistent with Path D. First, the reaction is not inhibited by hydroquinone (Fig. S29†), which has been reported to strongly inhibit radical chain hydrodehalogenation mediated by tin hydrides.53,61,62 Second, a freely diffusing ·CH2Cl radical would be expected to react with dichloromethane or acetonitrile solvent, but none of the products expected from radical reactivity—chloroform, isotopologues of dichloromethane and chloromethane, or chloromethyl complex 6—are observed. For comparison, ·CH3 free radical formation by photolysis of the iridium methyl complex [Cp*Ir(bpy)(CH3)]+ gave ethane via C–C radical coupling as well as H and D atom abstraction from solvents.63
The collected mechanistic data is most consistent with a photochemical hydrodechlorination via Path E. Scheme 7 depicts the sequence. Photoexcitation is followed by bimolecular self-quenching electron transfer and then rapid, sequential hydride ion transfer and hydrogen atom transfer to CH2Cl2. Key observations consistent with this mechanism are (a) the quantum yield that approaches (but does not exceed) the theoretical limit of 2; (b) the bimolecular self-quenching of the excited state indicated in iridium-dependent Stern–Volmer quenching studies; and (c) the highly selective reactivity without evident hallmarks of freely propagating radical chains. We speculate that the H–H bond formation reaction in the H2 evolution pathway occurs within the solvent cage, based on the highly exergonic and efficient H2 release step that follows self-quenching. By working in a substrate-rich reaction medium, part or all of the solvent cage itself becomes a reactive substrate, enabling PCET reactions between the reactive pair of metal hydrides and the surrounding CH2Cl2.
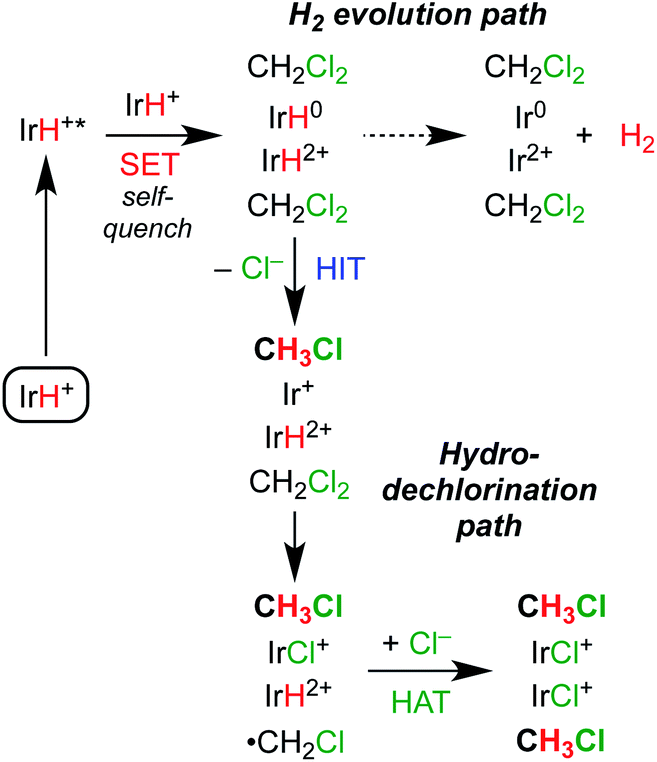 | ||
| Scheme 7 Proposed mechanisms of competing photochemical hydrodechlorination and H2 evolution. | ||
Conclusions
Tuning the reaction medium and conditions provides a method for changing the reactivity of an iridium hydride complex from H2 evolution towards hydride transfer. Photochemical reduction of dichloromethane to chloromethane is achieved with high quantum yield, even exceeding unity as the concentration of the hydride complex is increased. In neat dichloromethane solvent, H2 evolution—the dominant pathway in water and acetonitrile, even in the absence of acids—is completely suppressed. The high selectivity for hydride transfer is significant because most other transition metal hydride complexes produce only H2.Mechanistic studies of hydrodehalogenation are consistent with photochemical access to a triplet metal–ligand charge transfer excited state that undergoes self-quenching in a bimolecular reaction. The resulting reactive pair of oxidized and reduced organometallic hydrides is proposed to undergo sequential hydride ion transfer (HIT) and hydrogen atom transfer (HAT) PCET reactions with two dichloromethane molecules. Therefore, the quantum yield would have a theoretical maximum of two, because two equivalents of chloromethane could be produced per absorbed photon.
The highly efficient reduction is noteworthy because alkyl chlorides are traditionally challenging substrates in photoredox catalysis reactions that rely on SET pathways.7,8,64–66 The unusual reactivity with unactivated alkyl chlorides is attributed to the incorporation of a reactive metal–hydride bond in the photocatalyst, which engenders a long-lived charge-transfer excited state and enables a self-quenching pathway that effectively couples light absorption and bond-forming PCET steps.36,45
Although the net reaction is a hydride transfer from iridium to CH2Cl2, two different C–H bond-forming pathways are involved in the transformation. The possibility of direct excited state hydride transfer was considered, but despite the extremely potent thermodynamic hydricity predicted for the excited state of 1 (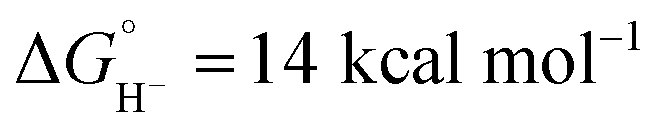 in CH3CN),44 the kinetic study shows that self-quenching electron transfer occurs preferentially. Then, hydride transfer occurs from the reduced hydride intermediate, Cp*Ir(bpy)H, with estimated
in CH3CN),44 the kinetic study shows that self-quenching electron transfer occurs preferentially. Then, hydride transfer occurs from the reduced hydride intermediate, Cp*Ir(bpy)H, with estimated 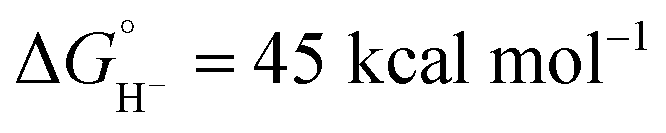 in CH3CN (see ESI Section V†). Although not as potent as the excited state hydride, neutral complex Cp*Ir(bpy)H is predicted to be almost 20 kcal mol−1 more hydridic than complex 1 (
in CH3CN (see ESI Section V†). Although not as potent as the excited state hydride, neutral complex Cp*Ir(bpy)H is predicted to be almost 20 kcal mol−1 more hydridic than complex 1 (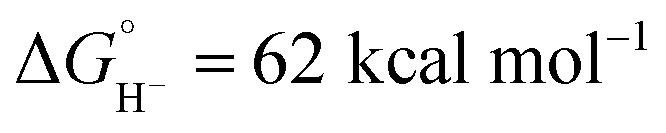 in CH3CN).44 The second C–H bond-forming pathway involves HAT, rather than hydride transfer.
in CH3CN).44 The second C–H bond-forming pathway involves HAT, rather than hydride transfer.
The present mechanistic study offers insight into future designs. The presence of reactive metal–hydride bonds is a key feature of this system, enabling rapid PCET reactivity immediately after excited state electron transfer. The step that controls the selectivity between H2 evolution and hydride transfer occurs directly after self-quenching. To maximize hydride transfer reactivity, a high concentration of substrate can be introduced to intercept the reactive hydride pair before H–H coupling can occur. Alternatively, the H–H coupling step could be slowed, for example by changing the structure to provide steric or geometric constraints. Improved understanding of metal hydride photochemical reactivity will open new avenues in photocatalytic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-SC0014255. S. M. B. acknowledges support from a NSF Graduate Research Fellowship (DGE-1144081). C. L. P. acknowledges the support of the Royster Society of Fellows. D. A. K. acknowledges the support of the Kenan Graduate Fellowship. Hydrogen gas detection was performed using a GC in the AMPED EFRC Instrumentation Facility established by the Alliance for Molecular PhotoElectrode Design for Solar Fuels, an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011.Notes and references
- Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Chichester, UK, 2017 Search PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- J. L. Dempsey, A. J. Esswein, D. R. Manke, J. Rosenthal, J. D. Soper and D. G. Nocera, Inorg. Chem., 2005, 44, 6879–6892 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. H. Shon and T. S. Teets, ACS Energy Lett., 2019, 4, 558–566 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends in Chemistry, 2019, 1, 111–125 CrossRef.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS.
- D. V Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- G. L. Geoffroy and M. S. Wrighton, Organometallic photochemistry, Academic Pr, 1979 Search PubMed.
- M. Wrighton, Chem. Rev., 1974, 74, 401–430 CrossRef CAS.
- C. E. Johnson, B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1983, 105, 7772–7774 CrossRef CAS.
- A. J. Kunin and R. Eisenberg, J. Am. Chem. Soc., 1986, 108, 535–536 CrossRef CAS PubMed.
- J. A. Maguire, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1989, 111, 7088–7093 CrossRef CAS.
- G. P. Rosini, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1994, 116, 9498–9505 CrossRef CAS.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS PubMed.
- S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647–651 CrossRef CAS PubMed.
- H.-Q. Do, S. Bachman, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 2162–2167 CrossRef CAS PubMed.
- Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed.
- J. M. Ahn, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 18101–18106 CrossRef CAS PubMed.
- C. D. Matier, J. Schwaben, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 17707–17710 CrossRef CAS PubMed.
- A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492–5496 CrossRef CAS PubMed.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS PubMed.
- K. R. Brereton, A. G. Bonn and A. J. M. Miller, ACS Energy Lett., 2018, 3, 1128–1136 CrossRef CAS.
- D. Sandrini, M. Maestri and R. Ziessel, Inorg. Chim. Acta, 1989, 163, 177–180 CrossRef CAS.
- J. C. Deaton, C. M. Taliaferro, C. L. Pitman, R. Czerwieniec, E. Jakubikova, A. J. M. Miller and F. N. Castellano, Inorg. Chem., 2018, 57, 15445–15461 CrossRef CAS PubMed.
- R. Ziessel, J. Chem. Soc., Chem. Commun., 1988, 16–17 RSC.
- R. Ziessel, Angew. Chem., 1991, 103, 863–866 CrossRef CAS.
- R. Ziessel, Angew. Chem., Int. Ed., 1991, 30, 844–847 CrossRef.
- K. J. Watson and R. Ziessel, Inorg. Chim. Acta, 1992, 197, 125–127 CrossRef CAS.
- R. Ziessel, J. Am. Chem. Soc., 1993, 115, 118–127 CrossRef CAS.
- C. L. Pitman and A. J. M. Miller, ACS Catal., 2014, 4, 2727–2733 CrossRef CAS.
- S. M. Barrett, S. A. Slattery and A. J. M. Miller, ACS Catal., 2015, 5, 6320–6327 CrossRef CAS.
- S. M. Barrett, C. L. Pitman, A. G. Walden and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14718–14721 CrossRef CAS PubMed.
- M. B. Chambers, D. A. Kurtz, C. L. Pitman, M. K. Brennaman and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 13509–13512 CrossRef CAS PubMed.
- A. J. M. Miller, D. M. Heinekey, J. M. Mayer and K. I. Goldberg, Angew. Chem., Int. Ed., 2013, 52, 3981–3984 CrossRef CAS PubMed.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed.
- W. B. Connick, D. Geiger and R. Eisenberg, Inorg. Chem., 1999, 38, 3264–3265 CrossRef CAS PubMed.
- W. B. Connick and H. B. Gray, J. Am. Chem. Soc., 1997, 119, 11620–11627 CrossRef CAS.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- T. Suenobu, D. M. Guldi, S. Ogo and S. Fukuzumi, Angew. Chem., Int. Ed., 2003, 42, 5492–5495 CrossRef CAS PubMed.
- S. Ogo, N. Makihara, Y. Kaneko and Y. Watanabe, Organometallics, 2001, 20, 4903–4910 CrossRef CAS.
- H. G. Kuivila, Acc. Chem. Res., 1968, 1, 299–305 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- J. V Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef.
- Y. Sun and C. Turro, Inorg. Chem., 2010, 49, 5025–5032 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS.
- M. Ladwig and W. Kaim, J. Organomet. Chem., 1992, 439, 79–90 CrossRef CAS.
- L. W. Menapace and H. G. Kuivila, J. Am. Chem. Soc., 1964, 86, 3047–3051 CrossRef CAS.
- Q.-Y. Chen and Z.-Y. Yang, J. Fluorine Chem., 1985, 28, 399–411 CrossRef CAS.
- C. L. Pitman and A. J. M. Miller, Organometallics, 2017, 36, 1906–1914 CrossRef CAS.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and thermochemical analysis. See DOI: 10.1039/d0sc00422g |
| This journal is © The Royal Society of Chemistry 2020 |