
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiomerically enriched tetrahydropyridine allyl chlorides†
Sedef
Karabiyikoglu
 a,
Alexandre V.
Brethomé
a,
Thomas
Palacin
a,
Robert S.
Paton
*b and
Stephen P.
Fletcher
*a
a,
Alexandre V.
Brethomé
a,
Thomas
Palacin
a,
Robert S.
Paton
*b and
Stephen P.
Fletcher
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stephen.fletcher@chem.ox.ac.uk
bDepartment of Chemistry, Colorado State University Fort Collins, Colorado 80523, USA. E-mail: robert.paton@colostate.edu
First published on 1st April 2020
Abstract
Enantiomerically enriched allyl halides are rare due to their configurational lability. Here we report stable piperidine-based allyl chloride enantiomers. These allyl chlorides can be produced via kinetic resolution, and undergo highly enantiospecific catalyst-free substitution reactions with C, N, O and S-based nucleophiles. DFT calculations and experiments with deuterium-labelled chloro-tetrahydropyridine, selectively prepared using H/D primary kinetic isotope effect, were used to investigate the mechanisms of resolution and substitution reactions. The allyl chlorides may also serve as valuable mechanistic tools for probing stereoselective reaction pathways.
Introduction
The stereoselective synthesis of molecules with chloro-substituted stereogenic centres is challenging. Despite considerable effort, few asymmetric chlorination methods have been reported.1–3 Synthesis of enantiomerically enriched allyl halides has proven to be especially difficult as they are generally prone to fast isomerization and racemization (Scheme 1(1)).4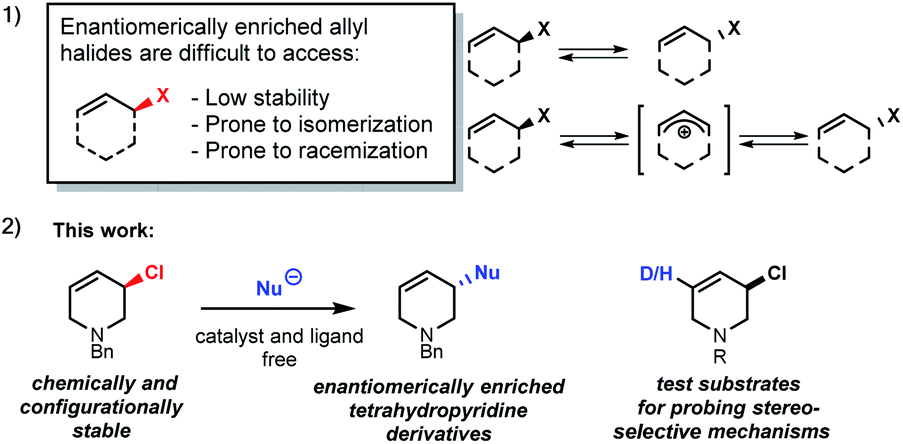 | ||
| Scheme 1 Properties of allyl halide enantiomers and this work. | ||
Enantioenriched allyl chlorides are virtually unknown, except as a feature of diastereomeric compounds, where other stereogenic elements exert control over the observed stable allyl halide configuration.2d,5 The selective preparation of enantiomerically enriched allyl chlorides5a could be important as stereogenic centres containing C–Cl bonds are found in pharmaceuticals,5b,6 and the allyl halides may undergo stereospecific reactions.1,6,7
Tetrahydropyridines (THPs) are a subgroup of nitrogen heterocycles found in biologically active molecules.8 THPs are also important precursors to functionalized piperidines. Piperidine is among the most common motifs found in licensed pharmaceuticals,8a and is frequently found in best-selling brand-name medicines.9,10 3-substituted piperidine derivatives are at the core of many potent therapeutic agents (Fig. 1),9,11,12 and methods for their preparation have attracted growing attention.13 However, asymmetric syntheses of 3-substituted piperidines via direct functionalization is scarce.14
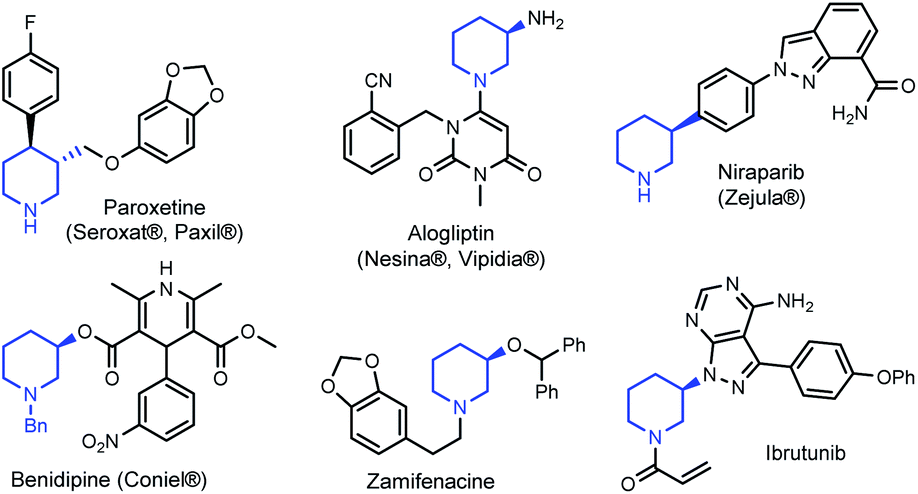 | ||
| Fig. 1 Examples of biologically active 3-substituted piperidines. | ||
Results and discussion
Due to the importance of THPs, racemic 3-chloro-1,2,3,6-tetrahydropyridines were examined in asymmetric allylic additions (AAAs) with Zr-nucleophiles and copper catalysts. To our surprise, we were able to isolate enantiomerically enriched allyl chlorides from the reaction mixture. The kinetic resolution of halides has received little attention,2e,15 with resolution of allyl halides being limited to a single report of activated allyl fluorides.16 Here we report the synthesis of enantioenriched allylic 3-chloro-THPs via kinetic resolution and investigate alternative preparation methods. The chemically and configurationally stable allyl chlorides can be used to prepare a wide range of THP derivatives that may be useful in synthesis and medicinal chemistry (Scheme 1(2)).Initial exploration17 of Cu-catalyzed AAA with chloro-tetrahydropyridines showed recovery of scalemic allyl chloride, indicating slow (or indeed no) interconversion between starting material enantiomers during the reaction. The reaction of 1-benzyl-3-chloro-1,2,3,6-tetrahydropyridine (rac-1) with 4-phenyl-1-butene (2) was followed in time (Scheme 2). After 30 minutes at −10 °C the reaction reached 16% conversion, giving product 3a in 94% ee, with the ee of 3a then decreasing ∼15% due to slow consumption of the less reactive starting enantiomer over time. The ee of 1 increased from 0 to 80% over 22 hours, where it remained unchanged (within experimental error).17 Allyl chlorides bearing different protecting groups (Boc, Cbz, Ts and Ms) were tested in Cu-catalyzed AAAs with 4-phenyl-1-butene,17 however; these reactions were low yielding and not stereoselective.
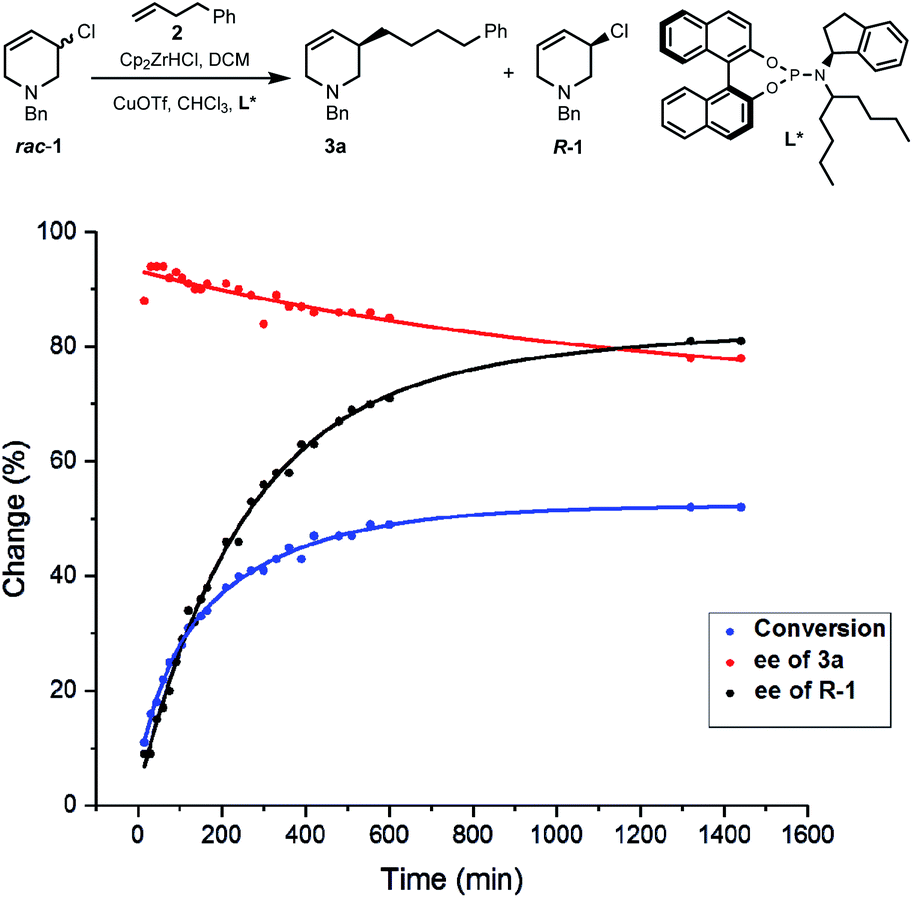 | ||
| Scheme 2 Kinetic resolution of rac-1 using a copper–ligand catalyst and an alkylzirconium reagent. | ||
We then investigated conditions to optimize the ee of 1.17 With styrene, diluting the reaction conditions, and using DCM as solvent, R-1 was obtained in useful yield with excellent selectivity (Scheme 3(1a)). Kinetic resolution was found to be much faster on larger scales (Scheme 3(1b and c)). These reactions also afforded alkylation product 3b in 49–65% yield and 84–88% ee. The mass balance in these reactions is good in that neither the small nor large scale reactions of rac-1 with styrene or 2 gave detectable amounts of side products. Conveniently, scalemic allylic chloride samples can be recycled, with resolution on such material giving up to 99% ee using lower equiv. of alkyl-Zr reagent (Scheme 3(2)).
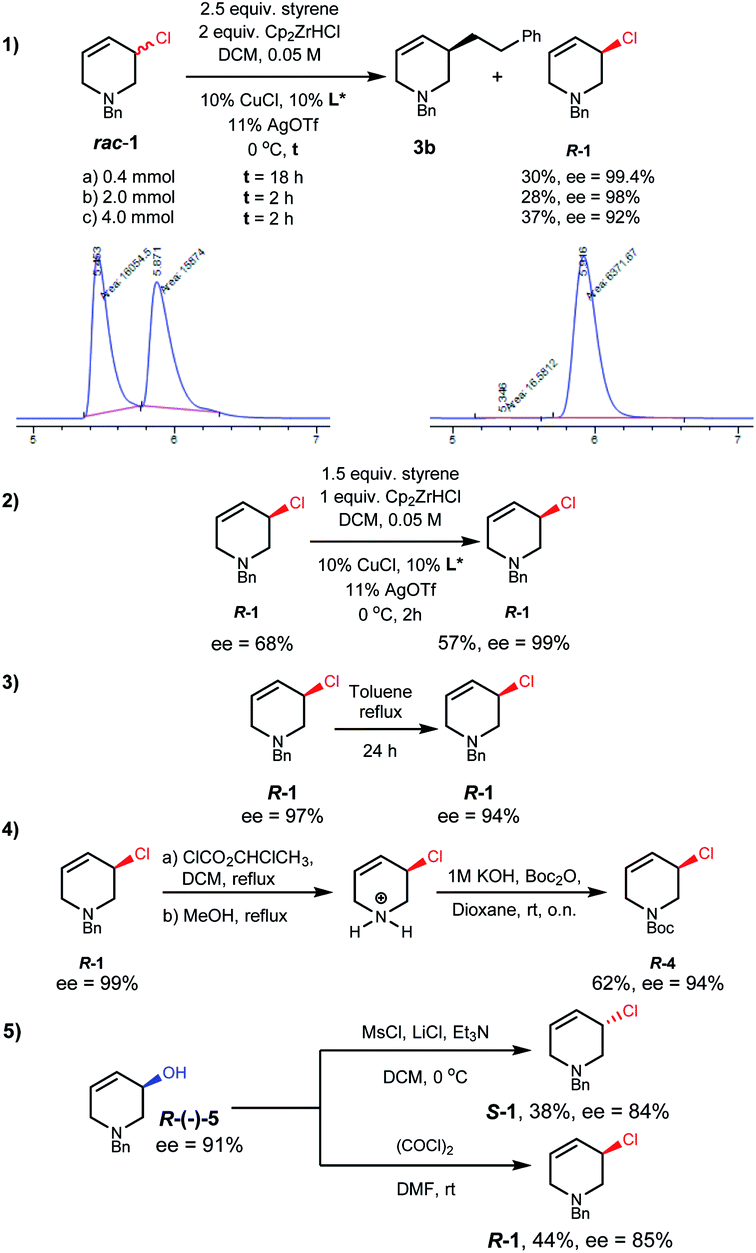 | ||
| Scheme 3 (1) Kinetic resolution of rac-1 and HPLC traces of racemic and enantiomerically enriched 1 using a chiral non-racemic stationary phase. (2) Kinetic resolution of scalemic 1. (3) Configurational stability of R-1 to heating. (4) Removal and addition of a protecting group to give N-Boc protected R-1 with little loss of enantiomeric excess (5) alternative synthesis of R- and S-1 from an enantiomerically enriched alcohol R-5via selective inversion (to give S-1) or selective retention (to give R-1). | ||
R-1 shows remarkable thermal stability; in toluene heated to reflux for 24 hours only 3% ee erosion was observed without any detectable side product formation (Scheme 3(3)). If desired the N-protecting group of R-1 can be exchanged, for example to N-Boc, through a pyridinium salt intermediate with only a small decrease in ee (Scheme 3(4)).17
Alternative routes to enantioenriched 1 were tested (see ESI†). Enantiomerically enriched (Scheme 3(5), 91% ee) alcohol R-5, after optimization,17 could be chlorinated with either inversion or retention, and reasonable stereospecificity, to give either enantiomer of 1.
Though thermally stable, R-1 is a versatile chiral non-racemic building block and undergoes a variety of highly stereospecific substitution reactions. Using mild conditions we were able to access a variety of THP derivatives including ethers (6), esters (7), vinylogous ethers (8), thioethers (9), malonates (10), fluorides (11) and amines (13) with very high stereospecificity (94–99% es, Scheme 4).
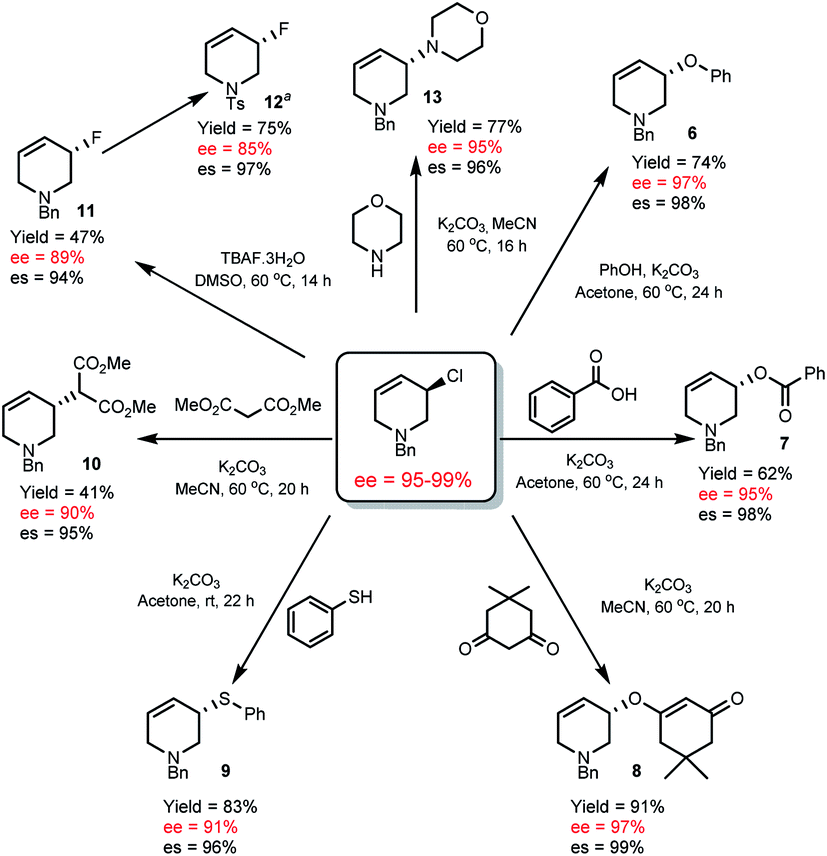 | ||
| Scheme 4 Enantiospecific substitution reactions of R-1. es = % enantiospecificity; es = [(ee of product/ee of R-1) × 100]. aThe absolute configuration of 12 is known, see ESI† for the conditions to prepare 12. | ||
The absolute configuration of R-1 was assigned17 by converting allyl fluoride 11 to 12 (Scheme 4), which has been determined by X-ray crystallographic analysis,18 and knowledge (vida infra) that R-1 to 11 occurs via an SN2 pathway.
To clarify the mechanism of stereospecific substitution we prepared isotopically labelled rac-1-d. Reduction of benzylpyridinium bromide in CD3OD selectively adds D to the C3 position of 14-d, which was epoxidized to 15-d. We used the primary kinetic isotope effect of H/D deprotonation19 as a strategy to prepare deuterium-labelled allyl alcohol 5-d. This sequence gave a 2.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D/H ratio at C5 after optimization.17Rac-1-d was obtained with 61–65% D saturation as judged by 1H and 2H NMR spectroscopic experiments after chlorination (Scheme 5(1)).20Acyclic allyl chlorides generally undergo SN2' substitutions as the carbon with the leaving group can freely rotate about the vicinal olefin,21 but mechanistic studies with cyclic allyl chlorides suggests SN2 substitutions are generally favoured followed by anti-SN2' and syn-SN2' pathways.4e–h,22
:1 D/H ratio at C5 after optimization.17Rac-1-d was obtained with 61–65% D saturation as judged by 1H and 2H NMR spectroscopic experiments after chlorination (Scheme 5(1)).20Acyclic allyl chlorides generally undergo SN2' substitutions as the carbon with the leaving group can freely rotate about the vicinal olefin,21 but mechanistic studies with cyclic allyl chlorides suggests SN2 substitutions are generally favoured followed by anti-SN2' and syn-SN2' pathways.4e–h,22
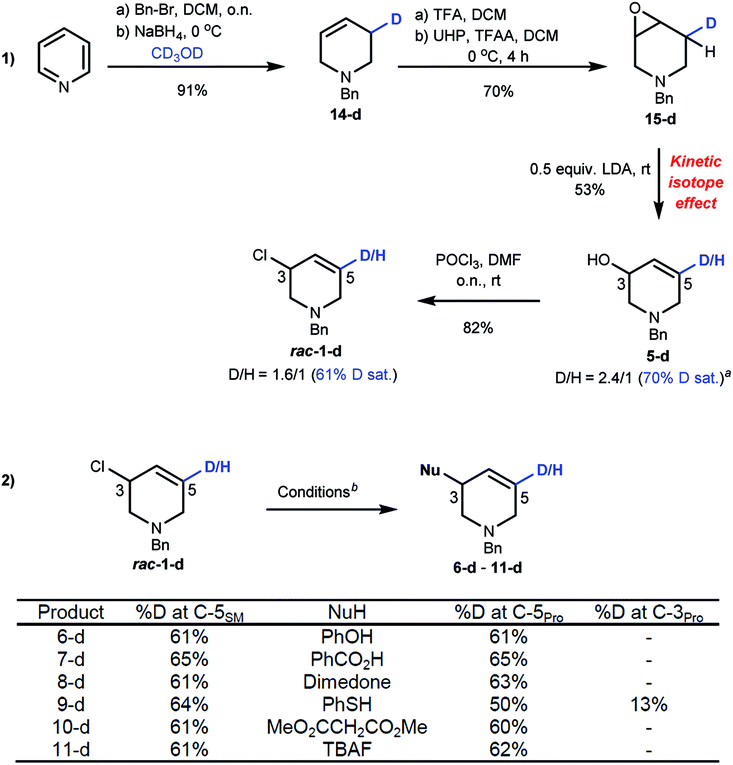 | ||
| Scheme 5 (1) Preparation of rac-1-d. a%D = [n5-d/(n5 + n5-d)] × 100. (2) Mechanistic studies on substitution reactions. bConditions for substitution reactions are the same as shown in Scheme 4. | ||
Our results (Scheme 5(2)) show that the %D saturation at C5 of rac-1-d is usually conserved throughout these substitutions, strongly suggesting SN2 reactions. An exception to this trend is seen with thioether 9-d which shows some D transfer to C3. As the es of R-1 to 9 is high (96% es), it suggests that the reaction occurs by a mixture of pathways, in favour of SN2, with both SN2 and syn-SN2' leading to the same enantiomer. Density Functional Theory (DFT) calculations excluded transannular aziridinium ion formation due to the large free energy difference between an azabicyclic intermediate and 1 (37.8 kcal mol−1).17 Such levels of energy necessarily imply unfavourable transition structures to access it (TS ≥ 37.8 kcal mol−1), thus eliminating the possibilities of an intra-nucleophilic reaction pathway with double inversion.
DFT studies were also used to probe the kinetic resolution of rac-1 (Scheme 6(1)). Geometry optimisations were carried out with M062X functional and the 6-31G(d) basis set for C, H, N, O, P, Cl atoms and the LANL2DZ effective core potential/valence double zeta basis set for Cu. Single point energy corrections were obtained at the M062X/def2-TZVPP level of theory with chloroform solvation described by an implicit Solvation Model based on Density (SMD), then corrected using D3-dispersion energy as developed by Grimme.17
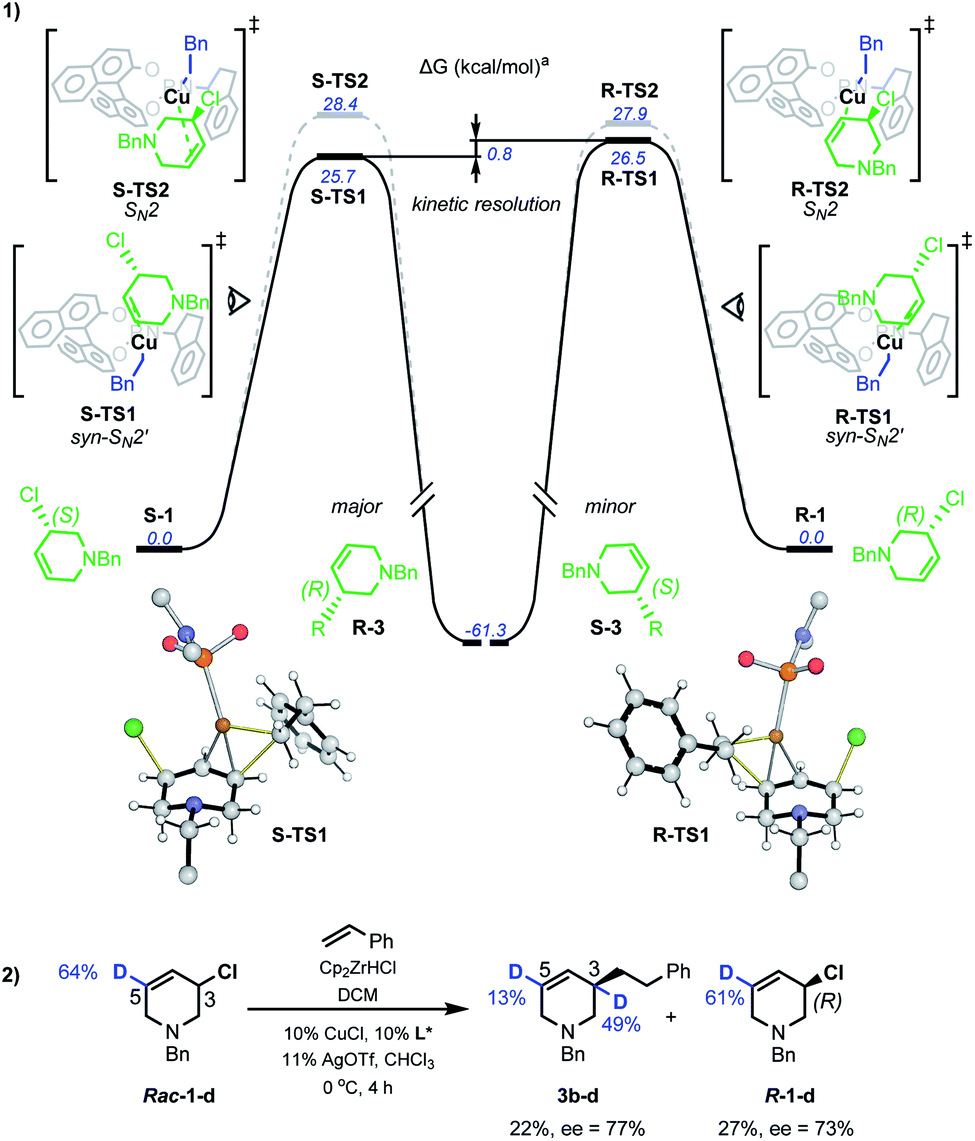 | ||
| Scheme 6 Mechanistic study of Cu-catalysed AAAs. aSMD-M062X/def2-TZVPP//M062X/6-31G(d)/LANL2DZ computed Gibbs energies at 298.15 K and 1 mol L−1. | ||
Starting from a Cu–L* complex in which the ligand n-alkyl groups were abbreviated in computational models,17rac-1 can bind to the metal centre at the N lone-pair. Two diastereomeric complexes can form with similar stabilities: of which S-C1 (0.8 kcal mol−1) is slightly less favourable than R-C1 (−0.2 kcal mol−1). Such a qualitative difference could potentially suggest a role in stereoselectivity and explain the specificity observed with N-benzyl-protected allyl chlorides.17 For displacement of chloride by the catalyst, five possible pathways were investigated: syn- and anti- oxidative addition, anti-SN2', SN2 and syn-SN2'. Alternative conformations were considered for each possibility.17 The syn-SN2' transition structure (TS) S-TS1 was the most favourable overall, proceeding from the reaction of S-1 with a barrier of 25.7 kcal mol−1 to give the (R)-product of alkylation. For the (S)-enantiomer, this was followed by the SN2 TS S-TS2 (28.4 kcal mol−1). Copper-catalyzed allylic alkylations are often described as occurring through complexation of a Cu(I) complex to the allylic olefin followed by oxidative addition to generate an allyl–Cu(III)23 complex, but here anti-SN2', syn- and anti-oxidative addition were found to be comparatively unfavourable, except with the anti-SN2'-oxidative addition pathway (S-TS3) that is only 0.1 kcal mol−1 higher than S-TS2.17 Our computational model is nevertheless qualitatively correct to disfavour S-TS3 over S-TS2 as it would otherwise give the opposite enantiomer than observed experimentally. Similarly to S-1, R-1 was found to preferentially react via a syn-SN2' pathway, with the barrier for R-TS1 at 26.5 kcal mol−1. As found for S-1, the next most stable was SN2 TS R-TS2 at 27.9 kcal mol−1. Based on these computations, the alkylation of R-1 is kinetically disfavoured vs.S-1 (ΔΔGsyn-SN‡2' = 0.8 kcal mol−1) via dominant syn-SN2' pathways for both enantiomers. This is consistent with the absolute sense of enantioenrichment observed experimentally, in which the (R)-enantiomer of both substrate and product accumulates. In addition to the major syn-SN2' pathway, our result possibly implicate involvement of a minor SN2 mechanism to give the same product stereochemistry.
D-labelled rac-1-d was subjected to Cu-catalyzed resolution (Scheme 6(2)), and in accordance with computation, both SN2' and SN2 pathways are operative: product 3b–d shows D-incorporation at the C3 (49%) and C5 (13%) positions consistent with syn-SN2' and then SN2 being the most favourable pathways. No deuterium-isomerization of starting 1-d was observed and R-1-d was isolated from the reaction mixture with 73% ee.
Conclusions
We have found allyl chlorides that are thermodynamically stable and can be prepared in highly enantioenriched form. A rare kinetic resolution of allylic chlorides formed piperidine-based allyl chlorides with high ee. The allyl chloride enantiomers can be separated by chromatography using a chiral non-racemic stationary phase, or prepared by stereospecific (with retention or inversion) chlorination of the corresponding alcohol. The allyl chloride undergoes highly enantiospecific substitution reactions with nucleophiles to give a wide range of tetrahydropyridine products which are important in biology and medicine. Experiments with D-labelled chloro-tetrahydropyridine and DFT calculations were used to investigate the mechanistic pathways of nucleophilic substitution and kinetic resolution reactions. This work provides new routes for the formation of enantiomerically enriched tetrahydropyridine derivatives. Further, rac-1, R- or S-1, R-1-d and other configurationally stable allyl halides may serve as probe substrates for investigating reaction pathways in mechanistic studies of reactions.24Conflicts of interest
Oxford University Innovation has filed a patent application (GB1815018.5) with S. P. F. and S. K. named as inventors. The remaining authors declare no competing financial interests. A version of this manuscript has been shared and posted on ChemRxiv.Acknowledgements
Financial support from the UK Engineering and Physical Sciences Research Council (EP/N022246/1) is gratefully acknowledged. A. V. B. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. T. P. thanks the People Programme (Marie Curie Actions) of the EU's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement 316955 for funding. The authors thank Dr Nader Amin for assistance in D-NMR. We acknowledge the RMACC Summit supercomputer, which is supported by the National Science foundation (ACI-1532235 and ACI1532236), the University of Colorado Boulder and Colorado State University, and the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation TGCHE180056.Notes and references
- For asymmetric α-chlorination of carbonyl compounds, see: (a) Y. H. Luo, Y. J. Ping, Z. R. Li, X. Gu, Z. J. Xu and C. M. Che, Synthesis, 2018, 50, 1105 CrossRef CAS; (b) X. Bao, S. Wei, L. Zou, Y. He, F. Xue, J. Qu and B. Wang, Chem. Commun., 2016, 52, 11426 RSC; (c) T. Sakai, S. Hirashima, K. Nakashima, C. Maeda, A. Yoshida, Y. Koseki and T. Miura, Chem. Pharm. Bull., 2016, 64, 1781 CrossRef CAS PubMed; (d) W. Zheng, Z. Zhang, M. J. Kaplan and J. C. Antilla, J. Am. Chem. Soc., 2011, 133, 3339 CrossRef CAS PubMed; (e) K. Shibatomi and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 5796 CrossRef CAS PubMed; (f) S. France, H. Wack, A. Taggi, A. M. Hafez, T. R. Wagerle, M. H. Shah, C. L. Dusich and T. Lectka, J. Am. Chem. Soc., 2004, 126, 4245 CrossRef CAS PubMed.
- For asymmetric chlorination of olefins, see: (a) U. Hennecke, Chem.–Asian J., 2012, 7, 456 CrossRef CAS PubMed; (b) A. Castellanos and S. P. Fletcher, Chem.–Eur. J., 2011, 17, 5766 CrossRef CAS PubMed; (c) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134 CrossRef CAS PubMed; (d) S. A. Snyder, Z. Y. Tang and R. Gupta, J. Am. Chem. Soc., 2009, 131, 5744 CrossRef CAS PubMed; (e) Y. Tan, S. Luo, D. Li, N. Zhang, S. Jia, Y. Liu, W. Qin, C. E. Song and H. Yan, J. Am. Chem. Soc., 2017, 139, 6431 CrossRef CAS PubMed; (f) M. L. Landry, D. X. Hu, G. M. McKenna and N. Z. Burns, J. Am. Chem. Soc., 2016, 138, 5150 CrossRef CAS PubMed.
- For asymmetric aziridine ring openings with chloride salts, see: K. Ohmatsu, Y. Hamajima and T. Ooi, J. Am. Chem. Soc., 2012, 134, 8794 CrossRef CAS PubMed.
- (a) X. Y. Cui, Y. Ge, S. M. Tan, H. Jiang, D. Tan, Y. Lu, R. Lee and C. H. Tan, J. Am. Chem. Soc., 2018, 140, 8448 CrossRef CAS PubMed; (b) J. Scoccia, S. J. Perez, V. Sinka, D. A. Cruz, J. M. Lopez-Soria, I. Fernandez, V. S. Martin, P. O. Miranda and J. I. Padron, Org. Lett., 2017, 19, 4834 CrossRef CAS PubMed; (c) E. Rideau, H. You, M. Sidera, T. D. W. Claridge and S. P. Fletcher, J. Am. Chem. Soc., 2017, 139, 5614 CrossRef CAS PubMed; (d) C. Li, Y. Zhang, Q. Sun, T. Gu, H. Peng and W. Tang, J. Am. Chem. Soc., 2016, 138, 10774 CrossRef CAS PubMed; (e) U. K. Wefelscheid and S. Woodward, J. Org. Chem., 2009, 74, 2254 CrossRef CAS PubMed; (f) E. W. Collington and A. I. Meyers, J. Org. Chem., 1971, 36, 3044 CrossRef; (g) H. L. Goering, T. D. Nevitt and E. F. Silversmith, J. Am. Chem. Soc., 1955, 77, 4042 CrossRef CAS; (h) W. G. Young, S. Winstein and H. L. Goering, J. Am. Chem. Soc., 1951, 73, 1958 CrossRef CAS.
- (a) For the single example only applied on 1H-indine, see: A. Lennartson, S. Olsson, J. Sundberg and M. Hakansson, Angew. Chem., Int. Ed., 2009, 48, 3137 CrossRef CAS PubMed; (b) F. Zhong, W. J. Yue, H. J. Zhang, C. Y. Zhang and L. Yin, J. Am. Chem. Soc., 2018, 140, 15170 CrossRef CAS PubMed; (c) C. V. Vogel, H. Pietraszkiewicz, O. M. Sabry, W. H. Gerwick, F. A. Valeriote and C. D. Vanderwal, Angew. Chem., Int. Ed., 2014, 53, 12205 CrossRef CAS PubMed.
- (a) J. R. Reyes and V. H. Rawal, Angew. Chem., Int. Ed., 2016, 55, 3077 CrossRef CAS PubMed; (b) D. X. Hu, F. J. Seidl, C. Bucher and N. Z. Burns, J. Am. Chem. Soc., 2015, 137, 3795 CrossRef CAS PubMed; (c) M. Oestreich, Angew. Chem., Int. Ed., 2005, 44, 2324 CrossRef CAS PubMed; (d) H. Ibrahim and A. Togni, Chem. Commun., 2004, 1147 RSC.
- (a) M. Bergeron-Brlek, T. Teoh and R. Britton, Org. Lett., 2013, 15, 3554 CrossRef CAS PubMed; (b) R. Britton and B. Kang, Nat. Prod. Rep., 2013, 30, 227 RSC.
- (a) Y. Dudognon, J. Rodriguez, T. Constantieux and X. Bugaut, Eur. J. Org. Chem., 2018, 2432 CrossRef CAS; (b) C. C. Chrovian, A. Soyode-Johnson, A. A. Peterson, C. F. Gelin, X. Deng, C. A. Dvorak, N. I. Carruthers, B. Lord, I. Fraser, L. Aluisio, K. J. Coe, B. Scott, T. Koudriakova, F. Schoetens, K. Sepassi, D. J. Gallacher, A. Bhattacharya and M. A. Letavic, J. Med. Chem., 2018, 61, 207 CrossRef CAS PubMed; (c) C. Allais and W. R. Roush, Org. Lett., 2017, 19, 2646 CrossRef CAS PubMed; (d) R. Watanabe, H. Mizoguchi, H. Oikawa, H. Ohashi, K. Watashi and H. Oguri, Bioorg. Med. Chem., 2017, 25, 2851 CrossRef CAS PubMed; (e) R. Aeluri, R. J. Ganji, A. K. Marapaka, V. Pillalamarri, M. Alla and A. Addlagatta, Eur. J. Med. Chem., 2015, 106, 26 CrossRef CAS PubMed; (f) T. Mesganaw and J. A. Ellman, Org. Process Res. Dev., 2014, 18, 1097 CrossRef CAS PubMed; (g) N. N. Mateeva, L. L. Winfield and K. K. Redda, Curr. Med. Chem., 2005, 12, 551 CAS.
- S. B. D. Jarvis and A. B. Charette, Org. Lett., 2011, 13, 3830 CrossRef CAS PubMed.
- ClinCalc: Clinical Tools and Calculators for Medical Professionals, http://clincalc.com/DrugStats/Top200Drugs.aspx, Oct 16, 2018 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) A. Akin, M. T. Barrila, T. A. Brandt, A. M. R. Dechert-Schmitt, P. Dube, D. D. Ford, A. S. Kamlet, C. Limberakis, A. Pearsall, D. W. Piotrowski, B. Quinn, S. Rothstein, J. Salan, L. Wei and J. Xiao, Org. Process Res. Dev., 2017, 21, 1990 CrossRef CAS; (b) P. Jones, S. Altamura, J. Boueres, F. Ferrigno, M. Fonsi, C. Giomini, S. Lamartina, E. Monteagudo, J. M. Ontoria, M. V. Orsale, M. C. Palumbi, S. Pesci, R. G. Roscilli, R. Scarpelli, C. Schultz-Fademrecht, C. Toniatti and M. Rowley, J. Med. Chem., 2009, 52, 7170 CrossRef CAS PubMed; (c) J. Feng, Z. Zhang, M. B. Wallace, J. A. Stafford, S. M. Kaldor, D. B. Kassel, M. Navre, L. Shi, R. J. Skene, T. Asakawa, K. Takeuchi, R. Xu, D. R. Webb and S. L. Gwaltney, J. Med. Chem., 2007, 50, 2297 CrossRef CAS PubMed; (d) J. G. Varnes, D. S. Gardner, J. B. Santella, J. V. Duncia, M. Estrella, P. S. Watson, C. M. Clark, S. S. Ko, P. Welch, M. Covington, N. Stowell, E. Wadman, P. Davies, K. Solomon, R. C. Newton, G. L. Trainor, C. P. Decicco and D. A. Wacker, Bioorg. Med. Chem. Lett., 2004, 14, 1645 CrossRef CAS PubMed; (e) J. Cossy, C. Dumas and D. M. Pardo, Bioorg. Med. Chem. Lett., 1997, 7, 1343 CrossRef CAS; (f) E. N. Petersen, E. Bechgaard, R. J. Sortwell and L. Wetterberg, Eur. J. Pharmacol., 1978, 52, 115 CrossRef CAS PubMed.
- For selected examples of synthesis of 3-substituted THPs via cycloadditions, see: ref. 1f and (a) Z. Wang, H. Xu, Q. Su, P. Hu, P. L. Shao, Y. He and Y. Lu, Org. Lett., 2017, 19, 3111 CrossRef CAS PubMed; (b) H. Wang, W. Zhou, M. Tao, A. Hu and J. Zhang, Org. Lett., 2017, 19, 1710 CrossRef CAS PubMed; (c) S. Yang, K. H. Rui, X. Y. Tang, Q. Xu and M. Shi, J. Am. Chem. Soc., 2017, 139, 5957 CrossRef CAS PubMed Via intramolecular coupling, see: (d) L. Hou, Y. Yuan and X. Tong, Org. Biomol. Chem., 2017, 15, 4803 RSC . Via aziridinium ring expention, see: ref. 11.
- (a) P. Schäfer, T. Palacin, M. Sidera and S. P. Fletcher, Nat. Commun., 2017, 8, 15762 CrossRef PubMed; (b) K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338 CrossRef CAS PubMed; (c) Y. Uozumi, Pure Appl. Chem., 2007, 79, 1481 CAS; (d) D. A. Evans, K. R. Campos, J. S. Tedrow, F. E. Michael and M. R. Gagne, J. Am. Chem. Soc., 2000, 122, 7905 CrossRef CAS.
- (a) K. Shibatomi, T. Okimi, Y. Abe, A. Narayama, N. Nakamura and S. Iwasa, Beilstein J. Org. Chem., 2014, 10, 323 CrossRef PubMed; (b) W. Chung, J. S. Carlson and C. D. Vanderwal, J. Org. Chem., 2014, 79, 2226 CrossRef CAS PubMed.
- T. Nishimine, K. Fukushi, N. Shibata, H. Taira, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2014, 53, 517 CrossRef CAS PubMed.
- For full details and supporting references see ESI.†.
- M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402 CrossRef CAS PubMed.
- (a) M. E. Wood, S. Bissiriou, C. Lowe, A. M. Norrish, K. Senechal, K. M. Windeatt, S. J. Coles and M. B. Hursthous, Org. Biomol. Chem., 2010, 8, 4653 RSC; (b) J. Clayden, J. H. Pink, N. Westlund and F. X. Wilson, Tetrahedron Lett., 1998, 39, 8377 CrossRef CAS; (c) D. Hoppe, M. Paetow and F. Hintze, Angew. Chem., Int. Ed. Engl., 1993, 32, 394 CrossRef.
- Deuterium erosion in the chlorination step, may be due to proto de-deuteration under acidic conditions and/or relocation of D by SN2' transformations. However, the latter pathway likely contributes only at minute levels as D atoms at C3 position of rac-1-d could only be detected by sensitive 2H-NMR spectrscopic analyses. See ESI.†.
- (a) K. N. Houk, M. N. Paddon-Row and N. G. Rondan, J. Mol. Struct., 1983, 103, 197 CrossRef; (b) R. M. Magid and O. S. Fruchey, J. Am. Chem. Soc., 1977, 8368 CrossRef CAS.
- B. L. Kormos and C. J. Cramer, J. Org. Chem., 2003, 68, 6375 CrossRef CAS PubMed.
- (a) J. B. Langlois, D. Emery, J. Mareda and A. Alexakis, Chem. Sci., 2012, 3, 1062 RSC; (b) A. Alexakis, J. E. Backvall, N. Krause, O. Pamies and M. Dieguez, Chem. Rev., 2008, 108, 2796 CrossRef CAS PubMed.
- For use of these molecules in elucidating the mechanism of Rh-catalyzed asymmetric additions see: L. Dijk, R. Ardkhean, M. Sidera, S. Karabiyikoglu, O. Sari, T. D. W. Claridge, R. Paton and S. P. Fletcher, ChemRxiv, 2019 DOI:10.26434/chemrxiv.8208617.v1.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and spectral data. See DOI: 10.1039/d0sc00377h |
| This journal is © The Royal Society of Chemistry 2020 |