
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing amidoxime-modified porous adsorbents with open architecture for cost-effective and efficient uranium extraction†
Zhangnan
Li
,
Qinghao
Meng
,
Yajie
Yang
,
Xiaoqin
Zou
 ,
Ye
Yuan
* and
Guangshan
Zhu
*
,
Ye
Yuan
* and
Guangshan
Zhu
*
Key Laboratory of Polyoxometalate Science of Ministry of Education, Northeast Normal University, Changchun 130012, China. E-mail: yuany101@nenu.edu.cn; zhugs@nenu.edu.cn
First published on 14th April 2020
Abstract
The dense structure of polymeric matrices exposes only 10–20% of adsorption (amidoxime) groups, thus detracting from the extraction efficiency of uranium from seawater. Herein, the amidoxime-modified building units were cross-linked via the Scholl reaction into porous aromatic frameworks (PAFs). Due to the formation of open architecture, PAF adsorbents reveal a larger utilization ratio (>60%) of amidoxime groups. Consequently, PAF samples enable an ultrahigh uranium capacity of 702 mg g−1, which creates a 16-fold capacity enhancement and gains a 7-fold adsorption rate improvement compared with polymer-based adsorbents. Notably, PAF solids are able to be integrated into various devices, thus realizing versatile and efficacious uranium extraction from real seawater (meeting the commercial standard ∼6 mg g−1 in 21 days). In addition, the final cost using our PAF-based adsorbent is US $189.77 per kg uranium, it is in accordance with the prevailing market cost ($100–335 per kg).
Introduction
Nuclear energy, as a clean, safe, and economical energy source, plays a vital role in ensuring the global energy supply and attenuating the requirement of fossil fuel.1,2 The element uranium is the principal ingredient for the development of the nuclear industry; however, the terrestrial reserves will be depleted in several decades due to the worldwide consumption.3 The largest uranium resource (4.5 billion tons) is found in the oceans; these deposits are approximately one-thousand times greater than the available land resources, which suggests an inexhaustible supply for nuclear power generation.4,5 Unfortunately, the ultralow concentration of UO22+ species (∼3.3 ppb) in addition to the presence of bulky interfering ions strongly reduces the extraction efficiency of uranium from real seawater.6The amidoxime group is widely considered to be the most promising group for uranyl ion capture because of its strong affinity and considerable selectivity. In the past several decades, some classical adsorbents, such as amidoxime/carboxyl polypropylene,7 amidoxime-modified polyethylene fibres,8 natural ore materials halloysite nanotubes,9 porous amidoxime fibers,10 and polyamidoxime fibres,11 have been developed based on polymeric solids. The soft nature of polymers leads to the dense structure, and only the active sites exposed on the exterior surfaces of the particles function effectively,12 thereby greatly impacting extraction efficiency (Scheme 1). Accordingly, some rigid fragments are introduced into the skeleton to obtain the open frameworks for improving the availability factors of amidoxime groups13–16 such as MNP-b-AMD-2 (60 mg g−1),17 POP-pNH2-AO (530 mg g−1),18 and PPN-6-PAN (65.2 mg g−1) (Scheme 1b).19 However, limited adsorption efficiency and the high preparation cost make industrialized mass production impossible. The development of a new strategy to construct commercially viable porous adsorbents with open architecture is urgently needed to realize amenability towards industrial-scale production, ease of casting mould, and low cost.
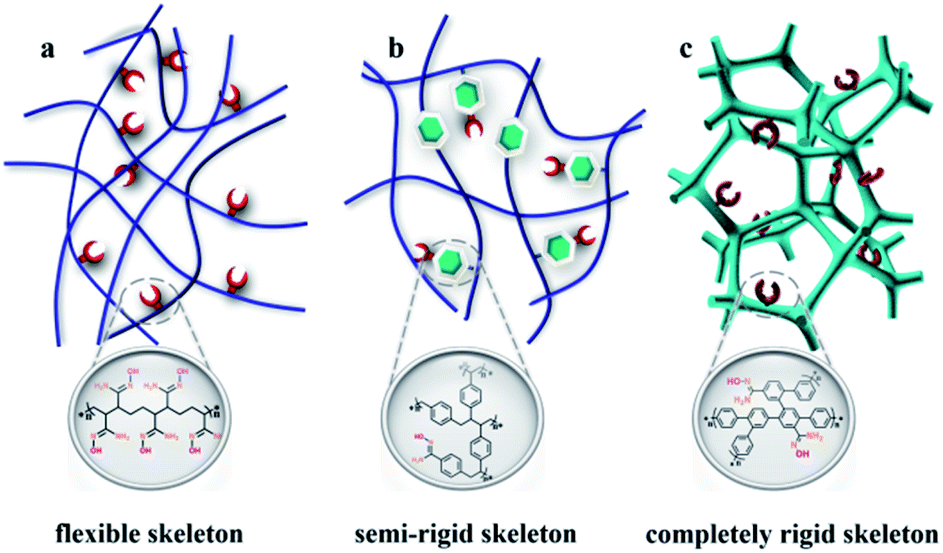 | ||
| Scheme 1 Schematic representation of amidoxime functionalized adsorbents including a (a) flexible skeleton, (b) semi-rigid skeleton, and (c) completely rigid skeleton. | ||
Herein, a cost-effective approach is utilized to synthesize the industrialized porous adsorbents by crosslinking low-cost cyanophenyl into the porous architecture through an AlCl3 catalysed Scholl reaction.20 After the post-treatment with NH2OH·HCl in an alkaline environment, the cyano groups were converted to the amidoxime units in the frameworks. Since the open architecture provided a large number of accessible adsorption sites, the PAF materials revealed an ultrahigh uranium capacity (702 mg g−1), which is ∼16 times higher than that of amidoxime-modified polymers/fibres. For industrial considerations, the resulting material could be integrated into an electrospun fibre membrane and filter screen for practical uranium adsorption from seawater.
Results and discussion
First, biphenyl and cyanophenyl were used to obtain the optimal proportion for the maximum uptake of uranium by considering a range of molar ratios (cyanophenyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biphenyl), including 1:4; 1:2; 1:1; 2:1; and 4:1, which generate the PAFs 170-1, 170-2, 170-3, 170-4, and 170-5, respectively. Then, the PAF-170 series was treated with triethylamine and hydroxylamine hydrochloride to accomplish the hydrogenation of –CN groups into amidoxime groups (specific methods in Fig. S2†). As studied by Fourier transform infrared spectroscopy (FT-IR), the –CN band disappeared at approximately 2225 cm−1 for the PAF-170 series. In addition, the emergence of N–H (3451 and 3364 cm−1), C
:biphenyl), including 1:4; 1:2; 1:1; 2:1; and 4:1, which generate the PAFs 170-1, 170-2, 170-3, 170-4, and 170-5, respectively. Then, the PAF-170 series was treated with triethylamine and hydroxylamine hydrochloride to accomplish the hydrogenation of –CN groups into amidoxime groups (specific methods in Fig. S2†). As studied by Fourier transform infrared spectroscopy (FT-IR), the –CN band disappeared at approximately 2225 cm−1 for the PAF-170 series. In addition, the emergence of N–H (3451 and 3364 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1652 cm−1), C–N (1390 cm−1) and N–O (930 cm−1) characteristic peaks for PAF-170-AO series confirmed the conversion of cyano groups into amidoxime groups (Fig. S4†). The porosity for PAF-170-AO series was measured at 77 K through N2 adsorption experiments. As shown in Fig. S5 and S6,† the Brunauer–Emmett–Teller (BET) surface areas decreased gradually from 478 (PAF-170-AO-1) to 126 m2 g−1 (PAF-170-AO-5), as the amidoxime group doping ratios increased. Nonlocal density functional theory (NL-DFT) calculations demonstrated that all amidoxime-modified PAFs possessed a wide pore size distribution in the range of 0.4–4 nm that favoured UO22+ ion (uranyl ion with an irregular shape possesses a maximum length of 6.04–6.84 Å) access into the internal space of particles (Fig. S6 and Table S1†).9 From the PXRD spectra, the PAF-170 series depicted amorphous structures (Fig. S8†). All PAF powders are aggregated by irregular particles with a diameter of 0.5–3 μm indicated by the SEM images (Fig. S9†). The TGA analysis showed the high thermostability of PAF materials and there was no weight loss under 300 °C in air (Fig. S10†). After heated to 600 °C, there was no residue left which suggested PAF materials were pure without any metal catalyst.
N (1652 cm−1), C–N (1390 cm−1) and N–O (930 cm−1) characteristic peaks for PAF-170-AO series confirmed the conversion of cyano groups into amidoxime groups (Fig. S4†). The porosity for PAF-170-AO series was measured at 77 K through N2 adsorption experiments. As shown in Fig. S5 and S6,† the Brunauer–Emmett–Teller (BET) surface areas decreased gradually from 478 (PAF-170-AO-1) to 126 m2 g−1 (PAF-170-AO-5), as the amidoxime group doping ratios increased. Nonlocal density functional theory (NL-DFT) calculations demonstrated that all amidoxime-modified PAFs possessed a wide pore size distribution in the range of 0.4–4 nm that favoured UO22+ ion (uranyl ion with an irregular shape possesses a maximum length of 6.04–6.84 Å) access into the internal space of particles (Fig. S6 and Table S1†).9 From the PXRD spectra, the PAF-170 series depicted amorphous structures (Fig. S8†). All PAF powders are aggregated by irregular particles with a diameter of 0.5–3 μm indicated by the SEM images (Fig. S9†). The TGA analysis showed the high thermostability of PAF materials and there was no weight loss under 300 °C in air (Fig. S10†). After heated to 600 °C, there was no residue left which suggested PAF materials were pure without any metal catalyst.
After confirming the formation of amidoxime groups, the uranyl ion sorption capability was examined at a sorbent/solvent ratio of 0.005 mg mL−1 at pH ∼ 6 (widely used in previous reports). The performance was monitored by the saturated adsorption capacity in the uranium concentration range of 1–85 ppm using inductively coupled plasma mass spectrometry (ICP-MS). As the gradient ratio increased (cyanophenyl:biphenyl from 1:4 to 4:1), compared with pure PAFs (without any functional groups in Fig. S31†), the uranium capacities were 251, 370, 533, 702, and 101 mg g−1, respectively (Fig. 3a and S12†). The utilization ratios of the amidoxime groups (79.4% for PAF-170-AO-1, 69.1% for PAF-170-AO-2, 65.0% for PAF-170-AO-3, 61.9% for PAF-170-AO-4, and 7.4% for PAF-170-AO-5) decreased with the increase in the ratio of amidoxime content. The uranium uptake first increased and then decreased because the availability of adsorption sites depended upon both the accessible surface and quantity of amidoxime groups. There was an increase in the quantity of accessible adsorption groups as the ratio increased from 1:4 to 2:1. As the amount of cyanophenyl increased, this compound served as the terminus of the skeleton, which inhibited the expansion of the porous network and resulted in fewer accessible adsorption groups within the internal space. Significantly, PAF-170-AO-4 was determined to possess the highest adsorption capacity of 702 mg g−1 and is denoted as PAF-170-AO herein to simplify the discussion.
After optimizing the ratio of benzonitrile and phenyl building blocks (2:1), the respective rigid monomers (p-terphenyl and 1,3,5-triphenylbenzene) with expanded structures were utilized to improve the porosity of adsorbents to allow for a high usage ratio of the adsorption sites. The resulting samples (PAF-171 and PAF-172) were processed through a similar procedure to prepare the amidoxime-modified porous adsorbents PAF-171-AO and PAF-172-AO, respectively (Fig. S3† and 1).
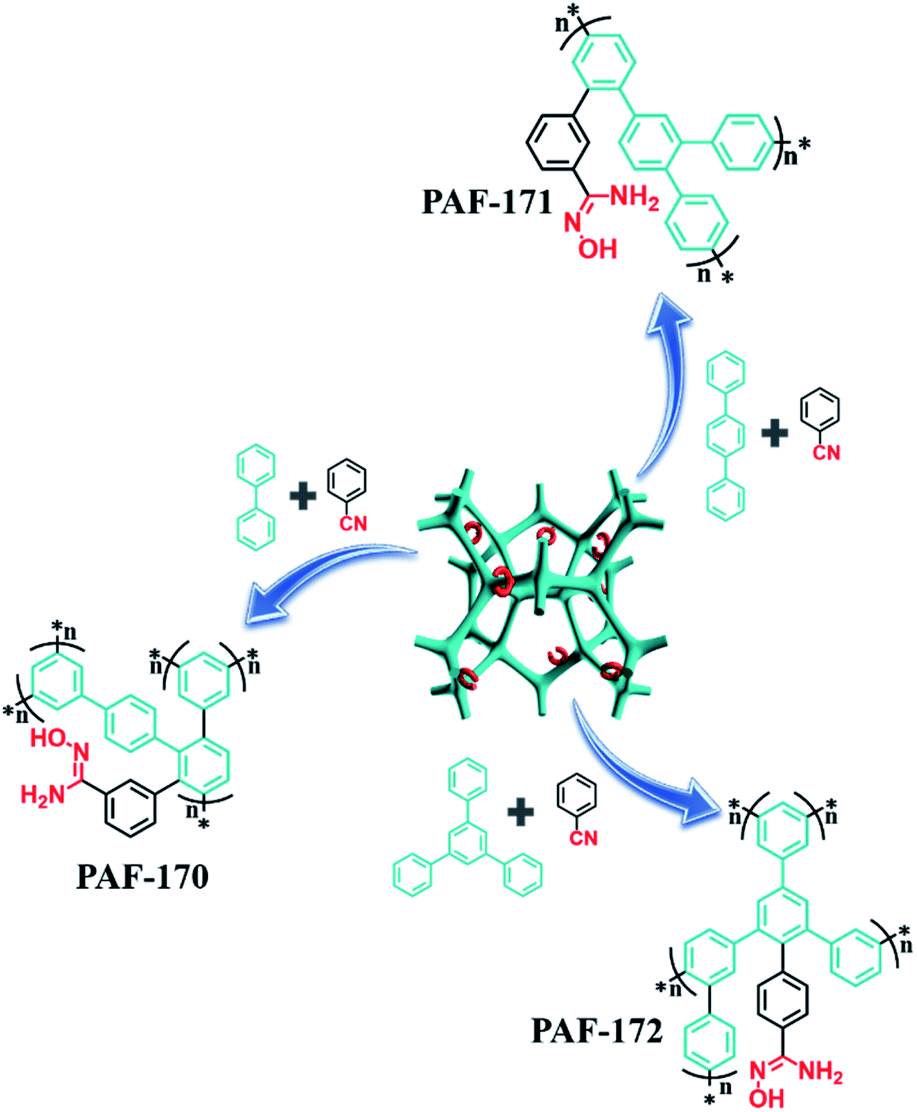 | ||
| Fig. 1 Scheme of synthesis for PAF-AOs: AlCl3/CHCl3, 60 °C, 48 h. NH2OH·HCl/N(CH2CH3)3/EtOH, 70 °C, 48 h. | ||
The conversion of cyano to amidoxime groups was confirmed by the absence of –CN group signal (2229 cm−1) and increased intensity of characteristic amidoxime group peaks at 1382 cm−1 (C–N); 3453 and 3360 cm−1 (N–H); 1650 cm−1 (CN); and 927 (N–O) (Fig. 2a). Meanwhile, the oxygen element in the elemental analysis also indicates the successful complete transformation of the amidoxime group (Table S2†). Additionally, the amidoxime carbon atom which appeared at 149.7 ppm in solid-state 13C NMR monitored the conversion of –CN to amidoxime groups in PAF-171/172-AO (Fig. 2b). PXRD spectra of PAF-170/171/172-AO showed that they were amorphous structures (Fig. S16†). Further, all amidoxime-modified PAFs had similar a morphology, as indicated by the SEM images (Fig. S17†); spherical particles composed of irregular aggregates. The TEM images showed that PAF-170/171/172-AO materials were pure and without metal components. This result was in agreement with the powder XRD results indicating that PAF-170/171/172-AO materials lack significantly ordered structures (Fig. S18†). As observed from TGA analysis (Fig. S18†), the curves revealed that there was nearly no weight loss under 300 °C in air, suggesting the good thermostability of PAF materials. After heating to 600 °C, ca. 2% residues were maintained due to the traces of AlCl3 catalyst left behind in the porous architecture.
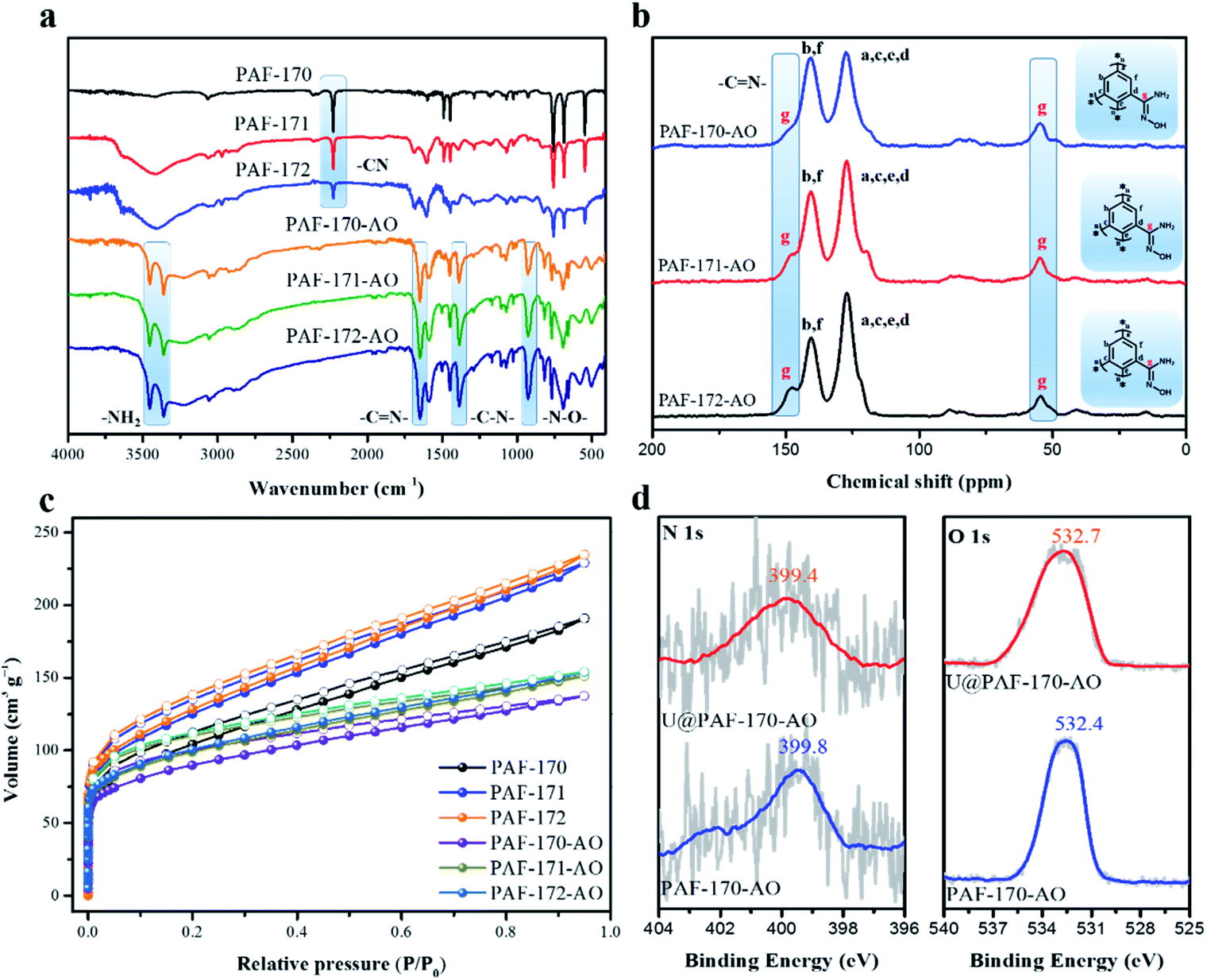 | ||
| Fig. 2 Structural characterization: (a) FT-IR spectra for PAF-170/171/172 and PAF-170/171/172-AO, respectively. (b) 13C CP/MAS NMR spectra for PAF-170/171/172-AO. (c) N2 sorption isotherms for PAF-170/171/172 and PAF-170/171/172-AO, respectively. (d) N 1s and O 1s XPS spectra for PAF-170-AO and uranium adsorbed PAF-170-AO. | ||
As indicated by the N2 sorption isotherms, the surface areas increased along with the expansion of the volume of phenyl monomers. The specific BET surface areas ranged from 312 m2 g−1 for PAF-170-AO to 425 and 541 m2 g−1 for PAF-171-AO and PAF-172-AO, respectively (Fig. 2c and S20†). After confirming the structural integrity, we examined the UO22+ ion sorption capability at a sorbent/solvent ratio of 0.005 mg mL−1, pH ∼ 6. The performance was monitored by the adsorption isotherms and adsorption kinetics in the concentration of 1, 5, 7, 10, 15, 20, 30, 40, 50, 60, 70, and 85 ppm by ICP-MS. The equilibrium adsorption capacity uptakes were 702, 608, and 569 mg g−1 for PAF-170-AO, PAF-171-AO, and PAF-172-AO, respectively, in ∼7 ppm simulated seawater (Fig. 3b). As calculated by the ratio of the actual used amount and the theoretical amount, the usage ratios were determined to be 62.96% (PAF-170-AO), 65.12% (PAF-171-AO), and 71.54% (PAF-172-AO). This result is ascribed to that the incorporated building blocks (p-terphenyl and 1,3,5-triphenylbenzene) increased the mass per structural unit.
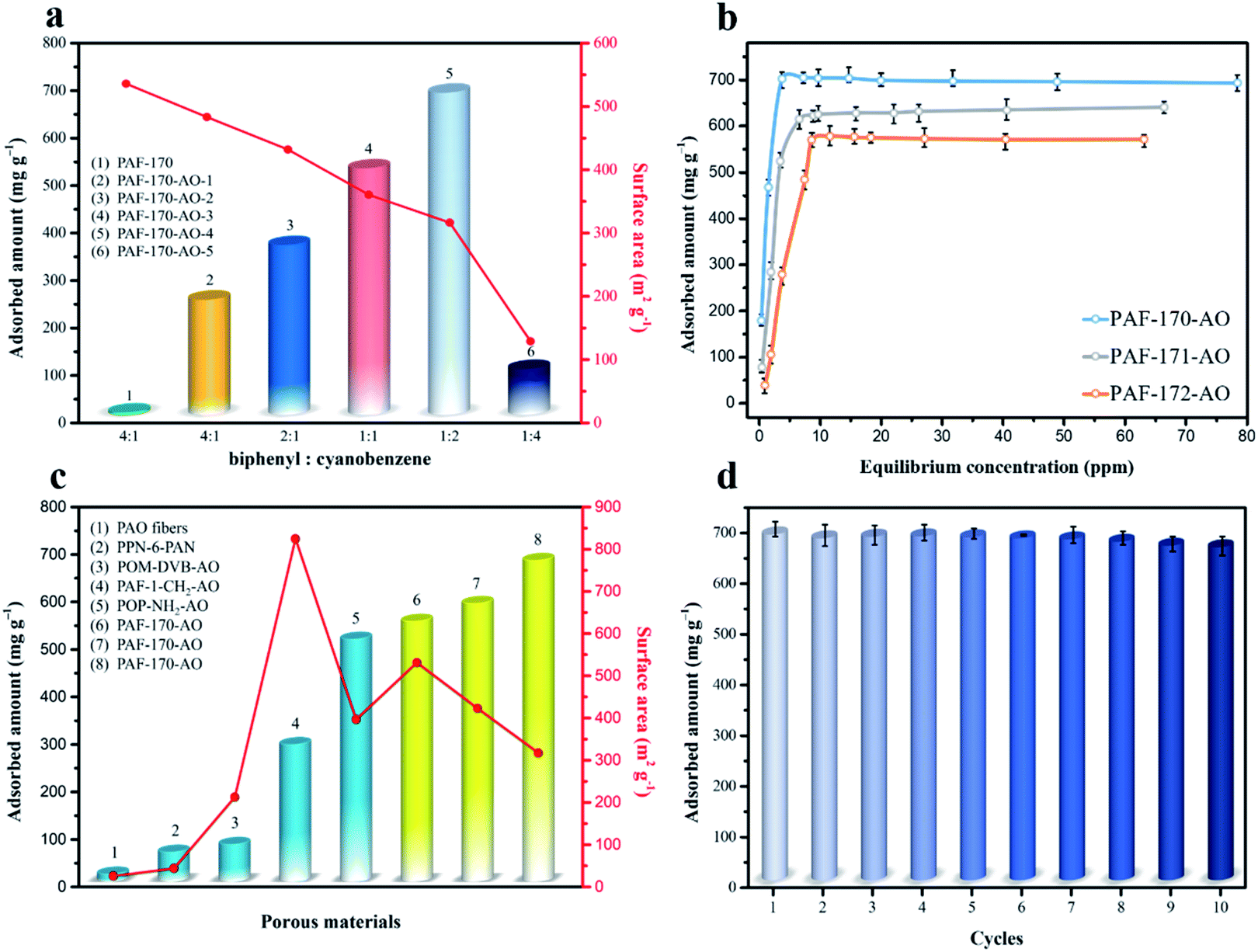 | ||
| Fig. 3 (a) Uranium adsorption and surface areas for PAF-170-AO series. (b) Uranium sorption isotherms for PAF-170/171/172-AO in the uranyl ion solution (7 ppm, pH ∼ 6). (c) Uranium adsorption and surface area comparison with other amidoxime-modified adsorbents in the simulated seawater system (7 ppm, pH ∼ 6). (d) Reusability of PAF-170-AO. | ||
For comparison sake, we prepared the polymer-based adsorbent (PVA-g-AO) with flexible skeleton21 and CMP-based adsorbent (CMP-DVB-AO) with semi-rigid skeleton (Fig. S1†). As depicted in Fig. 3c, PAF-170-AO revealed the highest uranium uptake of 702 mg g−1 among the reported amidoxime-modified adsorbents, which was nearly 16 times as capable as polymer-based adsorbent PVA-g-AO (42.84 mg g−1), and was much higher than CP-OMS (57 mg g−1),22 CMP-DVB-AO (101 mg g−1), PAO-PE (370 mg g−1),23 and PAF-1-CH2AO (300 mg g−1).24 This high performance is attributed to the open architecture providing a large quantity of accessible adsorption sites for uranyl ion binding.
With such encouraging results, the adsorption isotherm of PAF-170-AO was fitted by Langmuir and Freundlich models (Fig. S28a and c†). A higher correlation coefficient (R2 = 0.9964) based on the Langmuir model suggests a monolayer chemical adsorption is dominant during the adsorption process. To further study the complexation behaviour for uranyl ions, we carried out scanning electron microscope energy dispersive spectroscopy (EDS) analysis and acquired X-ray photoelectron spectroscopy (XPS) spectra. The EDS result exhibits a uniform distribution of the uranium elements in the UO22+ ion adsorbed PAF material (U@PAF-170-AO) (Fig. S26†). The XPS spectrum shows that the N 1s and O 1s binding energies are located at ∼399 and ∼532 eV, respectively (Fig. 2d). They are assigned to the –NH2 and amidoxime groups, respectively, indicating the successful amidoxination of –CN groups in PAF-170-AO. The spectrum for U@PAF-170-AO clearly shows the strong U 4f5/2 and U 4f7/2 signals centred at 398.1 and 384.0 eV, respectively (Fig. S24h†). After uranium adsorption, the O 1s peak shifts from 532.4 to 531.9 eV, which proves the chelation of uranyl ions with the amidoxime groups (Fig. S24 and S25†).
After confirming the binding patterns, the PAF-170-AO powder was shaken within the 7 ppm U(VI) solution, at pH ∼ 6. The adsorption curve shown in Fig. S23† reveals that there are several steps involved in the adsorption of uranyl ions. First, there is a sharp uptake because uranyl ions in solution are spread to the external surfaces of particles. Second, the sorption rate for the UO22+ ions gradually decreases, which is ascribed to the situation in which the target ions initially pass through the larger cavities rapidly and then diffuse into the smaller pores slowly. Finally, the accessible adsorption sites are completely occupied, producing an equilibrium state. The pseudo-first-order and pseudo-second-order models have been adopted to study the applicable ion sorption behaviours. As shown in Fig. S28b and d,† the correlation coefficient (R2 = 0.9990) according to the pseudo-second-order model implies that chemical interaction between the adsorption sites and uranyl ions.
Because the open architecture provides a high speed channel for mass transfer, the adsorption rate of PAF-170-AO is much higher (351 mg g−1 h−1) than those of the other amidoxime polymers such as PVA-g-AO (50.4 mg g−1 h−1),22 BAP (0.68 mg g−1 h−1),25 and CMP-DVB-AO (50 mg g−1 h−1). This result also demonstrates that the open architecture boosts the ion migration into the inner space of particles. This substantial improvement reveals that the PAF materials overcome the limitations of flexible polymers, suggesting promising utilization for uranyl ion extraction.
Selectivity is another important factor for evaluating the performance of an adsorbent. Various metal ions (V5+, Zn2+, K+, Ca2+, Na+, Mg2+, Pb2+, Ni3+, Fe3+, Cu2+, Co2+, Li+, Mn2+, Cr3+ and Cd2+) at the same concentration (5 ppm) associated with uranyl competition (5 ppm) in natural seawater were used to investigate the binding competition assay. The distribution coefficient (Kd) is the partition coefficient for comparing the affinity of the adsorbent for a certain ion. The affinity of PAF-170-AO for various ions was verified by calculating the Kd ratio. PAF-170-AO had the ultra-large partition coefficient (Kd = 9.37 × 106) for U(VI), with a selectivity coefficient over 14.89, as determined based on the distribution coefficient for U/V (Table S3†). This high selectivity of PAF-170-AO for U(VI) in the presence of diversified competing ions suggested excellent potential for uranium extraction from natural seawater (Fig. S29†).
The process of capturing uranium is the first step in using it as a nuclear fuel source. The uranium must also be eluted from the adsorbent effectively for it to be fully utilized while also regenerating the adsorbent for multiple capture cycles. PAF-170-AO is able to recover the uranium from the adsorption sites using a simple treatment with a sodium carbonate solution (1 mol L−1). This easy process could be expanded on a large scale with minimal environmental impact. Further, the regenerated PAF-170-AO was able to maintain its capacity with near complete recovery and an uptake of 677 mg g−1 after ten regeneration cycles (Fig. 3d). This finding indicates the potential of PAF adsorbent to be used for repetitive cycles of uranium capture and elution.
Given that uranium elements exist in a soluble state (uranyl species) in seawater, there is a strong motivation to develop adsorbent devices that efficiently seize uranium stocks from seawater. PAF-170-AO powder can be readily dispersed in various organic solvents and exhibits excellent compatibility with conventional polymers. The simple doping of PAF-170-AO allows for the facile fabrication of devices based on various substrates. (i) A composite fibre was obtained by electrospinning the mixture of PAF material and polyacrylonitrile (PAN) with the mass ratio of PAF material in the range of 1–10% (Fig. S30†). (ii) PAF-170-AO powder was able to be uniformly coated on the porous ceramic sheet to prepare the PAF-loaded porous ceramic sheet (PAF-CS) with the mass ratio ranged from 1 to 10% (Fig. 4a).
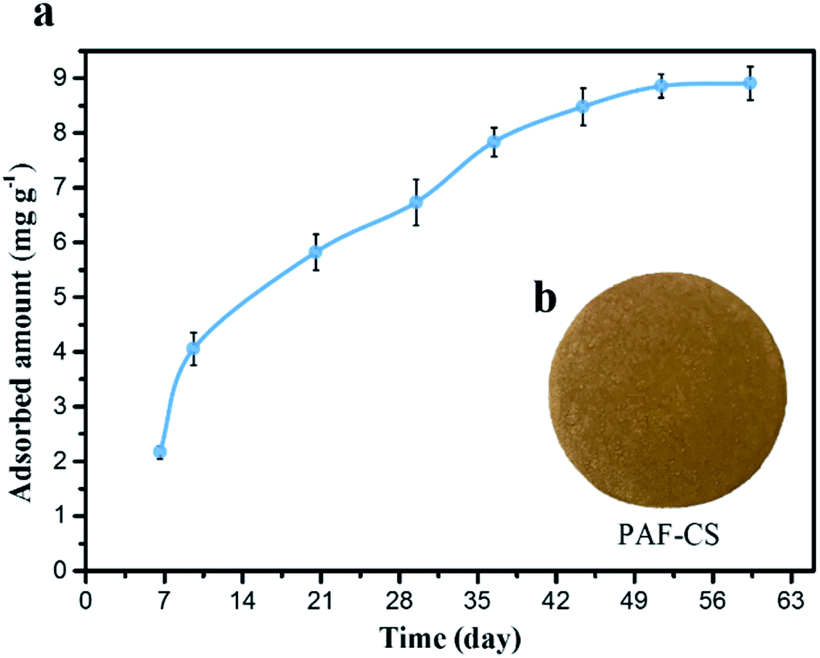 | ||
| Fig. 4 (a) Photograph of PAF-CS prepared in the laboratory. (b) Uranium adsorption isotherms for PAF-CS in real seawater (∼3 ppb) obtained at a flow rate of 5 L min−1 for 60 days. | ||
Based on these devices, we proposed that our PAF materials could be potentially applied for uranium collection by different routes to achieve real-world application; for example, these materials could be employed with the electrically driven pumping approach. Here, we performed the electrically driven seawater testing of PAF-CS in real seawater (∼3.3 ppb, flow rate of 5 L min−1) for 60 days. As shown in Fig. 4b, the adsorption capacity scaled up along with the increase in the number of days, finally reaching the equilibrium of 8.92 mg g−1 after 60 days of testing. Its extraction capability satisfies the strict commercial standard set by the economic evaluation of uranium extraction from seawater (UES) in 21 days, with total capacity of 6 mg g−1 and a service life of 10 adsorption–desorption cycles (3% loss per cycle). The PAF adsorbents are superior to AO-UHMWPE-1 (2.93 mg g−1),8 UiO-66-AO (2.68 mg g−1),26 MIL-101-OA (4.61 mg g−1),27 PVC-co-CPVC (5.22 mg g−1),28 and ORNL (3.3 mg g−1).29
Our techno-economic analysis showed that the use of inexpensive raw materials (biphenyl, nitrile, aluminium chloride, etc.) dramatically lowered the synthesis cost and provided excellent reusability of the synthesized materials.30–32 According to Lindner and Schneider et al. theory, capacity, selectivity, and operational cost were selected as basic parameters to prepare a cost estimate. First, the porous ceramic sheet used exhibited hierarchical porosity with low mass transfer resistance. Using gravitational potential (1 m) is sufficient for maintaining seawater current through the sheet. To achieve the 1 kg uranium extraction requires processing ∼3.03 × 105 m3 seawater through our PAF-CS sheet; therefore the cost for the electricity for processing ∼3.03 × 105 m3 seawater should be RMB 660 (US $97). Adding the cost for the preparation of PAF adsorbents, the final cost using our PAF-modified porous ceramic sheet is ca. US $189.77 per kg for uranium. This price is in accordance with the prevailing market cost for production from land uranium reserves ($100–335 per kg uranium).30,33,34
Conclusion
In summary, a series of porous adsorbents with open architecture was constructed from the inexpensive building units and catalyst to make vast amidoxime groups accessible. The resulting materials exhibited some advantages in terms of kinetics, capacity, and renewability which were not encountered in the conventional polymer-based adsorbents. After the fabrication, the excellent performance was promising for the industrialization process of UES. This work illustrates the preparation of porous adsorbents based on low-priced integrants, an inexpensive catalyst, and simple operation, thus indicating potential directions for the high-performance industrial products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21604008, 21531003 and 91622106) and the “111” project (B18012), and Natural Science Foundation of Jilin Province of China (grant no. 20180520144JH).Notes and references
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Sep. Sci. Technol., 2013, 48, 367–387 CrossRef CAS.
- Y. Yue, R. T. Mayes, G. Gill, L. J. Kuo, J. Wood, A. Binder, S. Brown and S. Dai, RSC Adv., 2015, 5, 50005–50010 RSC.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076–6083 CrossRef CAS.
- T. Saito, S. Brown, S. Chatterjee, J. Kim, C. Tsouris, R. T. Mayes, L. J. Kuo, G. Gill, Y. Oyola, C. J. Janke and S. Dai, J. Mater. Chem. A, 2014, 2, 14674–14681 RSC.
- J. Kim, Y. Oyola, C. Tsouris, C. R. Cole, R. T. Mayes, J. C. Janke and S. Dai, Ind. Eng. Chem. Res., 2013, 52, 9433–9440 CrossRef CAS.
- Y. Yuan, Q. H. Meng, M. Faheem, Y. J. Yang, Z. N. Li, Z. Y. Wang, D. Deng, F. X. Sun, H. M. He, Y. H. Huang, H. Y. Sha and G. S. Zhu, ACS Cent. Sci., 2019, 5, 1432–1439 CrossRef CAS PubMed.
- J. Xiong, S. Hu, Y. Liu, J. Yu, H. Yu, L. Xie, J. Wen and X. L. Wang, ACS Sustainable Chem. Eng., 2017, 5, 1924–1930 CrossRef CAS.
- C. J. Ling, X. Y. Liu, X. J. Yang, J. T. Hu, R. Li, L. J. Pang, H. J. Ma, J. Y. Li, G. Z. Wu, S. M. Lu and D. L. Wang, Ind. Eng. Chem. Res., 2017, 56, 1103–1111 CrossRef CAS.
- T. Wang, M. Xu, X. Han, S. Yang and D. Hua, J. Hazard. Mater., 2019, 368, 214–220 CrossRef CAS PubMed.
- X. Xu, H. J. Zhang, J. X. Ao, L. Xu, X. Y. Liu, X. J. Guo, J. Y. Li, L. Zhang, Q. N. Li, X. Y. Zhao, B. J. Ye, D. L. Wang, F. Shen and H. J. Ma, Energy Environ. Sci., 2019, 12, 1979–1988 RSC.
- A. P. Ladshaw, A. S. Ivanov, S. Das, V. S. Bryantsev, C. Tsouris and S. Yiacoumi, ACS Appl. Mater. Interfaces, 2018, 10, 12580–12593 CrossRef CAS PubMed.
- H. H. Zhao, X. Y. Liu, M. Yu, Z. Q. Wang, B. W. Zhang, H. J. Ma, M. Wang and J. Y. Li, Ind. Eng. Chem. Res., 2015, 54, 3101–3106 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- (a) I. Tabushi, Y. Kobuke and T. Nishiya, Nature, 1949, 280, 665–666 CrossRef; (b) R. V. Davies, J. Kennedy, R. W. Mcilroy, R. Spence and K. M. Hill, Nature, 1964, 203, 1110–1115 CrossRef; (c) D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935 CrossRef CAS PubMed.
- A. S. Ivanov, C. J. Leggett, B. F. Parker, Z. Zhang, J. Arnold, S. Dai, C. W. Abney, V. S. Bryantsev and L. Rao, Nat. Commun., 2017, 8, 1560 CrossRef PubMed.
- M. Eva, S. Jérémy, H. Ahmed Sami, M. Alvaro, D. Philippe, L. Rémi, L. Claude, A. Souad, G. Thierry Le and H. Miryana, J. Nanosci. Nanotechnol., 2019, 19, 4911–4919 CrossRef PubMed.
- Q. Sun, B. Aguila, J. Perman, A. S. Ivanov, V. S. Bryantsev, L. D. Earl, C. W. Abney, L. Wojtas and S. Q. Ma, Nat. Commun., 2018, 9, 1644 CrossRef PubMed.
- Y. F. Yue, C. X. Zhang, Q. Tang, R. T. Mayes, W. P. Liao, C. Liao, C. Tsouris, J. J. Stankovich, J. H. Chen, D. K. Hensley, C. W. Abney, D. E. Jiang, S. Brown and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4125–4129 CrossRef CAS.
- B. Y. Li, Z. H. Guan, X. J. Yang, W. D. Wang, W. Wang, I. Hussain, K. P. Song, B. E. Tan and T. Li, J. Mater. Chem. A, 2014, 2, 11930–11939 RSC.
- F. T. Chi, S. Hu, J. Xiong and X. L. Wang, Sci. China: Chem., 2013, 56, 1495–1503 CrossRef CAS.
- C. Gunathilake, J. Górka, S. Dai and M. Jaroniec, J. Mater. Chem. A, 2015, 3, 11650–11659 RSC.
- A. P. Ladshaw, A. S. Ivanov, S. Das, V. S. Bryantsev, C. Tsouris and S. Yiacoumi, ACS Appl. Mater. Interfaces, 2018, 10, 12580–12593 CrossRef CAS PubMed.
- B. Y. Li, Q. Sun, Y. M. Zhang, C. W. Abney, B. Aguila, W. B. Lin and S. Q. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 12511–12517 CrossRef CAS PubMed.
- M. Piechowicz, C. W. Abney, N. C. Thacker, J. C. Gilhula, Y. F. Wang, S. S. Veroneau, A. G. Hu and W. B. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 27894–27904 CrossRef CAS PubMed.
- L. Chen, Z. L. Bai, L. Zhu, L. J. Zhang, Y. W. Cai, Y. X. Li, W. Liu, Y. L. Wang, L. H. Chen, J. Diwu, J. Q. Wang, Z. F. Chai and S. A. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32446–32451 CrossRef CAS PubMed.
- H. Y. Wu, F. T. Chi, S. Zhang, J. Wen, J. Xiong and S. Hu, Microporous Mesoporous Mater., 2019, 288, 109567 CrossRef CAS.
- S. Brown, Y. F. Yue, L. J. Kuo, N. Mehio, M. J. Li, G. Gill, C. Tsouris, R. T. Mayes, T. Saito and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4139–4148 CrossRef CAS.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076–6083 CrossRef CAS.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076 CrossRef CAS.
- H. Lindner and E. Schneider, Energy Econ., 2015, 49, 9 CrossRef.
- L. J. Kuo, H. B. Pan, C. M. Wai, M. F. Byers, E. Schneider, J. E. Strivens, C. J. Janke, S. Das, R. T. Mayes and J. R. Wood, Ind. Eng. Chem. Res., 2017, 56, 11603 CrossRef CAS.
- Technology and applied R&D Needs for nuclear fuel resources, 2010, Nuclear Fuel Resources Workshop, Norwood, Massachusetts, USA, October 13–15, 2010 Search PubMed.
- T. Saito, S. Brown, S. Chatterjee, J. Kim, C. Tsouris, R. T. Mayes, L. J. Kuo, G. Gill, Y. Oyola, C. J. Janke and S. Dai, J. Mater. Chem. A, 2014, 2, 14674–14681 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00249f |
| This journal is © The Royal Society of Chemistry 2020 |