
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical biomass valorization on gold-metal oxide nanoscale heterojunctions enables investigation of both catalyst and reaction dynamics with operando surface-enhanced Raman spectroscopy†
Nina
Heidary
 and
Nikolay
Kornienko
*
and
Nikolay
Kornienko
*
Department of Chemistry, Université de Montréal, Roger-Gaudry Building, Montreal, Quebec H3C 3J7, Canada. E-mail: Nikolay.kornienko@umontreal.ca
First published on 4th February 2020
Abstract
The electrochemical oxidation of biomass platforms such as 5-hydroxymethylfurfural (HMF) to value-added chemicals is an emerging clean energy technology. However, mechanistic knowledge of this reaction in an electrochemical context is still lacking and operando studies are even more rare. In this work, we utilize core–shell gold-metal oxide nanostructures which enable operando surface-enhanced Raman spectroelectrochemical studies to simultaneously visualize catalyst material transformation and surface reaction intermediates under an applied voltage. As a case study, we show how the transformation of NiOOH from ∼1–2 nm amorphous Ni layers facilitates the onset of HMF oxidation to 2,5-furandicarboxylic acid (FDCA), which is attained with 99% faradaic efficiency in 1 M KOH. In contrast to the case in 1 M KOH, NiOOH formation is suppressed, and consequently HMF oxidation is sluggish in 10 mM KOH, even at highly oxidizing potentials. Operando Raman experiments elucidate how surface adsorption and interaction dictates product selectivity and how the surface intermediates evolve with applied potential. We further extend our methodology to investigate NiFe, Co, Fe, and CoFe catalysts and demonstrate that high water oxidation activity is not necessarily correlated with excellent HMF oxidation performance and highlight catalytic factors important for this reaction such as reactant–surface interactions and the catalysts' physical and electronic structure. The insights extracted are expected to pave the way for a deepened understanding of a wide array of electrochemical systems such as for organic transformations and CO2 fixation.
Introduction
As effects of fossil fuel consumption are increasingly prevalent, renewable energy technologies and green chemistry are coming more into focus. Of particular interest is the use of renewable-derived electricity (e.g. from solar, wind, hydro…) to generate fuels and value-added chemicals in a sustainable fashion.1 Aside from the relatively well-established water electrolysis and CO2 reduction reactions, the electrochemical conversion of increasingly abundant biomass platforms into fuels and components of polymers, fertilizers, organic electronics and pharmaceuticals is rather nascent yet growing rapidly.2 These reactions carry high economic potential because the products that can be generated have a much higher value than the simple products generated from water electrolysis and CO2 reduction (e.g. H2, CO, HCOOH…). In addition, biomass oxidation can replace water oxidation in the aforementioned systems to simultaneously co-generate value-added products on the anodic and cathodic sides.3,4The oxidation of 5-hydroxymethylfurfural (HMF), an acid-catalyzed dehydration product of biomass-derived hexose and pentose sugars, to value-added chemicals is one of the most prominent reactions studied in the field (Fig. 1a).5–7 A series of different products can be obtained through sequential electrochemical oxidation of the alcohol and aldehyde groups of HMF, with 2,5-furandicarboxylic acid (FDCA) being sought after as a substitute for fossil-based terephthalic acid (typically generated from naphtha in an energy demanding fashion) in the fabrication of polyesters, amongst other polymers. While the conversion of HMF to FDCA has long been established through thermal catalysis,8 its generation via electrochemical oxidation is much less known. Both metals and metal oxides have been investigated, with nickel oxides and nickel-containing materials generally being reported to the most active material in terms of selectivity and efficiency.9–18 Despite its potential, mechanistic studies of electrochemical HMF oxidation on metal oxides to understand surface dynamics and key factors that render certain materials highly active, especially through spectroscopic means, are few and there are many knowledge gaps left to fill. In comparison, operando spectroscopy on reactions such as water electrolysis and CO2 reduction has greatly enriched the field's knowledge of these systems and consequently, accelerated progress.19,20
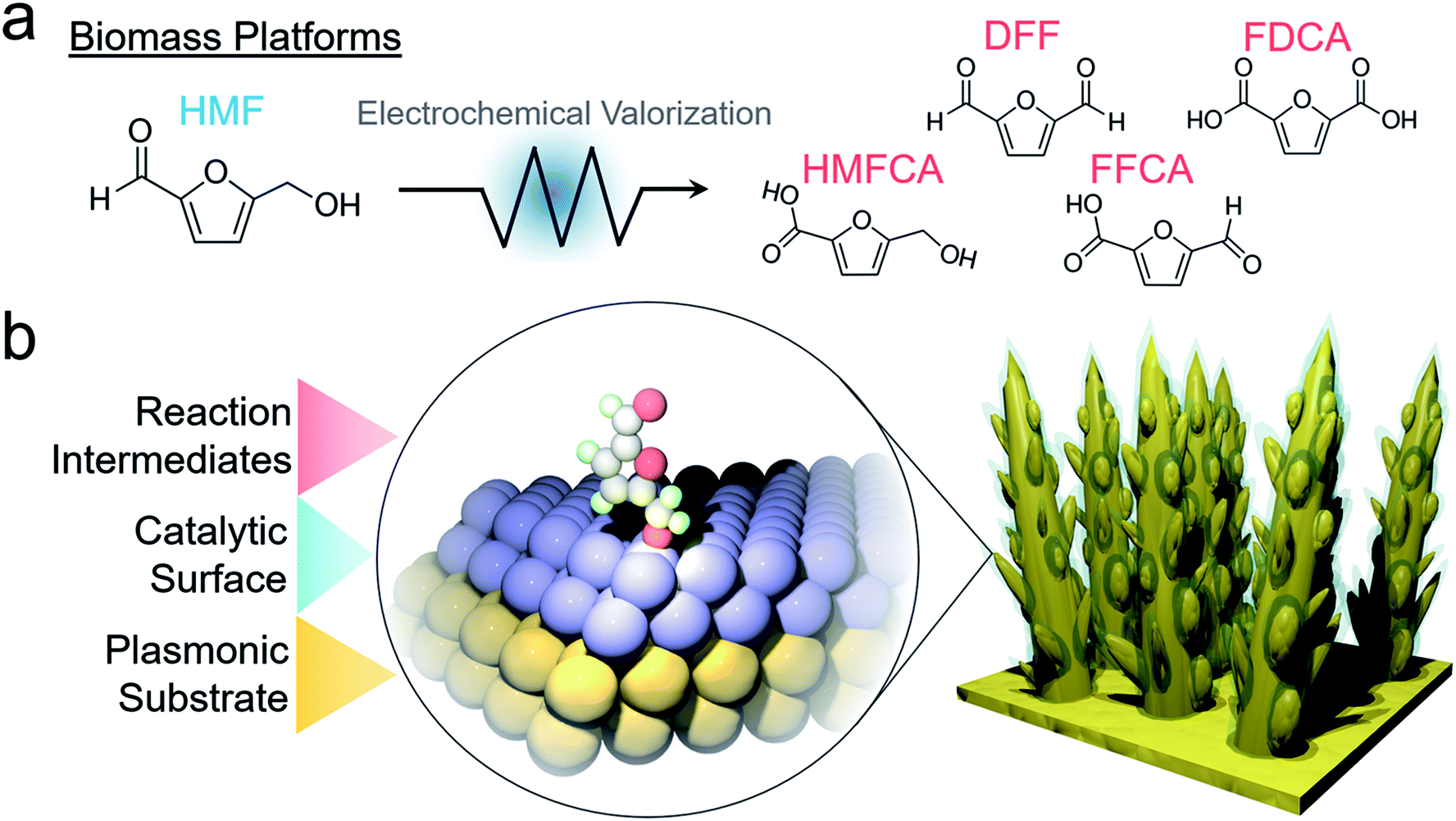 | ||
| Fig. 1 Possible reaction products of electrochemical oxidation of HMF (a). The designed system utilizes a plasmonic substrate for SERS enhancement to view catalyst material transformations as well as the reaction pathway on the surface (b). | ||
Recent infrared spectroscopic studies of nickel borate catalysts elucidated that HMF oxidation proceeded via the 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) intermediate.11 Other infrared spectroscopic studies also hinted at potential- and scan direction-dependent reactivity of glycerol oxidation on Pd–Bi catalysts.21 Sum frequency generation spectroscopy similarly revealed that HMF is first oxidized to HMFCA prior to 5-formyl-2-furancarboxylic acid (FFCA) and FDCA formation.22
Operando surface-enhanced Raman spectroscopic (SERS) studies probed HMF conversion to 2,5-diformylfuran (DFF) on a Au surface and provided evidence for an oxygen-bound surface intermediate.16 SERS is enabled by enhanced local electromagnetic fields at the surface of plasmonic materials with a high amount of free electron density (e.g. Au and Ag) and from charge transfer effects between substrate and chemisorbed molecule, all of which may increase the resultant Raman intensity by orders of magnitude.23,24 As such, this technique is uniquely suited for probing heterogeneous catalysis because the signal enhancement decays rapidly with distance away from the substrate (∼2 nm), rendering SERS an inherently surface-sensitive probe.
Though Au is an excellent model system and SERS substrate, transition metal oxides are more active for this electrochemical HMF oxidation in alkaline solutions. In this work, we thus set out to probe HMF oxidation on nickel oxide surfaces with operando SERS. To utilize the SERS effect on a non-plasmonic substrate, we designed an electrocatalytic system comprised of electrochemically fabricated plasmonic gold dendrites with a thin (∼1–2 nm) shell of nickel. This would then enable the simultaneous detection of catalyst material dynamics and surface reaction pathways (Fig. 1b). The insights derived through the investigation of this system, and other transition metal analogues, shed crucial mechanistic information regarding the influence of surface chemistry on HMF oxidation and how the interplay of HMF and water oxidation dictates electrocatalyst efficacy.
Results and discussion
As a well-established SERS substrate that can be readily deposited on conductive surfaces, gold dendrites were grown on carbon paper through reductive electrodeposition from an aqueous gold solution following previously established literature recipes.25,26 Following the growth of the gold, a sacrificial Zn layer was electrodeposited, which was subsequently converted to Ni (and later other metals) through a galvanic exchange reaction by soaking the electrode in a nickel acetate solution (Fig. 2a). Electron microscopy was then used to characterize the morphology and composition of the resultant electrodes. Scanning electron microscopy (SEM) analysis illustrated that rough, dendritic structures were grown on the carbon fibers of the electrode (Fig. 2b). The Au, as investigated with transmission electron microscopy (TEM), featured a ∼1–2 nm coating of amorphous nickel (Fig. 2c and S1†). Energy-dispersive X-ray spectroscopy (EDS) showed only peaks arising from Au, Ni, and Cu (with the Cu coming from the Cu TEM grid used) (Fig. 2d). The conformal nature of the Ni coating was further demonstrated with an EDS linescan and elemental map (Fig. 2e–g).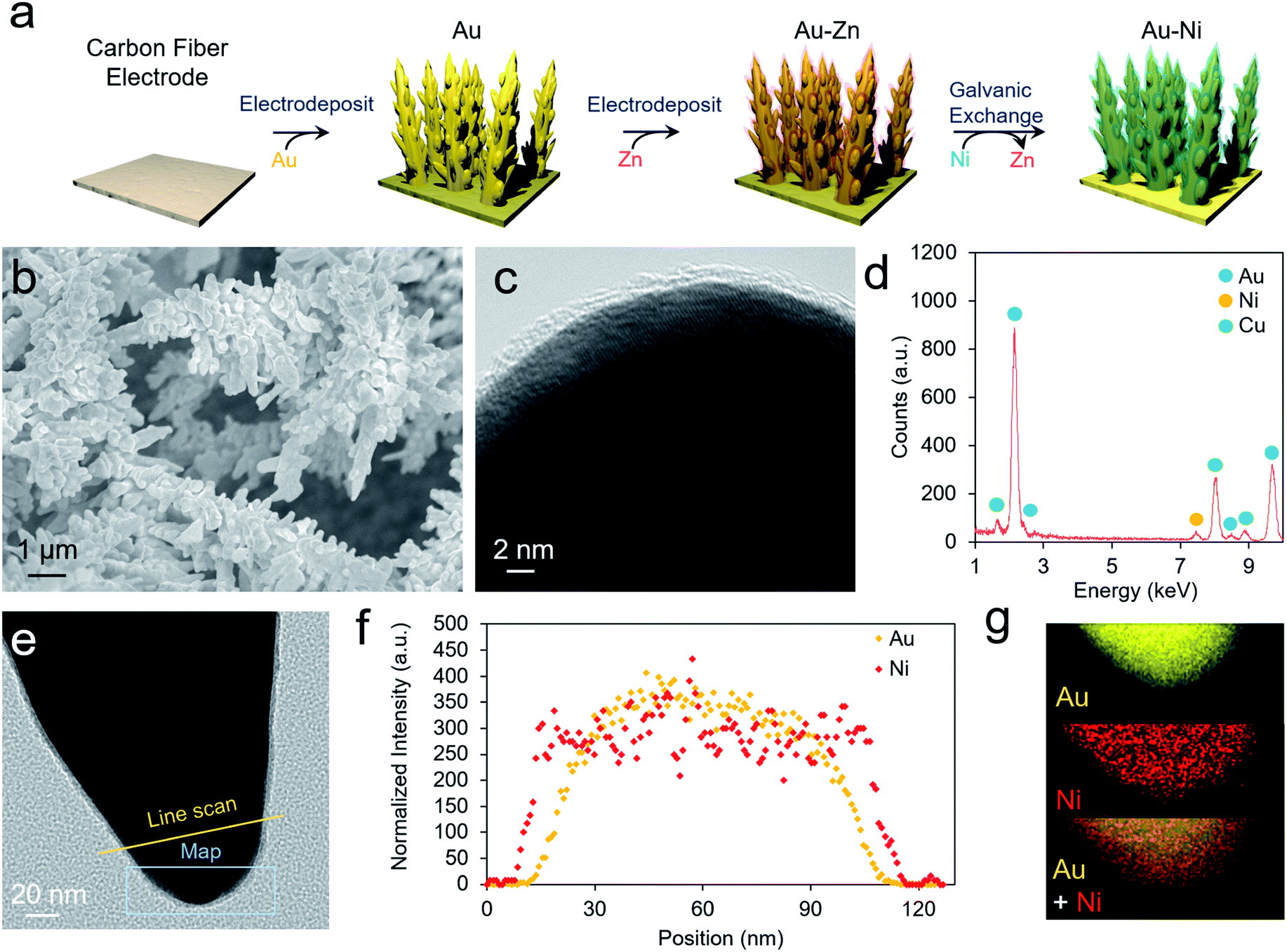 | ||
| Fig. 2 The AuNi catalysts are prepared through a facile electrodeposition and galvanic exchange method (a). The resultant materials are high surface area Au dendrites (b) with a 1–2 nm shell of amorphous Ni (c). EDS analysis only shows peaks stemming from Au, Ni and Cu, the latter originating from the Cu TEM grid employed in this work (d). A catalyst tip (e) was further evaluated with an EDS linescan (f) and elemental map (g) to show the conformal nature of the Ni layer. | ||
We next moved to study the electrochemical performance of the AuNi catalysts, using Au as a comparison, in 10 mM KOH and 1 M KOH electrolytes. Generally, the reaction kinetics are faster in highly alkaline solutions, but 10 mM KOH was also used as HMF is stable for longer durations in this environment. When measuring the open circuit potential and injecting 10 mM HMF into the electrolyte, the open circuit potential for the AuNi rapidly drops from ∼0 to −0.7 V vs. Ag/AgCl (and to −0.2 V for Au only), indicating a substantial change of the electrode potential equilibrium (Fig. 3a). This can be caused by the exchange of adsorbates and ions within the Helmholtz layer possibly hinting to a strong HFM surface adsorption process.
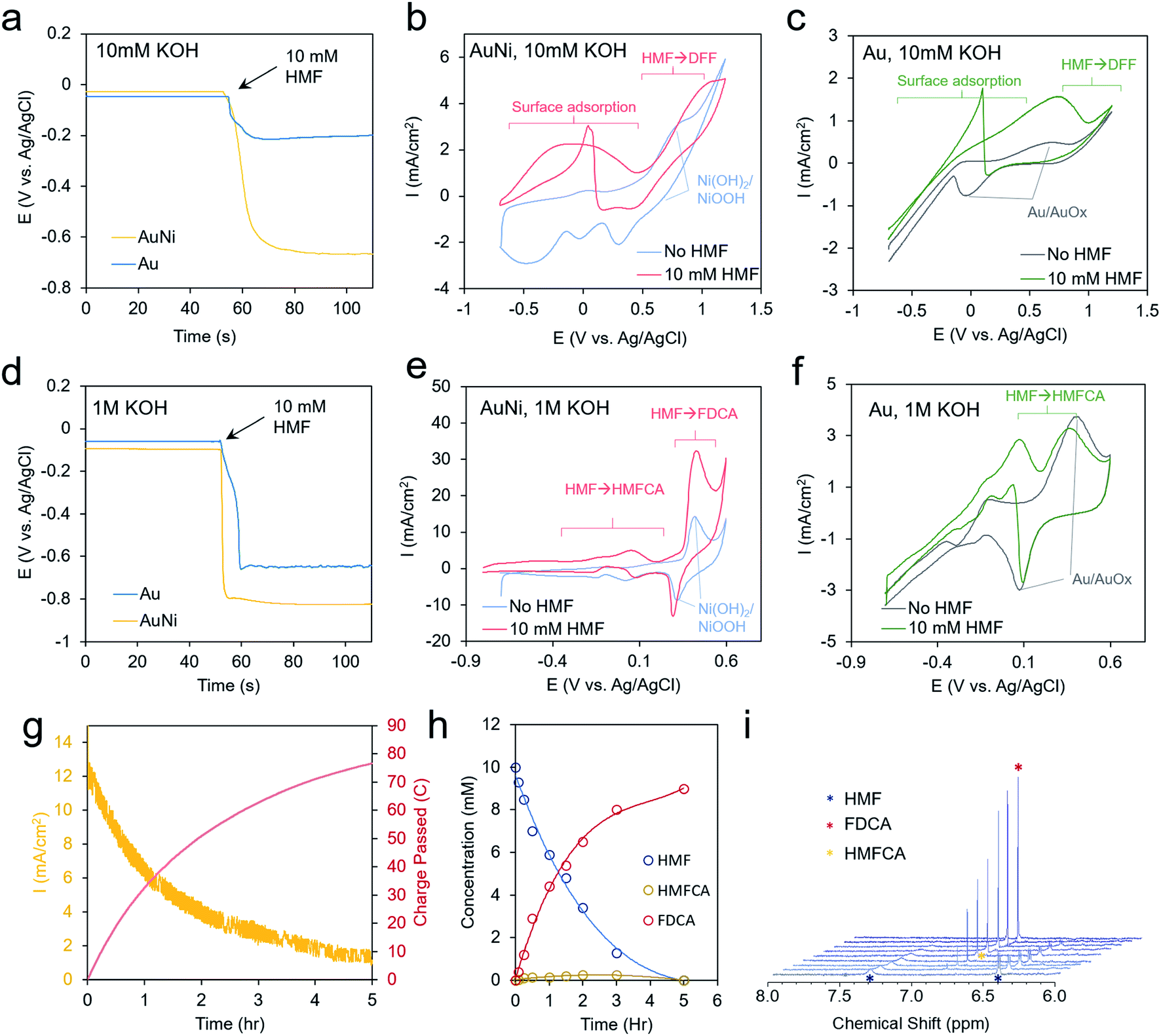 | ||
| Fig. 3 Electrochemical analysis. In 10 mM KOH, strong affinity of HMF to the Ni surface is evidenced by the change in open circuit potential upon injection of 10 mM HMF into the electrolyte solution (a). Enhanced current is noted from −0.6 to 0.5 V AuNi (b) and from −0.2 V to 1.0 V for Au (c). Larger shifts in the open circuit potential are seen in 1 M KOH (d) and a current enhancement is especially pronounced at 0.4 V for AuNi (e) and at 0 V for Au (f). A chronoamperometric scan at 0.4 V shows a decreasing current as HMF is depleted (g) and converted into FDCA (h and i). | ||
Cyclic voltammetry (CV) measurements show only the redox of the Ni and water oxidation in 10 mM KOH and a broad wave centered at 0 V when 10 mM HMF is added to the solution (Fig. 3b). A decrease of current upon progressing to higher potentials may stem from HMF fully covering the surface and blocking the OH− adsorption, which may act as a reactant in the catalytic process. This is also evident by the sharp jump in current in the reverse scan at 0 V as some of the HMF desorbs allowing the remainder to react with OH−.27
Interestingly, the redox peaks tentatively attributed to NiOOH formation at 0.8 V are also suppressed in the presence of HMF, potentially from its high degree of surface binding (Fig. 3b). At potentials from 0 to 0.5 V, the current typically decayed to 0 within tens of minutes in a chronoamperometric scan and no reaction products were detected. At higher potentials of 1.2 V, DFF is detected in low yields after extended electrolysis (Fig. S6†). This indicates that the current below 0.5 V is due to HMF adsorption but not significant catalytic turnover. For reference, the bare Au shows a Au/AuOx redox peak centered at 0.3 V, a catalytic wave at 0.8 V, and the same current jump at 0 V as with the Ni (Fig. 3c).
In 1 M KOH even larger open circuit potential drops are evident with HMF addition (Fig. 3d). The Ni shows enhanced currents from −0.8 to 0.2 V (Fig. 3e). At 0.4 V, the NiOOH formation is concurrent with a large catalytic wave and the reverse reduction of NiOOH is still seen in the presence of HMF. At lower potentials, HMFCA is the most abundant product (Fig. S7†), indicating that the oxidation of the aldehyde group preferentially proceeds first. This is in contrast to the case in 10 mM KOH, where the oxidation of the alcohol group occurred, and a much higher voltage was necessary to generate reaction products. OH− availability, both from the electrolyte and/or on the catalyst surface, may well be the main factor behind this diverging behavior as an oxygen atom is consumed in the aldehyde to carboxylic acid transformation step. The CVs of Au in these conditions are also given for reference (Fig. 3f). In contrast to minimal product formation at lower potentials or at less-alkaline solutions, the reaction with AuNi at 0.4 V maintained high current densities for several hours, until the HMF reactant was used up (Fig. 3g). The reaction product was mainly FDCA, which, at least in part, proceeded via an HMFCA intermediate as this species was detected as well (Fig. 3h and i). However, the DFF pathway cannot be completely excluded just because DFF was not detected in the solution. The faradaic efficiency for FDCA formation, calculated from the amount of charge passed and quantity of product formed, was approximately 99%, as this reaction could be driven on a NiOOH surface prior to the onset of water oxidation.
To obtain a further level of mechanistic insights into the HMF oxidation process on the Ni surface, we turned to operando SERS, executed with the use of a custom-built spectroelectrochemical cell and a water-immersion objective (Fig. 4a). The solution spectra of HMF with the main bands assigned is presented in Fig. 4b. The bands are assigned on the basis of established experimental and theoretical studies.28 In general, surface-enhanced Raman bands shift upon changes in the molecule's electron density as, for example, through bonding or charge transfer processes, and their relative intensities can vary with distance and molecular orientation to the surface.23,29 The enhanced intensity of a particular vibrational mode declines rapidly with distance from the surface and to an extent with an angle-dependent deviation from the direction of the electric field oscillation. In 10 mM KOH, the formation of NiOOH is readily seen beginning at 0.6 V, evidenced by the characteristic marker bands for the Ni–O vibrational modes at 490 and 561 cm−1 of similar intensities (Fig. 4c).30 This matches the redox wave observed in the CV in Fig. 3b. However, NiOOH formation is largely suppressed when 10 mM HMF is present possible due to surface HMF binding (Fig. 4d). At the high-frequency region, both, symmetric and asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bands at 1510 cm−1 and 1570 cm−1, respectively, are red-shifted by 12 cm−1 and 8 cm−1 relative to the solution spectra. The symmetric CC stretching mode of the ring is most pronounced and broadened. The C–O band of the alcohol group is also red-shifted by 8 cm−1 to 1022 cm−1 (Fig. S11†). Due to an HMF adsorption process, the electronic charge redistributes within the molecule, and consequently band positions and relative intensities are affected. Hence, this indicates an adsorption motif of the HMF in which its electron density is reduced by the interaction with the Ni surface through the furan ring and the alcohol group. Furthermore, the shift to lower frequency may indicate electron density being transferred from HMF to the Ni surface from the alcohol group in the binding process. The aldehyde group, however, does not seem to be affected to as significant of an extent. In solution, it is commonly detected as a doublet band representing two conformers.31 Its invariant band positions centered at 1664 cm−1, suggests that the carbonyl group is not significantly involved in the binding to the Ni surface.32 The red-shift of 8 cm−1 also suggests that the alcohol group does not undergo a change in protonation as much larger shifts would be expected if that were the case. As the potential is poised more positive to 0.4 V, the 1022 and 1510 cm−1 bands disappear and a new band at 1577 cm−1 emerges, which blue-shifts to 1582 cm−1 beginning at 1.0 V, when DFF formation begins to occur. This band appears as the alcohol 1022 cm−1 band disappears, indicating that this is a signature of the intermediate en route to DFF, pointing the surface transformation of the alcohol group to the aldehyde. At this potential, the carbonyl band also blue-shifts from 1664 to 1666 cm−1.
C bands at 1510 cm−1 and 1570 cm−1, respectively, are red-shifted by 12 cm−1 and 8 cm−1 relative to the solution spectra. The symmetric CC stretching mode of the ring is most pronounced and broadened. The C–O band of the alcohol group is also red-shifted by 8 cm−1 to 1022 cm−1 (Fig. S11†). Due to an HMF adsorption process, the electronic charge redistributes within the molecule, and consequently band positions and relative intensities are affected. Hence, this indicates an adsorption motif of the HMF in which its electron density is reduced by the interaction with the Ni surface through the furan ring and the alcohol group. Furthermore, the shift to lower frequency may indicate electron density being transferred from HMF to the Ni surface from the alcohol group in the binding process. The aldehyde group, however, does not seem to be affected to as significant of an extent. In solution, it is commonly detected as a doublet band representing two conformers.31 Its invariant band positions centered at 1664 cm−1, suggests that the carbonyl group is not significantly involved in the binding to the Ni surface.32 The red-shift of 8 cm−1 also suggests that the alcohol group does not undergo a change in protonation as much larger shifts would be expected if that were the case. As the potential is poised more positive to 0.4 V, the 1022 and 1510 cm−1 bands disappear and a new band at 1577 cm−1 emerges, which blue-shifts to 1582 cm−1 beginning at 1.0 V, when DFF formation begins to occur. This band appears as the alcohol 1022 cm−1 band disappears, indicating that this is a signature of the intermediate en route to DFF, pointing the surface transformation of the alcohol group to the aldehyde. At this potential, the carbonyl band also blue-shifts from 1664 to 1666 cm−1.
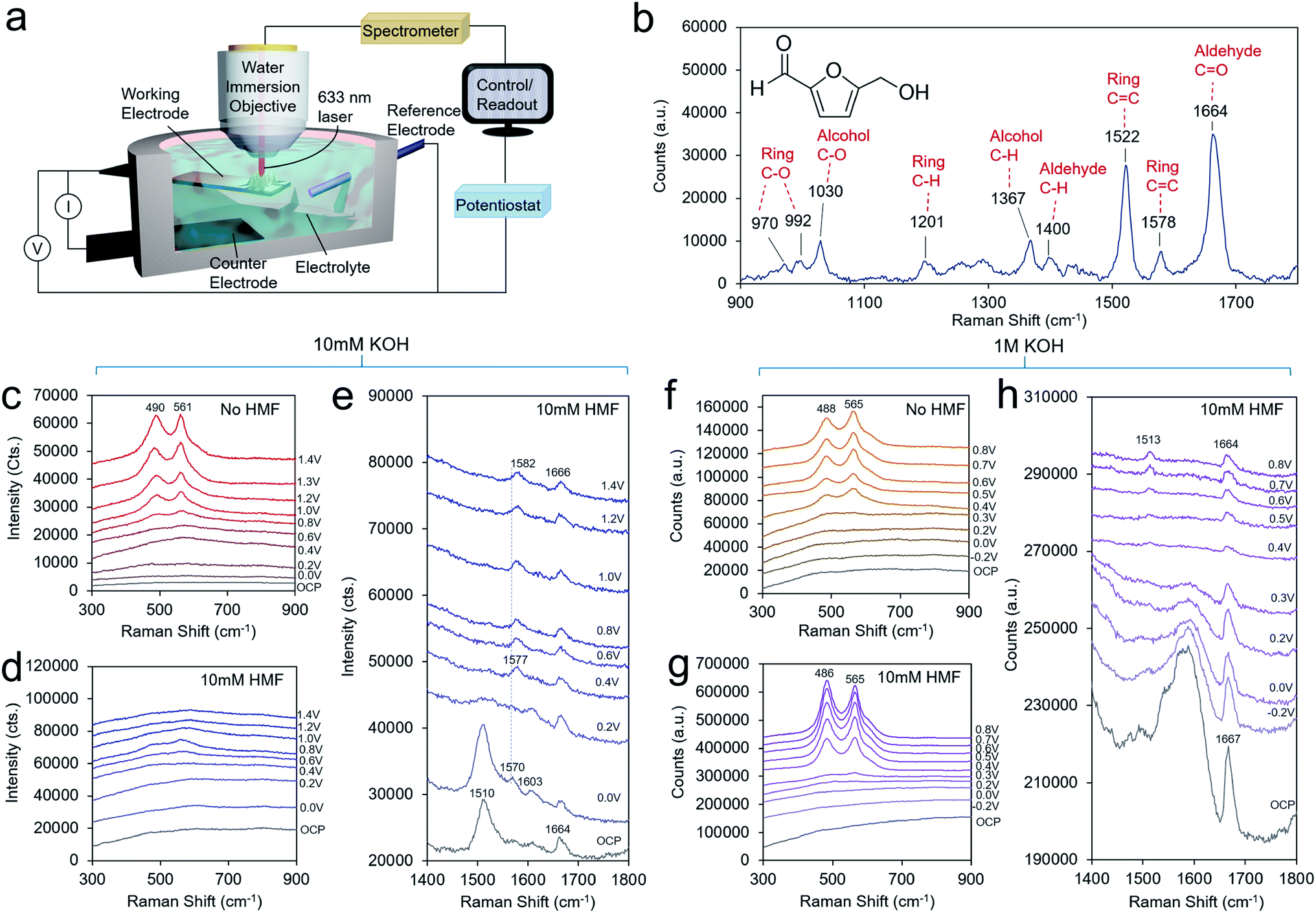 | ||
| Fig. 4 Operando SERS. A custom-built reaction cell and water immersion objective were employed (a). A spectrum of a 500 mM aqueous solution of HMF is illustrated in (b) for reference. In 10 mM KOH, NiOOH readily forms beginning at 0.6 V (c) but when 10 mM HMF is added to the same solution, its formation is largely suppressed, shown in the low frequency region (d). The high frequency region illustrates the HMF adsorption and reaction pathway on the surface en route to DFF (e). In 1 M KOH, NiOOH for both without HMF (f) and with HMF (g) present, albeit with an altered structure. The high frequency spectra portray the surface intermediates as HMF is converted to HMFCA and to FDCA at potentials of 0.4 V and above (h). | ||
In 1 M KOH, the NiOOH readily forms at 0.4 V both with, and without HMF in the solution (Fig. 4f and g). A higher OH− concentration drives the NiOOH formation here, even in the presence of bound HMF. HMF may also be bound in a different fashion in 1 M KOH. However, the ratio of the two bands is different in the presence of HMF on an identically prepared electrode, indicating that the physical structure of the NiOOH, such as through the presence of strain or interlayer spacing and intercalated, species is altered in these conditions.30,33,34 At open circuit conditions, a strong band 1667 cm−1 is seen (Fig. 4h). This vibrational mode at 1667 cm−1 corresponding to the aldehyde group is very intense and its blue-shift relative to that of the solution spectrum hints at an interaction between this group and HMF surface or even the formation of an intermediate upon HMF interaction (possibly in the HMFCA reaction pathway) with the Ni surface. In contrast, the C–O mode of the alcohol group is found at an identical band position (1027 cm−1) relative to the solution spectrum, pointing to minimal interaction between the alcohol and Ni surface (Fig. S12†). Note, spectral features in the region between 1500–1650 cm−1 are not assigned here, due to strong overlap of broad features that stem from the underlying carbon support that do not allow further deconvolution. At potentials >0.3 V, the interfering features decrease due to the oxidation of the carbon surface. As the potential is applied at 0.4 V, a new band at 1513 cm−1 emerges. At the same potentials, this band is accompanied by the formation of NiOOH and the disappearance of the band at 1027 cm−1 representing the alcohol group due to its oxidation. Thus, this band is assigned to a CC ring stretching mode of a surface intermediate en route to FDCA that is likely now interacting with the NiOOH surface via its alcohol group, or at least has this group in close enough proximity to be oxidized. For reference, the Raman spectra of FFCA and FDCA in 1 M KOH feature strong bands at 1515 and 1513 cm−1, respectively (Fig. S10†).
We finally proceeded to extend our investigation to a series of other metal-oxides with varying water oxidation behavior to see if it correlates with activity for HMF oxidation and to see if HMF oxidation exhibits the same mechanism and kinetics on other transition metal oxide surfaces. We synthesized Au with Co, CoFe, NiFe and Fe shells through the same procedure as that for AuNi synthesis, except the galvanic exchange was executed in the different solutions. Upon HMF addition, the Co exhibited increased current densities beginning at −0.8 V and a small peak at 0.25 V (Fig. 5a). The AuCo catalyst produced both HMF and FDCA as detectable products (Fig. S13†). Interestingly, CoFe and NiFe, exhibited the best water oxidation performance but qualitatively less of a propensity for HMF oxidation, judging from the relatively smaller changes in the CV upon HMF addition. Though the exact active site of these materials (e.g. Fe or Co/Ni) and factors affecting their water oxidation performance is still under debate, high water oxidation performance generally stems from a smooth energy landscape for all steps of the reaction pathway. As HMF oxidation proceeds through an alternate mechanism, featuring HMF-derived intermediates, it is not surprising that the criteria for efficient HMF oxidation would be different. This is in contrast to a study using NiFe layered double hydroxide materials for HMF oxidation, which were found to be highly active for FDCA generation.9 A potential explanation is that the active structure of our material is under an applied bias may not be the same and thus, the reactivity also differs. Fe, on the other hand, featured enhanced currents mostly at potentials more negative than 0.2 V.
 | ||
| Fig. 5 Extension to other transition metal thin films. Co (a), CoFe (b), NiFe (c), and Fe (d) catalysts were screened for their activity towards HMF oxidation. A closer look at AuCo catalysts with operando Raman spectroscopy in the low frequency region reveals the formation of an amorphous CoOx surface under oxidizing conditions35 (e), and the high-frequency region gives insight on the nature of the surface intermediates (f). | ||
Operando SERS measurements on AuCo in 1 M KOH both with and without HMF showed that an amorphous CoOx layer forms on the surface, as opposed to crystalline phases such as Co3O4 or CoOOH, evidenced by a broad spectral feature that is pronounced at 616 cm−1 that interconverts to 578 cm−1 under an applied potential (Fig. 5e). This behavior is similar to that of few-layer Co deposited on gold substrates which red-shift as the Co becomes progressively more oxidized.35 Like that seen above for AuNi, the AuCo surface oxide is slightly altered in the presence of HMF, evidenced from a smaller potential-dependent shift of the CoOx band at ∼600 cm−1 (88 cm−1/V without HMF vs. 63 cm−1/V with HMF).
The high frequency region of the shows several potential-dependent changes. At open circuit conditions, peaks at 1027, 1157, 1506, 1533, 1580, 1609, and 1664 cm−1 are visible (Fig. 5F and S14†). Several bands in the 1500–1600 cm−1 region where CC and CO modes are typically found are present in the spectrum at open circuit that are not in the solution spectrum. As in the case with Ni, the C–O band at 1027 cm−1 is at the same position as that of the HMF solution, pointing to a probable lack of interaction of the alcohol group and the surface. At 0 V, bands at 1235, and 1296 cm−1 become visible, and the band at 1609 cm−1 grows in intensity. This is coupled with a peak in the CV as illustrated in Fig. 5a. By 0.3 V, this band decreases in intensity and the spectrum is dominated by the bands at 1335 and 1580 cm−1. The 1580 band again is likely a CO band from a surface bound intermediate in which the C–O bond order is between 1 and 2. At 0.6 V, all bands generally decrease in intensity and mainly, bands at 1440, 1518, 1580 and 1664 cm−1 are visible. As the primary reaction with CoOx is the oxidation of HMF to HMFCA, these bands likely correspond to an intermediate in this reaction pathway. The broadening of the CO band is indicative of this group interacting with the CoOx surface. The 1518 and 1664 cm−1 bands are similar to that spectrum of the AuNi under the same conditions, through the 1580 and 1440 cm−1 bands may indicate a separate intermediate.
Putting together the sum of the data, several important insights can be obtained regarding the factors affecting HMF oxidation selectivity and kinetics. A highly alkaline environment both dictates selectivity and rate of this reaction. In 10 mM KOH, DFF is the primary product obtained. A similar behavior was seen on bare Au surfaces in 10 mM KOH. The propensity of oxidizing the alcohol group rather the aldehyde may stem from a relatively lower OH− concentration necessary to oxidize the aldehyde group to the carboxylic acid and from the preferential interaction of the HMF alcohol group with the catalyst surface (Fig. 6a). Furthermore, the presumable high degree of HMF surface binding to the Ni surface under these conditions does not allow for the active NiOOH species to form in substantial quantities, as evidenced by CV and operando SERS measurements. While it is certainly possible that NiOOH does form as a metastable active species and is rapidly reduced as it oxidizes HMF, the lack of product accumulation suggests that this is not highly prevalent in these conditions.
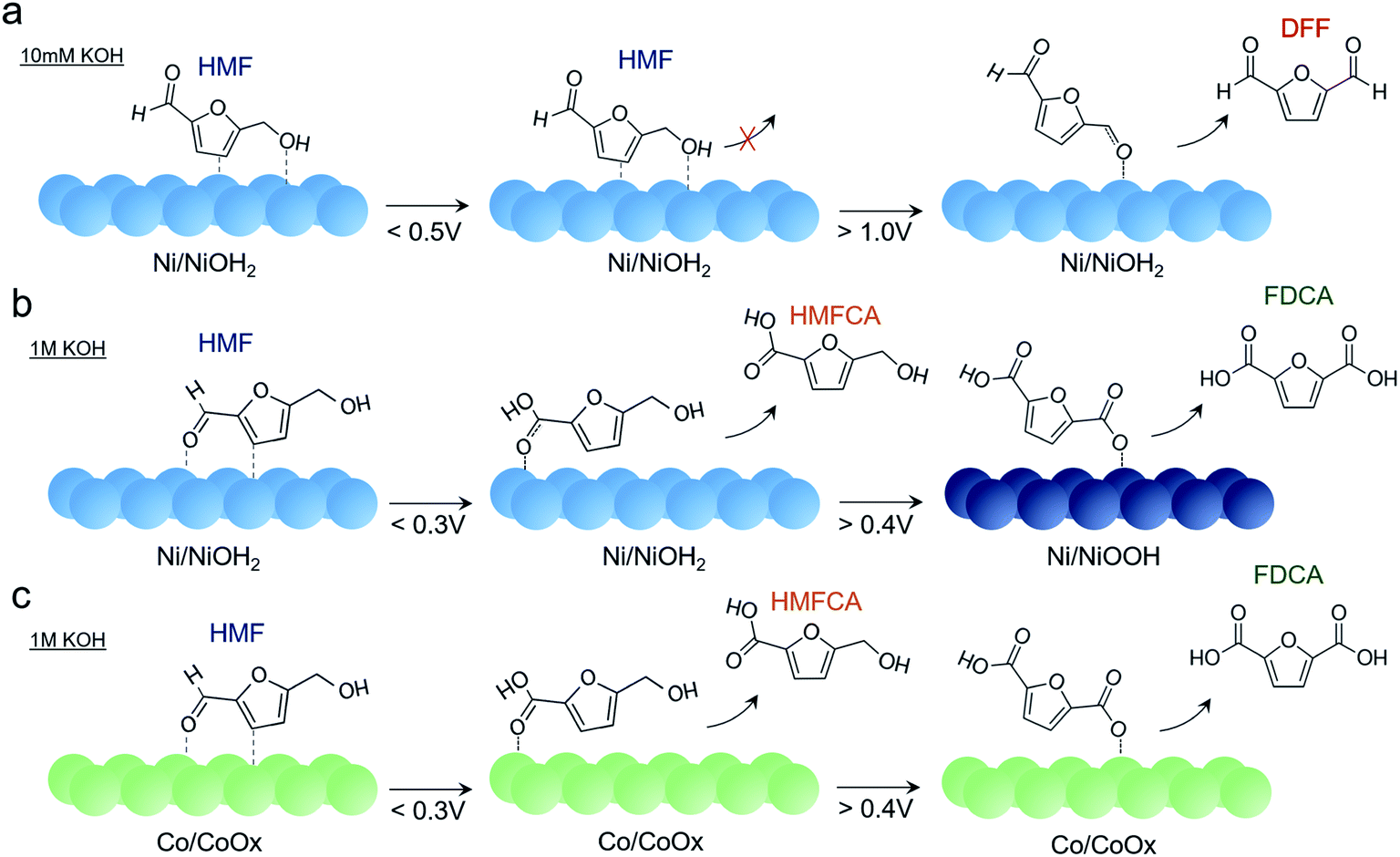 | ||
| Fig. 6 Reaction pathways of HMF oxidation on Ni in 10 mM KOH (a), 1 M KOH (b), and on Co in 1 M KOH (c), as determined from electrochemical and spectroscopic analysis. | ||
At highly alkaline conditions of 1 M KOH, both HMFCA and, to a lesser degree, FDCA, are generated even prior to NiOOH formation (Fig. 6b). This means that the Ni surface (presumably NiOH2) prefers to react with the aldehyde group but as HMFCA is built up, reactions with the alcohol group are still do not occur. This is also evidenced in the Raman spectrum that hints at an interaction between the aldehyde group and Ni surface. In contrast to the case in 10 mM KOH, NiOOH formation is not suppressed in 1 M KOH and once NiOOH forms, FDCA generation greatly accelerates. The NiOOH active phase can therefore readily oxidize both alcohol and aldehyde groups of HMF.
Other transition metals were tested for this reaction as well, and Ni was still the most active species in our conditions. NiFe and CoFe exhibited the highest activity for water oxidation but appeared to be rather poor HMF oxidation catalysts. Co also oxidized HMF to HMFCA, though it did not form the CoOOH phase and remained as a thin, amorphous CoOx layer (Fig. 6c). As such, the HMFCA was built up over time with relatively minor FDCA amounts forming at 0.25 V, and substantial FDCA was only generated after prolonged electrolysis (24 h) at 0.4 V (Fig. S15†) which indicates that the oxidation of the alcohol group is also sluggish with this material. However, previous investigations of CoOOH (though in 0.1 M KOH) also demonstrated that FDCA was only formed in limited amounts.10 Similarities in the Raman spectrum indicate a shared intermediate, but in contrast to the case with NiOOH, HMFCA is seen building up on the surface to a greater extent.
Despite the insights unveiled through this work, a plethora of questions remain. The exact role of solvent alkalinity is not fully solved and precisely, why in 10 mM KOH the oxidation of the alcohol group is favored whereas the aldehyde is preferentially oxidized in 1 M KOH. Furthermore, exact HMF binding motifs to the catalyst surfaces are not fully resolved. While we see preferential interactions with various functional groups that is surface chemistry and pH dependent, exact geometries of both the substrate and reactant are not determined. A previous study indicated that HMF preferentially adsorbs via its aldehyde group at the water–NiO interface, through there is much more to be investigated.36 The exact role of solvent hydrogen bonding networks, surface defects and undercoordinated sites, co-adsorbed species, and solvent cations all likely influence this to an extent. Site-specific reactivity could perhaps be probed with electrochemical microscopy and techniques such as operando X-ray absorption could correlate catalyst oxidation state and structural changes with HMF oxidation performance in a manner complementary to this work.
While NiOOH rapidly oxidizes HMF to FDCA once it is formed, the exact mechanism behind this observation is still to be determined, whether the Ni3+ oxidation state, the NiOOH layered structure, or both are critical for this. NiOOH did not form in 10 mM KOH and did not yield much FDCA. In addition, Co did not form the CoOOH phase, even at very positive potentials, and did not as efficiently generate FDCA, though it likely did contain Co3+ and Co4+ in these conditions.35 Surface chemistry is also postulated to play a role in the catalysts' activity. Recent investigations pointed to varying levels of oxygenic intermediates being present on Co, Ni, and Fe surfaces.37 In particular, the high reactivity of adsorbed *OH with methanol led to enhanced catalytic currents when CH3OH was added to the electrolyte. This occurred as Ni(OH)2 oxidized to NiOOH. As we observe HMF oxidation current that initiates at the same potential, a very plausible scenario is that surface *OH is a driving factor behind efficient HMF oxidation to FDCA. Methanol oxidation is also found to inhibit NiOOH formation in 10 mM KOH, but not in 1 M KOH (Fig. S18†). In the latter case, methanol oxidation occurs once NiOOH forms and is likely driven by the presence of *OH or *O surface-bound species on this material, which are more prevalent in highly alkaline conditions. The enhanced current observed prior to NiOOH formation stems from the oxidation of the aldehyde group as HMFCA is the primary product found at 0.1 V. Surface *OH was also postulated to be found at a higher density on Ni than Co surfaces and could also help explain the discrepancy between the Ni and Co catalysts' activity found in this work. While we do not believe that exposed Au to be the catalytic species in this work because of the much small reactivity of Au, in terms of steady state current and product generation, as well as because of several Raman bands in the Au spectra not visible in the AuNi spectra (Fig. S19†), there could indeed be electronic effects imparted by Au onto the overlaying transition metal catalyst. For example, the activities of Co, Mn, and Ni oxides/oxyhydroxides were enhanced by underlying Au layers and so could the Ni, Co and Fe here.35,38,39
Concluding remarks
In summary, we present the first operando Raman study of metal oxide – catalyzed HMF oxidation. The execution of this work was enabled by the use of nanoscopic gold-transition metal heterojunctions that give surface-enhanced Raman signals from catalysis on non-plasmonic materials and illustrate both catalyst transformation and surface reactivity under operating conditions. The results indicate how electrolyte alkalinity and surface binding dictate the catalytic mechanism and how the HMF oxidation reaction proceeds on transition metal surfaces. While many questions still remain, the methods developed and insights extracted in this work represent a significant step forward towards understanding the electrochemical oxidation of HMF, and in a larger context, the electrocatalytic transformation of organic molecules.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
N. K. and N. H. acknowledge the NSERC Grant R-N401-02839. The authors thank Samir Elouatik for help with Raman measurements, Philippe Plamondon with SEM measurements, and Jean-Philippe Masse with TEM measurements.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- K. Li and Y. Sun, Chem.–Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: volume I--results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab., Golden, CO (US), 2004 Search PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- S. R. Kubota and K.-S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- N. Heidary and N. Kornienko, Chem. Commun., 2019, 55, 11996–11999 RSC.
- D.-H. Nam, B. J. Taitt and K.-S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- X. Deng, X. Kang, M. Li, K. Xiang, C. Wang, Z. Guo, J. Zhang, X.-Z. Fu and J.-L. Luo, J. Mater. Chem. A, 2020, 8, 1138–1146 RSC.
- N. Heidary, K. H. Ly and N. Kornienko, Nano Lett., 2019, 19, 4817–4826 CrossRef CAS.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937–3945 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- W.-H. Park and Z. H. Kim, Nano Lett., 2010, 10, 4040–4048 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382 CrossRef CAS PubMed.
- M. B. Ross, Y. Li, P. De Luna, D. Kim, E. H. Sargent and P. Yang, Joule, 2019, 3, 257–264 CrossRef CAS.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 CrossRef CAS.
- T. Kim, R. S. Assary, L. A. Curtiss, C. L. Marshall and P. C. Stair, J. Raman Spectrosc., 2011, 42, 2069–2076 CrossRef CAS.
- C. L. Haynes, A. D. McFarland and R. P. Van Duyne, Anal. Chem., 2005, 77, 338 A–346 A CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- M. Rogojerov, G. Keresztury and B. Jordanov, Spectrochim. Acta, Part A, 2005, 61, 1661–1670 CrossRef CAS PubMed.
- T.-j. Jia, P.-w. Li, Z.-g. Shang, L. Zhang, T.-c. He and Y.-j. Mo, J. Mol. Struct., 2008, 873, 1–4 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- R. Kostecki and F. McLarnon, J. Electrochem. Soc., 1997, 144, 485–493 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- Y. Gorlin, C.-J. Chung, J. D. Benck, D. Nordlund, L. Seitz, T.-C. Weng, D. Sokaras, B. M. Clemens and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 4920–4926 CrossRef CAS PubMed.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00136h |
| This journal is © The Royal Society of Chemistry 2020 |