
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrostatically-directed Pd-catalysis in combination with C–H activation: site-selective coupling of remote chlorides with fluoroarenes and fluoroheteroarenes†
William A.
Golding
 and
Robert J.
Phipps
*
and
Robert J.
Phipps
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: rjp71@cam.ac.uk
First published on 18th February 2020
Abstract
Systems incorporating catalyst–substrate non-covalent interactions are emerging as a versatile approach to address site-selectivity challenges in remote functionalization reactions. Given the achievements that have been made in this regard using metals such as iridium, manganese and rhodium, it is surprising that non-covalent catalyst direction has not been utilized in reactions incorporating palladium-catalyzed C–H activation steps, despite palladium being arguably the most versatile metal for C–H activation. Herein, we demonstrate that electrostatically directed, site-selective C–Cl oxidative addition is compatible with a subsequent C–H activation step, proceeding via a concerted metalation deprotonation-type mechanism. This results in site-selective cross-coupling of dichloroarenes with fluoroarenes and fluoroheteroarenes, with selectivity controlled by catalyst structure. This study demonstrates that Pd-catalyzed C–H activation can be used productively in combination with a non-covalently-directed mode of catalysis, with important implications in both fields.
Introduction
In recent years, non-covalent interactions have been increasingly explored as a powerful tool for modulating positional selectivity in transition-metal catalyzed reactions.1 Examples of reactions that have been investigated using this approach include hydroformylation,2 alkyne hydrometalation,3 cross-coupling4 and C–H activation.5 The latter is an area of chemistry in which control of positional selectivity is one of the defining challenges and which arguably has much to gain from the application of non-covalent strategies.6 To this end, iridium-catalyzed borylation of arenes has received the most attention to date, possibly due to its relatively mild reaction conditions, high functional group tolerance and compatibility with non-polar solvents.7 A broad spectrum of non-covalent interactions including hydrogen bonding,8 ion-pairing9 and electrostatic interactions10 have all been employed to direct the reactive iridium catalyst through substrate–ligand interactions.11 But it is conspicuous that little progress has been made on applying analogous non-covalent approaches to control site-selectivity in reactions involving palladium-catalyzed C–H activation processes.12,13 This is despite the rapid popularization of the use of ‘transient’ directing groups, which combine with the substrate in a reversible but covalent manner, to direct the Pd metal center in the C–H activation step.14,15 The inherent reversibility of non-covalent interactions makes them ideal to explore in this context. However, there is relatively little precedent for non-covalent catalyst direction being used in tandem with palladium-catalyzed C–H activation. An interesting recent example from Crimmin and co-workers suggested dispersion interactions can influence regioselectivity in the palladium-catalyzed C–H alumination of toluene.16 If compatibility could be more broadly demonstrated it would pave the way for new and innovative design strategies incorporating non-covalent design elements for controlling site-selectivity in Pd-catalyzed C–H activation reactions.We recently reported that sulfonated phosphine ligands can be used to direct site-selective palladium-catalyzed cross coupling on substrates featuring remote chlorides that would be very challenging to differentiate using existing methods.4sSPhos is a commercially available, water-soluble phosphine that we repurposed such that the sulfonate group engages in a non-covalent interaction with the substrate.17 Experiments provided support for a scenario wherein the potassium cation of the deprotonated substrate interacts with the sulfonate group of the ligand, leading to oxidative addition being directed to the C–Cl bond at the substrate meta position (Fig. 1, upper pathway). Following this, transmetalation or amine coordination/deprotonation was followed by reductive elimination to typically give a single regioisomer as product. At the outset of this work we questioned whether it might be possible to replace the transmetalation step with a CMD step to enable C–H bond activation to occur on the coupling partner. This would not only increase the efficiency of the C–C bond formation by avoiding prefunctionalization of one reactant, but more importantly would demonstrate proof-of-concept that CMD is compatible with non-covalent catalyst direction, in this case to control site-selectivity in oxidative addition to the C–Cl bond. Proof that this is viable may have broader implications for palladium-catalyzed C–H activation as it could act as a stepping-stone to non-covalent catalysis being applied for control of site-selectivity in the challenging C–H activation step of a catalytic cycle (vide supra). To test our proposal, we sought to examine fluoroarenes and fluoroheteroarenes as coupling partners (Fig. 1, lower pathway). In addition to being well suited to CMD with Pd-catalysis,18 success with this substrate class would also provide complementarity to our previous work. We had attempted to use polyfluorophenyl boronic acids, trifluoroborate salts and MIDA-boronates as coupling partners under our previous conditions but observed no conversion in all cases.19 This was also the case when using the sSPhos G2 pre-catalyst (see ESI† for full details). Hence, development of a C–H activation variant would be of practical utility, allowing access to these fluorinated biaryl products in a selective manner.
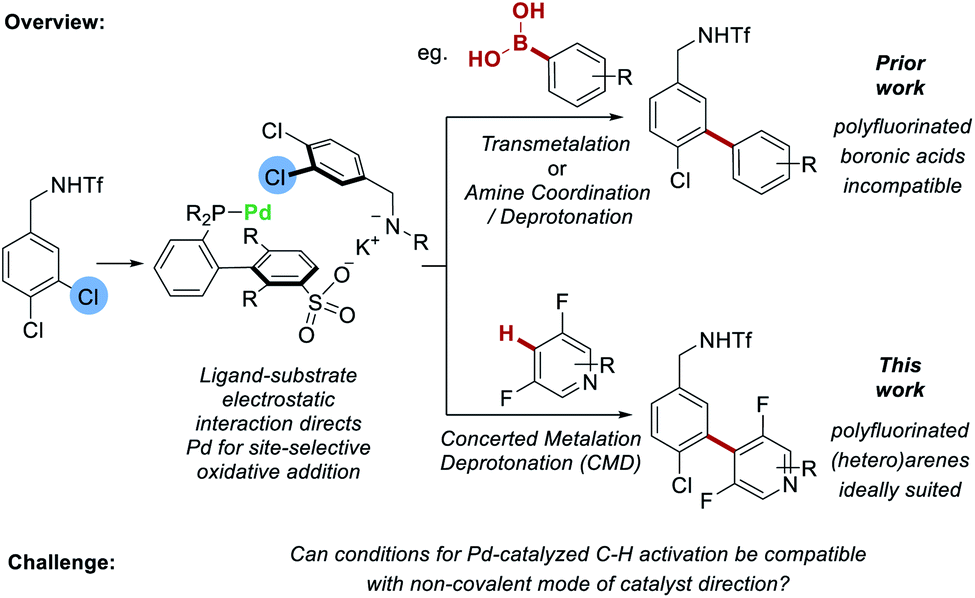 | ||
| Fig. 1 Proposed integration of C–H activation of fluoroarenes with an electrostatically-directed site-selective oxidative addition. | ||
Results and discussion
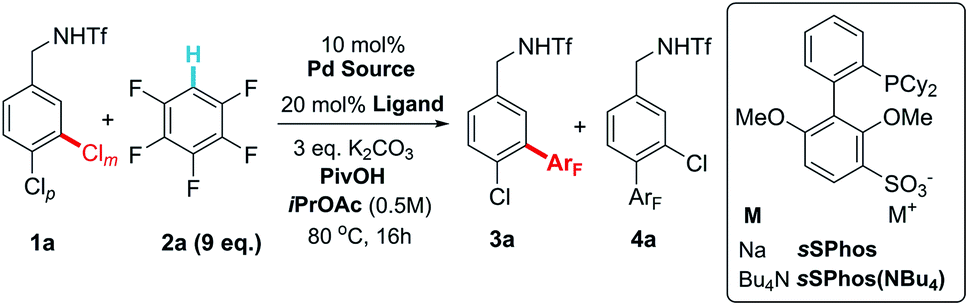
When approaching the optimization of the proposed reaction, our primary concern was whether the conditions required for effective C–H activation might interfere with the electrostatic interaction presumed to be responsible for the site-selectivity in the oxidative addition step. We commenced our investigations with triflate-protected dichlorobenzylamine 1a and pentafluorobenzene (2a) (Table 1). Under the conditions that we had previously used for site-selective Suzuki coupling, no product formation was observed (entry 1). Consulting the conditions optimized by Fagnou and co-workers in their seminal work on perfluoroarene C–H activation, we switched the solvent to iPrOAc but still observed no product formation (entry 2).18a,18b Addition of substoichiometric amounts of pivalic acid was found to be beneficial, in accordance with previous observations.20 Good yield was obtained with 60 mol% PivOH and, importantly, excellent levels of site-selectivity for coupling of the chloride at the meta position was obtained (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entries 3 and 4). The yield could be improved by switching from the typical sodium salt of sSPhos to the more soluble tetrabutylammonium salt, sSPhos(NBu4) (entry 5). Finally, an evaluation of Pd sources showed that [(Cinnamyl)PdCl]2 gave improved yield (entry 6). When switching the ligand to standard SPhos the site-selectivity dropped to 1.2:1, in line with the hypothesis that the sulfonate group on the ligand is crucial for this outcome (entry 7). To probe the effect of having an arene sulfonate moiety present but detached from the ligand structure, we carried out the reaction using 20 mol% SPhos with 20 mol% potassium 2,4-dimethoxybenzenesulfonate as an additive (entry 8). This showed similarly low site-selectivity as observed when using SPhos. The equivalents of pentafluorobenzene could be reduced to three equivalents before significant effect on yield was observed (entries 9 and 10). However, for the purposes of scope exploration, nine equivalents were retained in the remainder of the studies, due to anticipated lower reactivity of some lesser fluorinated arenes.
:1, entries 3 and 4). The yield could be improved by switching from the typical sodium salt of sSPhos to the more soluble tetrabutylammonium salt, sSPhos(NBu4) (entry 5). Finally, an evaluation of Pd sources showed that [(Cinnamyl)PdCl]2 gave improved yield (entry 6). When switching the ligand to standard SPhos the site-selectivity dropped to 1.2:1, in line with the hypothesis that the sulfonate group on the ligand is crucial for this outcome (entry 7). To probe the effect of having an arene sulfonate moiety present but detached from the ligand structure, we carried out the reaction using 20 mol% SPhos with 20 mol% potassium 2,4-dimethoxybenzenesulfonate as an additive (entry 8). This showed similarly low site-selectivity as observed when using SPhos. The equivalents of pentafluorobenzene could be reduced to three equivalents before significant effect on yield was observed (entries 9 and 10). However, for the purposes of scope exploration, nine equivalents were retained in the remainder of the studies, due to anticipated lower reactivity of some lesser fluorinated arenes.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd Source | Ligand | Eq. PivOH | % Conv. |
3:4 |
| a Ratios and conversions determined by 1H-NMR analysis with internal standard. b THF as solvent. c Potassium 2,4-dimethoxybenzenesulfonate included as additive (20 mol%). d 6 eq. 2a used. e 3 eq. 2a used. | |||||
| 1b | Pd(OAc)2 | sSPhos | 0 | 0 | N/A |
| 2 | Pd(OAc)2 | sSPhos | 0 | 0 | N/A |
| 3 | Pd(OAc)2 | sSPhos | 0.3 | 48 | 14:1 |
| 4 | Pd(OAc)2 | sSPhos | 0.6 | 56 | 14:1 |
| 5 | Pd(OAc)2 | sSPhos(NBu4) | 0.6 | 78 | 16:1 |
| 6 | [(Cin)PdCl]2 | sSPhos(NBu4) | 0.6 | 100 | 15:1 |
| 7 | [(Cin)PdCl]2 | SPhos | 0.6 | 100 | 1.2:1 |
| 8c | [(Cin)PdCl]2 | SPhos | 0.6 | 100 | 1.2:1 |
| 9d | [(Cin)PdCl]2 | sSPhos(NBu4) | 0.6 | 88 | 16:1 |
| 10e | [(Cin)PdCl]2 | sSPhos(NBu4) | 0.6 | 82 | 13:1 |
To provide support for the proposed electrostatic interaction, in which the potassium cation is thought to play a crucial role, we carried out experiments wherein stoichiometric amounts of various crown ethers are added under the optimized reaction conditions (Scheme 1). These showed that addition of 18-Crown-6, which is best able to bind potassium, resulted in no selectivity and poor conversion (entry 2). As the crown ether was made smaller and less able to bind potassium, selectivity and reactivity were restored (entries 3 and 4).
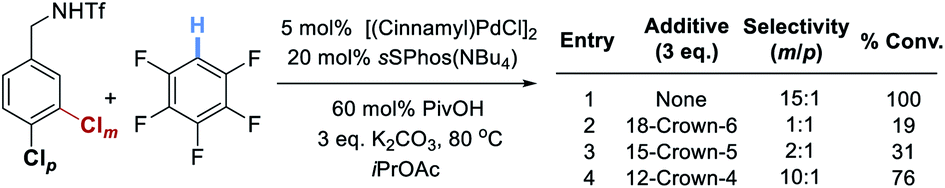 | ||
| Scheme 1 Evaluation of various sizes of crown ether as additives under the optimized conditions. | ||
Having optimized conditions for the site-selective coupling of 1a with pentafluorobenzene, we next evaluated the scope of the perfluoroarene component (Scheme 2). Undesired proteodechlorination of the reaction products was observed in some cases, however this deleterious pathway could be largely avoided by shortening the reaction time (see ESI† for full details). Isomeric tetrafluoroarenes underwent efficient coupling in high yield and with excellent site-selectivity (3b and 3c). 1,3,5-Trifluorobenzene represented the limit of reactivity (3d); whilst selectivity was high, conversion was moderate and such a drop-off of reactivity is precedented.18a Tetrafluorobenzenes bearing various substituents reacted smoothly and the scope encompassed methoxy (3e), trifluoromethyl (3f), alkynyl (3g), ester (3h) and acetamide (3i) groups. A further perfluoroarene could be incorporated without issue (3j). It is interesting to note that the site-selectivity, whilst typically >10:1, does vary to some degree with the fluoroarene partner, despite the selectivity-determining step occurring in the oxidative addition. We attribute this small variation to a solvent effect – as the reactions are relatively concentrated (0.5 M) and nine equivalents of arene are being used, this typically means a ∼1:1 ratio by volume of fluoroarene:solvent. It is therefore foreseeable that changes in the fluoroarene component could be manifested in minor fluctuations in oxidative addition regioselectivity.
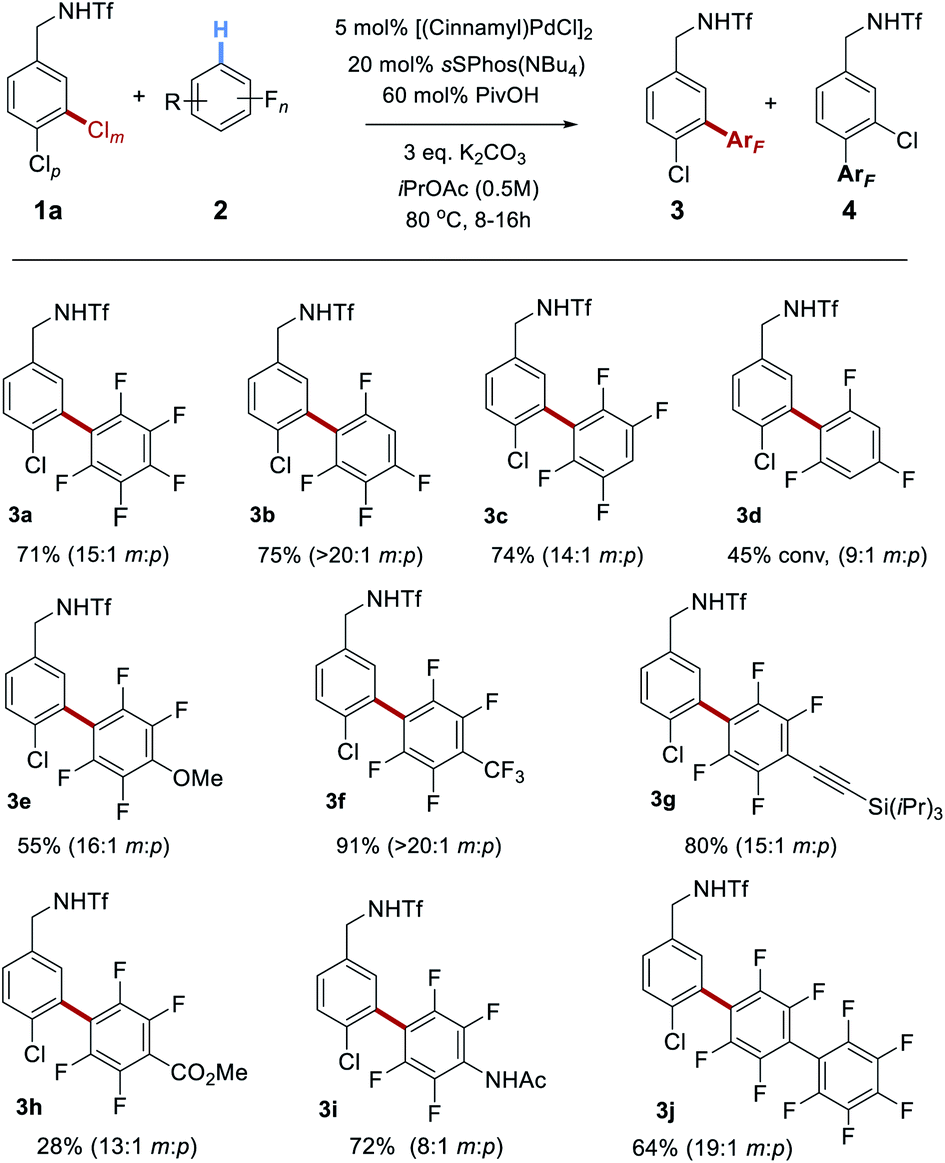 | ||
| Scheme 2 Scope of the perfluoroarene reaction component in coupling with 1a. | ||
We next sought to determine whether the reaction would be compatible with fluorinated heteroarenes and so evaluated several variously fluorinated pyridines (Scheme 3). Pleasingly, 2,3,5,6-tetrafluoropyridine was found to give excellent yield and site-selectivity (5a). Two different constitutional isomers of trifluorinated pyridine also reacted well, with C–H activation viable at either the C4- (5b) or C3- (5c) position relative to the pyridine nitrogen, dependant on the nature of the fluorination. Even 3,5-difluoropyridine reacted, albeit with low conversion (5d). In this case presumably the inductive withdrawal of the pyridine nitrogen compensates for the low degree of fluorination, still permitting some reactivity to be obtained, and representing the current limit of reactivity according to the present protocol.
 | ||
| Scheme 3 Evaluation of fluorinated pyridines as coupling partners with 1a. | ||
We next explored the scope of the aryl chloride component (Scheme 4). Extending the chain length to encompass phenethylamine (5e) and phenylpropylamine (5f) derivatives was tolerated, with reduced but still synthetically useful levels of site-selectivity. We found that benzylic substitution poses no problem for the catalyst control with both methyl (5g) and phenyl (5h) groups at that position giving excellent results.
 | ||
| Scheme 4 Evaluation of various Brønsted acidic groups able to interact with catalyst resulting in selectivity for reaction at the meta-chloride. | ||
Whilst the triflyl-protected benzylamines can be manipulated post-reaction, obtaining the free amine is challenging. We demonstrate that the parent benzylamine can be readily converted to the corresponding potassium sulfamate salt by treatment with chlorosulfonic acid followed by potassium hydroxide.11a This sulfamate salt 1b undergoes highly site-selective coupling with tetrafluoropyridine (13:1). The product was subjected to acid-promoted sulfamate cleavage followed by trifluoroacetylation, to enable isolation as amide 5i. As found previously with cross-coupling, a sulfonate as well accommodated as an electrostatic directing group in this C–H activation reaction, giving excellent selectivity for coupling at Clm (5j, after conversion to the corresponding sulfonamide). Herein, we also demonstrate that a mono-basic benzyl phosphonate also results in excellent site-selectivity, with the coupled product isolated after methylation as 5k. Perhaps the most commonly encountered Brønsted acidic functional group, the carboxylic acid, functions as an excellent electrostatic directing group, tolerating a variety of chain lengths. Chlorinated phenyl acetic acid (5m), hydrocinnamic acid (5n) and phenylbutyric acid (5o) all resulted in excellent site-selectivity. Furthermore, an alkene could be tolerated in the chain without side reactions and with no erosion of site-selectivity (5p). We next sought to evaluate the ability of our ligand to override a substrate's innate selectivity. As anticipated, coupling of 3,4-dichlorobenzoic acid with tetrafluoropyridine using SPhos as ligand gave very high selectivity for the para-chloride (6l), presumably due to substrate electronics rendering this the most electron deficient of the two C–Cl bonds. In contrast, simply switching the ligand to sSPhos rendered a switch in site-selectivity giving a remarkable 4:1 ratio of coupling at Clmvs. Clp, and 5l as major product.
Finally, we disclose that thiazole N-oxides and pyrazine N-oxides are viable C–H activation partners to be used in place of the fluoroarenes (Scheme 5).21 These were demonstrated on two different substrate classes of aryl chloride, triflamide 1a and sulfonate 1c. Although the conversions were only moderate under the conditions optimized for the perfluoroarenes, importantly site-selectivity was excellent in both cases and these promising results demonstrate the broader potential of this strategy.
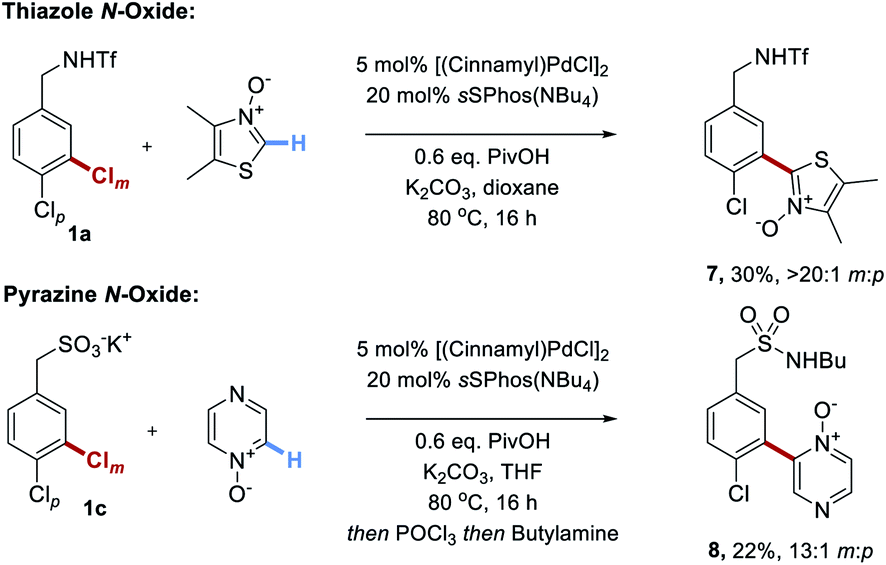 | ||
| Scheme 5 Unoptimized couplings of two examples of heterocyclic N-oxides with two different classes of substrate. | ||
Conclusions
In summary, we have demonstrated that electrostatically-directed palladium catalysis using sulfonylated phosphine ligands is compatible with CMD-type C–H activation in a single catalytic cycle. This results in a highly site-selective fluoroarylation of a number of substrates bearing two undifferentiated C–Cl bonds, with anionic handles for the electrostatic interaction encompassing triflamide, carboxylate, phosphonate, sulfonate and sulfamate. From a practical viewpoint, this work expands the scope of coupling partners able to engage in this site-selective coupling, since fluoroaryl boronic acids were incompatible with the cross-coupling protocol. More importantly though, this is a powerful demonstration that non-covalently-directed catalysis for control of site-selectivity can be used in conjunction with C–H activation. This paves the way for future, related developments in which site-selectivity may be controlled in the C–H activation step itself.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to AstraZeneca for a PhD studentship through the AZ-Cambridge PhD program (W. A. G.), the Royal Society for a University Research Fellowship (R. J. P.), the EPSRC (EP/N005422/1) and the ERC (NonCovRegioSiteCat, Starting Grant 757381). Thanks to Iain Cumming (AstraZeneca) for useful discussion.Notes and references
- (a) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (c) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (d) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- (a) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311–315 CrossRef PubMed; (b) P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed.
- S. M. Rummelt, K. Radkowski, D.-A. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506–5519 CrossRef CAS PubMed.
- W. A. Golding, R. Pearce-Higgins and R. J. Phipps, J. Am. Chem. Soc., 2018, 140, 13570–13574 CrossRef CAS PubMed.
- (a) R. Breslow, X. Zhang and Y. Huang, J. Am. Chem. Soc., 1997, 119, 4535–4536 CrossRef CAS; (b) S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed.
- F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef CAS PubMed.
- (a) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (b) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (c) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (d) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- (a) P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2012, 134, 11350–11353 CrossRef CAS PubMed; (b) S. M. Preshlock, D. L. Plattner, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, Angew. Chem., Int. Ed., 2013, 52, 12915–12919 CrossRef CAS PubMed; (c) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (d) H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef CAS PubMed; (e) M. R. Smith, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka and B. Chattopadhyay, ACS Catal., 2018, 8, 6216–6223 CrossRef CAS PubMed; (f) X. Lu, Y. Yoshigoe, H. Ida, M. Nishi, M. Kanai and Y. Kuninobu, ACS Catal., 2019, 9, 1705–1709 CrossRef CAS; (g) S. T. Bai, C. B. Bheeter and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 13039–13043 CrossRef CAS PubMed.
- (a) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed; (b) M. T. Mihai, H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764–3769 CrossRef CAS; (c) B. Lee, M. T. Mihai, V. Stojalnikova and R. J. Phipps, J. Org. Chem., 2019, 84, 13124–13134 CrossRef CAS PubMed.
- (a) B. Chattopadhyay, J. E. Dannatt, I. L. Andujar-De Sanctis, K. A. Gore, R. E. Maleczka, D. A. Singleton and M. R. Smith, J. Am. Chem. Soc., 2017, 139, 7864–7871 CrossRef CAS PubMed; (b) M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed; (c) R. Bisht, M. E. Hoque and B. Chattopadhyay, Angew. Chem., Int. Ed., 2018, 57, 15762–15766 CrossRef CAS PubMed.
- (a) M. T. Mihai, B. D. Williams and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 15477–15482 CrossRef CAS PubMed; (b) J. R. Montero Bastidas, T. J. Oleskey, S. L. Miller, M. R. Smith and R. E. Maleczka, J. Am. Chem. Soc., 2019, 141, 15483–15487 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (c) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (e) N. Della Ca', M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (g) L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (b) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef PubMed; (c) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed; (d) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (f) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- (a) For reviews on use of transient covalent directing groups in Pd-catalyzed C–H activation, see:; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (c) S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC; (d) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC; (e) For selected examples, see:; (f) Y. Liu and H. Ge, Nat. Chem., 2016, 9, 26 CrossRef; (g) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 CrossRef CAS PubMed; (h) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS PubMed; (i) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed; (j) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073–1076 CrossRef CAS PubMed; (k) X. Zhang, H. Zheng, J. Li, F. Xu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2017, 139, 14511–14517 CrossRef CAS PubMed; (l) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564–3568 CrossRef CAS PubMed.
- (a) For examples of templates attached through reversible metal–ligand covalent interactions for the remote C–H functionalization of arenes, see:; (b) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538 CrossRef CAS PubMed; (c) T. K. Achar, K. Ramakrishna, T. Pal, S. Porey, P. Dolui, J. P. Biswas and D. Maiti, Chem.–Eur. J., 2018, 24, 17906–17910 CrossRef CAS PubMed; (d) K. Ramakrishna, J. P. Biswas, S. Jana, T. K. Achar, S. Porey and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13808–13812 CrossRef CAS PubMed.
- T. N. Hooper, M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS PubMed.
- (a) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed; (b) M. Lafrance, D. Shore and K. Fagnou, Org. Lett., 2006, 8, 5097–5100 CrossRef CAS PubMed; (c) J. Guihaumé, E. Clot, O. Eisenstein and R. N. Perutz, Dalton Trans., 2010, 39, 10510–10519 RSC; (d) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed; (e) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- (a) J.-P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781–7786 CrossRef CAS PubMed; (b) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3276–3277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. See DOI: 10.1039/d0sc00105h |
| This journal is © The Royal Society of Chemistry 2020 |