
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A palladium-catalyzed C–H functionalization route to ketones via the oxidative coupling of arenes with carbon monoxide†
Taleah M.
Levesque
,
R. Garrison
Kinney
and
Bruce A.
Arndtsen
 *
*
Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: bruce.arndtsen@mcgill.ca
First published on 20th February 2020
Abstract
We describe the development of a new palladium-catalyzed method to generate ketones via the oxidative coupling of two arenes and CO. This transformation is catalyzed by simple palladium salts, and is postulated to proceed via the conversion of arenes into high energy aroyl triflate electrophiles. Exploiting the latter can also allow the synthesis of unsymmetrical ketones from two different arenes.
Introduction
Metal-catalyzed arene C–H bond functionalization has emerged as a powerful technique for the efficient and atom-economical build-up of organic products from feedstock chemicals.1 One important class of these reactions is to form carbon–carbon bonds from the oxidative coupling of two separate C–H bond containing substrates.2 In contrast to classical synthetic methods employing pre-functionalized substrates (e.g. cross-coupling reactions), this strategy offers a platform to assemble biaryls and related structures directly from arenes with high step efficiency and minimal waste (Fig. 1a).3 A potentially useful variant would be to perform these in concert with reactive units such as carbon monoxide. Carbon monoxide is a broadly available C1 building block, and its coupling with organic fragments leads to the formation of valuable carbonyl-containing products.4 Moreover, unlike inert aryl–aryl bonds, carbonyl derivatives such as ketones number among the most synthetically versatile functionalities in organic chemistry. The palladium-catalyzed carbonylative functionalization of aryl or even alkyl C–H bonds is well established, and dates back to early work by Fujiwara and others.5 However, these reactions commonly lead to carboxylic acids or their derivatives (e.g. esters, amides), and are rarely applicable to the assembly of ketones.6 This limitation has been attributed in part to the favored reaction of Pd-acyl intermediates with the carboxylate ligands that are often required for the C–H palladation step, therefore leading instead to the formation of anhydrides.7 As shown by Lei, this can be avoided by performing oxidative C–H functionalization in an intramolecular fashion (Fig. 1b).8 Nevertheless, the ability to assemble aryl ketones via the simple coupling of its two most fundamental building blocks, arenes and carbon monoxide, has to date presented an unmet challenge.9,10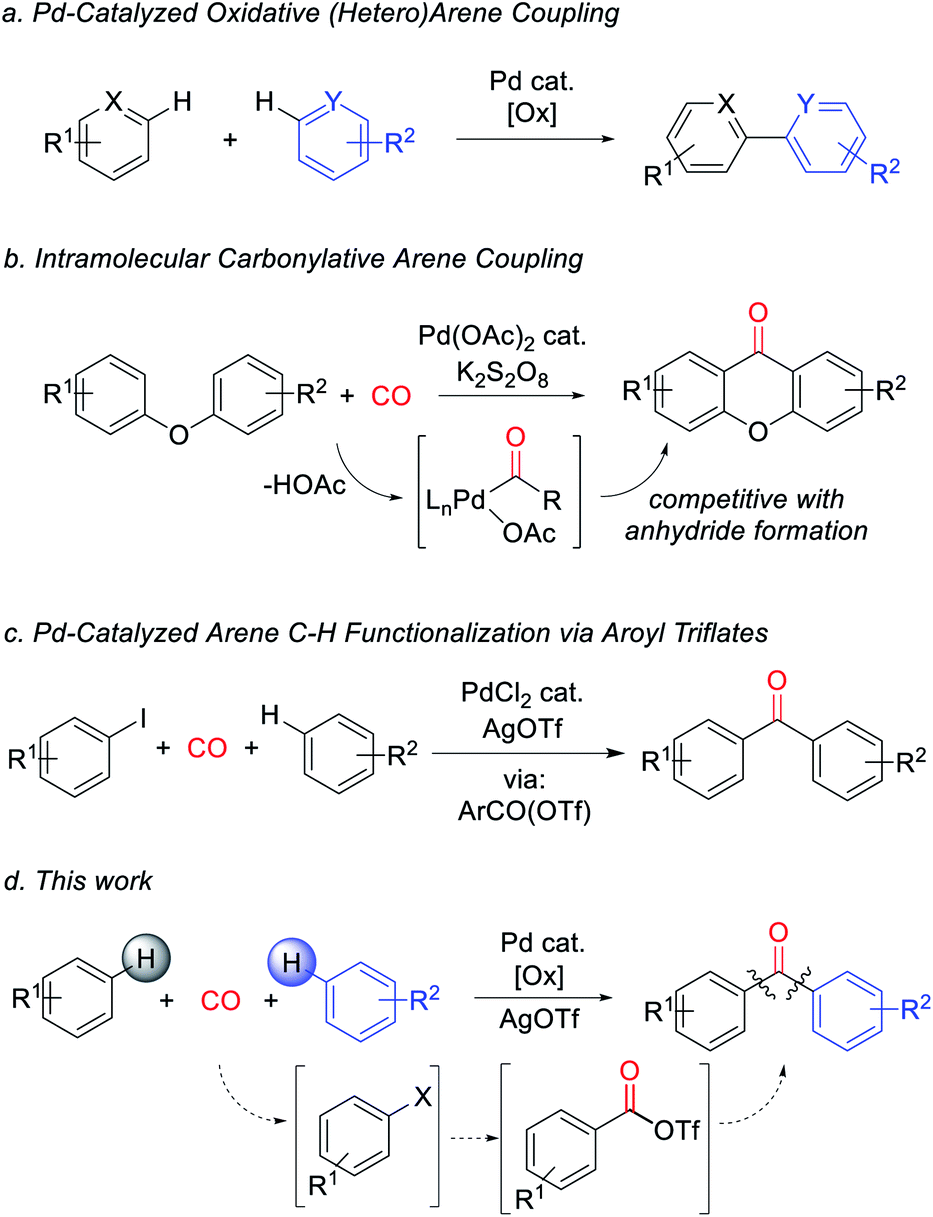 | ||
| Fig. 1 Approaches to intra- and intermolecular oxidative C–H functionalization and application to ketone synthesis. (a) Oxidative arene coupling. (b) Intramolecular carbonylative arene coupling. (c) Pd-catalyzed arene functionalization with aroyl triflates. (d) This work. | ||
We have recently described an alternative approach to carbonylative C–H functionalization, wherein palladium catalysts can mediate the in situ formation of reactive aroyl electrophiles such as acyl iodides and even acyl triflates.11 The latter have the ability to react with unactivated arenes and allowed the development of a carbonylative method to functionalize arenes to form ketones (Fig. 1c). In considering this chemistry, we questioned if this approach could be taken even further and offer a pathway to perform carbonylative arene/arene coupling with carbon monoxide. In principle, this might be accomplished by devising a method to replace aryl iodides with an oxidative carbonylative platform to convert arenes themselves to high energy acyl triflate electrophiles (Fig. 1d). We describe herein our studies towards such a system. This has led to the discovery of the first catalytic route to assemble ketones directly from two arenes and carbon monoxide. Mechanistic studies suggest this reaction proceeds via a series of steps that ultimately convert arenes into high energy aroyl triflate electrophiles. Exploitation of the latter has opened a novel route to generate heterocoupled ketones from two separate arenes.
Results and discussion
Our research on this topic grew out of a study on the mechanism of the Pd(0)-catalyzed carbonylative formation of aroyl triflate electrophiles from aryl iodides.11d This reaction can be performed using PdCl2 and related Pd(II) catalyst precursors (Fig. 1c), but it was unclear how these Pd(II) sources are reduced to Pd(0) to mediate the reaction. Carbon monoxide has been established to reduce Pd(II) to Pd(0).12,13 However, this often occurs in the presence of nucleophiles (e.g. water) that can trap the oxidized carbon monoxide fragment, and the reaction conditions did not include an obvious nucleophile, or even a dative ligand (e.g. phosphines) that might be oxidized by Pd(II). To probe this, the stoichiometric reaction of PdCl2 with carbon monoxide and AgOTf was examined in benzene. To our surprise, this leads to the formation of an aromatic product, benzophenone, in up to 71% yield (Scheme 1).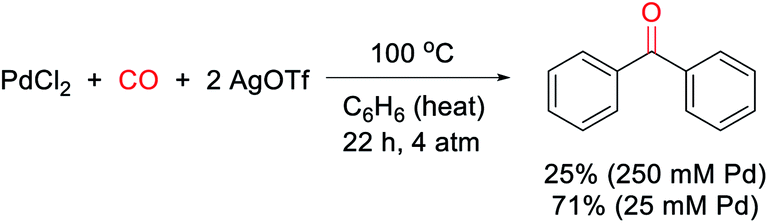 | ||
| Scheme 1 Generation of benzophenone via stoichiometric, PdCl2-mediated arene/CO coupling. | ||
The ability of metal salts such as PdCl2 to mediate the oxidative carbonylation of arenes to ketones is to our knowledge unknown.13 While there are several pathways by which this stoichiometric reaction may occur (vide infra), the formal by-product of this reaction is Pd(0), suggesting that a suitable oxidant may transform this stoichiometric chemistry into a catalytic route to form aryl ketones. As shown in Table 1, the addition of various metal salt oxidants that are commonly employed in related oxidative coupling chemistry to the reaction of benzene and CO with 10 mol% PdCl2 catalyst leads to minimal reaction (entries 1–3), and also inhibits the stoichiometric formation of benzophenone. However, we were pleased to find that simple iodine is an effective oxidant and allows the catalytic formation of benzophenone in good yield (79%, entry 4). Further experimentation showed that the reaction does not require the use of arene as solvent and proceeds in high yield with a near stoichiometric amount of arene in 1,2-dichloroethane (92%, entry 5) and at temperatures as low as 60 °C with 1 atm CO (entries 6–10). The reaction here was performed with t-butylbenzene to aid in situ characterization, but is equally efficient with benzene (entry 11). Simple [Pd(allyl)Cl]2 is the most effective catalytic precursor under these conditions,14 and the reaction can be performed with as little as 0.1 mol% [Pd(allyl)Cl]2 at elevated temperatures (entry 12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd cat. | Ligand | Temp. | Solvent | [Ox] | Yield (%) |
| a Conditions: arene (0.53 mmol), silver triflate (176 mg, 0.68 mmol), Ox (0.25 mmol), 5 mol% Pd, 1 atm CO, 22 h, R = tBu. b 10 mol% Pd, 4 atm CO, R = H (entries 1–4), tBu (entry 5). c 10 mol%. d 0.75 mmol arene, R = tBu (R = H in brackets). e 0.1 mol% [Pd(allyl)Cl]2. | ||||||
| 1b | PdCl2 | — | 100 °C | C6H6 | Ce(OTf)4 | 0 |
| 2b | PdCl2 | — | 100 °C | C6H6 | Na2S2O8 | 2 |
| 3b | PdCl2 | — | 100 °C | C6H6 | CuCl2 | 4 |
| 4b | PdCl2 | — | 100 °C | C6H6 | I2 | 79 |
| 5b | PdCl2 | — | 100 °C | 1,2-DCE | I2 | 92 |
| 6 | PdCl2 | — | 60 °C | 1,2-DCE | I2 | 23 |
| 7 | Pd(OAc)2 | — | 60 °C | 1,2-DCE | I2 | 60 |
| 8 | [Pd(allyl)Cl]2 | PPh3c | 60 °C | 1,2-DCE | I2 | 78 |
| 9 | [Pd(allyl)Cl]2 | PtBu3c | 60 °C | 1,2-DCE | I2 | 57 |
| 10 | [Pd(allyl)Cl]2 | — | 60 °C | 1,2-DCE | I2 | 87 |
| 11 | [Pd(allyl)Cl]2 | — | 60 °C | 1,2-DCE | I2 | 93 (94)d |
| 12 | [Pd(allyl)Cl]2e | — | 100 °C | 1,2-DCE | I2 | 72 |
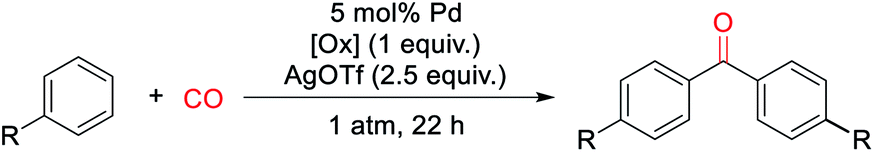
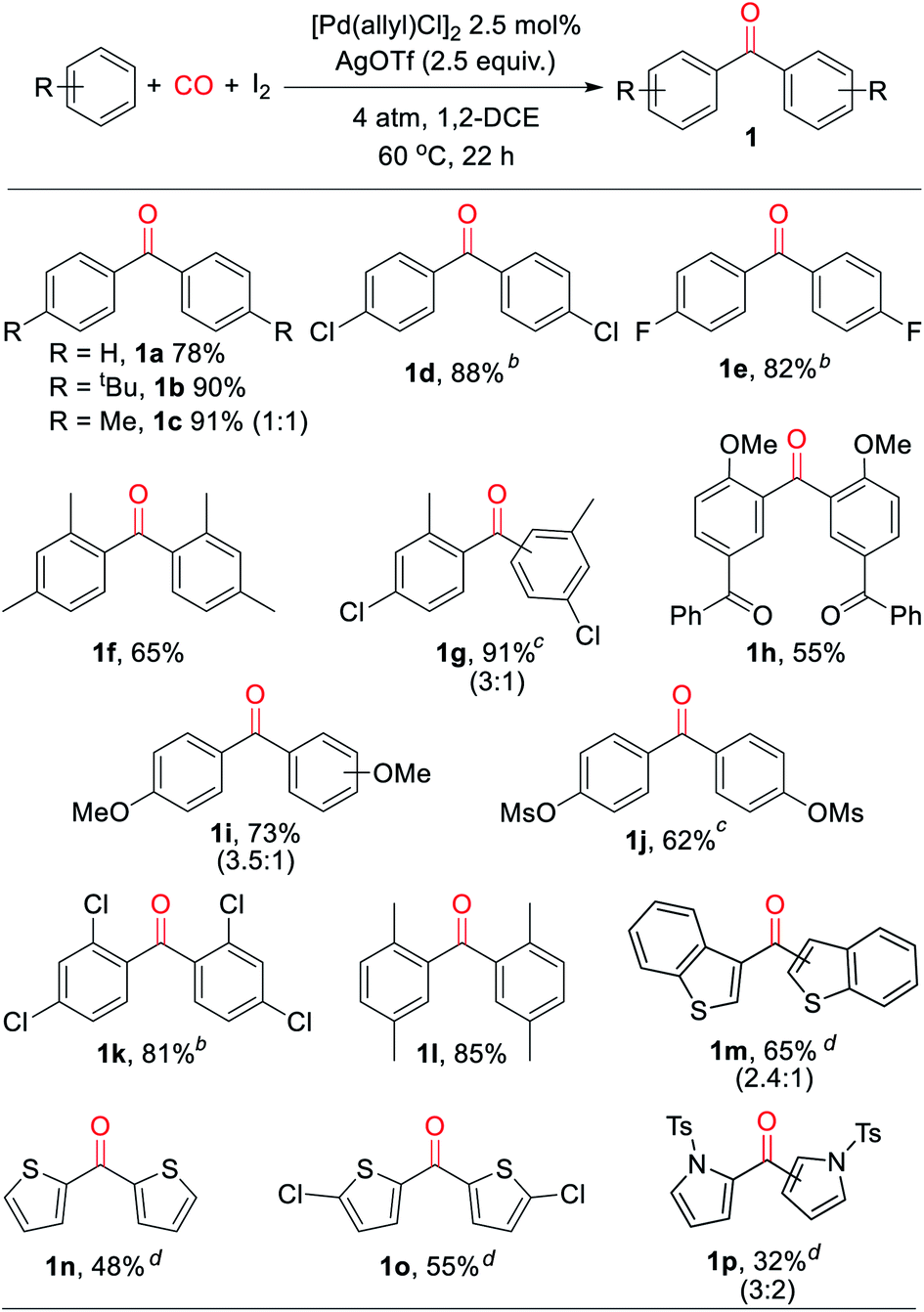
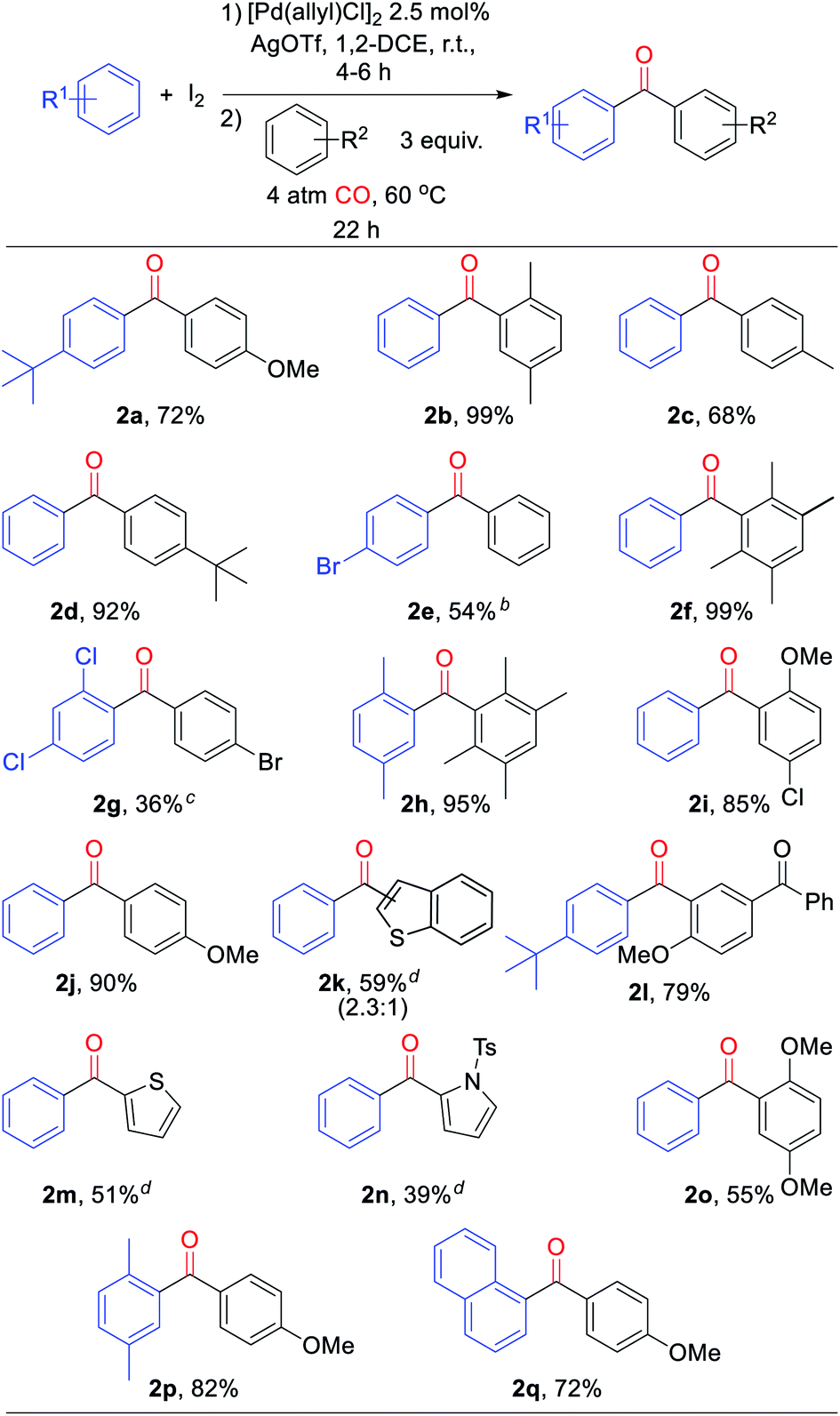
With a palladium-catalyzed method to assemble ketones from arenes and carbon monoxide in hand, we next turned to probing its generality. As shown in Table 2, various electron neutral and electron rich arenes can be employed in this reaction and form ketones in high yields. These include alkyl-substituted arenes (1b, 1c, 1f) as well as more electron rich anisole derivatives (1i, 1j). In each of the mono-substituted arenes, we observed 4,4′-substituted ketones as the major isolated product, consistent with an electrophilic mechanism for functionalization. Electron deficient substrates, such as mono- and di-halogenated arenes, react under more pressing conditions to form ketones in neat arene (1d, 1e, 1k). More sterically hindered 1,3- and 1,4-di-substituted arenes can also be converted to ketones (1f–h, 1k, 1l). In these cases, functionalization proceeds again at the most electron rich site on the arene. This oxidative carbonylation can also be applied to the synthesis of ketones from heteroarenes, with examples including thiophenes (1n, 1o), benzothiophene (1m), and N-tosyl pyrrole (1p). The addition of 2,6-tbutylpyridine base is required for these latter examples to inhibit substrate decomposition. Overall, this provides what is to our knowledge the first general catalytic method to assemble aryl ketones in one step from their two most fundamental building blocks, arenes and carbon monoxide, and without the required use of the synthetic, expensive, and/or unstable (hetero)aryl iodides (and organometallic reagents) commonly employed in carbonylative ketone synthesis.
a Conditions: arene (1.50 mmol), AgOTf (350 mg, 1.36 mmol), I2 (126 mg, 0.50 mmol), [Pd(allyl)Cl]2 (5 mg, 0.013 mmol), 1,2-dichloroethane (2 mL), 4 atm CO, 22 h, 60 °C (see ESI for details). Isolated yield (ratio of 4′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2′ isomers).
b At 150 °C in arene solvent.
c At 100 °C.
d With 2,6-di-tert-butylpyridine (258 mL, 1.15 mmol) (ratio of 3′:2′ isomers). :2′ isomers).
b At 150 °C in arene solvent.
c At 100 °C.
d With 2,6-di-tert-butylpyridine (258 mL, 1.15 mmol) (ratio of 3′:2′ isomers).
|
|---|
|
We next turned to examine how this transformation proceeds. As mechanistic possibilities, the stoichiometric reaction of PdCl2 (Scheme 1) suggests the potential in situ formation of phosgene equivalents upon reduction of Pd(II) by CO (Fig. 2a, path A),13 or the build-up of electrophilic Pd(II) triflate intermediates that may directly palladate arenes (path B).15 These could each lead to aroyl triflate intermediates for subsequent Friedel–Crafts acylations,11d and are consistent with the regioselectivity in the ketone products formed. The re-oxidation of Pd(0) by I2 would then make these viable cycles for catalysis. However, iodine is also established to react with arenes in the presence of silver triflate to form aryl iodides.16 It is therefore possible that the catalytic arene carbonylation occurs via an alternative Pd(0) cycle to form aroyl triflate products (path C). Monitoring the reaction by in situ1H and 13C NMR analysis show that the latter mechanism is likely the case during catalysis, and we observe the rapid, high yield build-up of aryl iodide within 1 h under the catalytic conditions (Fig. 2b, 94% yield). Continued heating converts this aryl iodide to ketone 1a. Similarly, control experiments show that the AgOTf mediated iodination of the arene is rapid (30 min at r.t.), relative to catalytic ketone formation (23% after 22 h at r.t.; Fig. 2c). Thus, in the presence of I2 as oxidant, these data suggest that the direct iodination of the arene is the most rapid pathway (path C). Nevertheless, the ability of Pd(II) salts themselves to mediate the direct carbonylative C–H functionalization of arenes also appears to be relevant to catalysis, as it allows the activation of the Pd(II) pre-catalyst (e.g.Table 1). The compatibility between Pd(0) catalysis and I2 is unusual, since Pd(0) can also undergo rapid oxidation. This presumably reflects the rapid rate of arene iodination relative to the slower carbonylation and build-up of Pd(0), as well as the ability of any in situ generated Pd(II) to re-enter the cycle (e.g. via paths A or B). The lack of potentially oxidizable donor ligands in the reaction further enhances this compatibility. Together, this suggests a new avenue to use Pd(0) catalysis in oxidative coupling chemistry with I2.
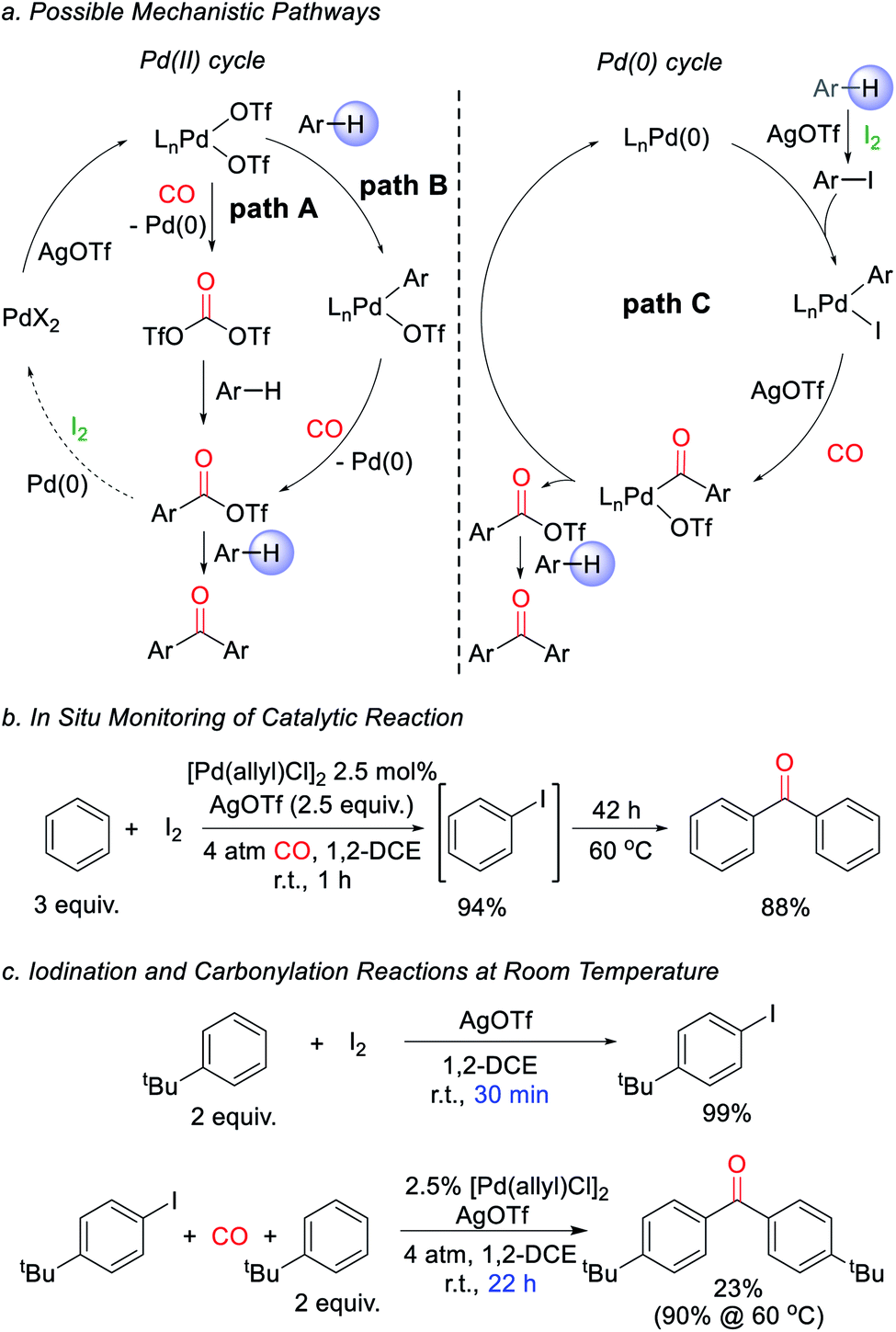 | ||
| Fig. 2 Proposed catalytic pathway for ketone formation. (a) Potential mechanisms. (b) In situ monitoring of reaction. (c) Relative rates of iodination and carbonylation. | ||
From a synthetic perspective, the generation of halogenated arene intermediates can have useful implications. Since these intermediates are formed more rapidly than carbonylation, the rate difference can be exploited to perform cross arene/arene coupling to build up more structurally diverse ketones. The latter can present a challenge with many oxidative arene/arene coupling reactions, and typically require significant differences in the electronic, steric, or coordinating ability of the two substrates.2 After examination of various reaction conditions, we were pleased to find that the ambient temperature reaction of arene, [Pd(allyl)Cl]2 catalyst, I2, and AgOTf, followed by the subsequent addition of a second, more electron rich arene and carbon monoxide leads to the clean overall heterocoupling of arenes to ketones (Table 3). Both electron neutral and electron rich arenes can be easily cross-coupled to form unsymmetrical ketones in high yield, including sterically encumbered tetra-substituted arenes and an activated benzophenone (2f, 2h, 2l). Employing more pressing conditions allows for the use of deactivated arenes such as bromo- or dichlorobenzene (2e, 2g), and does so without secondary activation of these carbon–halogen bonds. Thiophene, N-substituted pyrrole, and benzothiophene can also be incorporated into this reaction to create mixed (hetero)aryl ketones (2k, 2m, 2n).
|
a Conditions: arene (1.00 mmol), AgOTf (339 mg, 1.32 mmol), I2 (126 mg, 0.49 mmol), [Pd(allyl)Cl]2 (5 mg, 0.013 mmol), 1,2-dichloroethane (2 mL), 4–6 h, r.t., then second arene (1.50 mmol), 4 atm CO, 22 h, 60 °C (see ESI for details). Isolated yields (3′:2′ ratio).
b Both steps at 100 °C.
c Step 1 at 100 °C, 22 h; step 2 at 150 °C w/10 eq. bromobenzene.
d With 2,6-di-tert-butylpyridine (1.15 mmol).
|
|---|
|
Finally, we have examined the use of this system to assemble more structurally elaborate products. As an example, crystal violet (3, Scheme 2) is a common dye exploited in textiles. Crystal violet is typically generated starting from reactive, synthetic acylating agents (e.g. phosgene), in a sequence that proceeds through the initial build-up of ketone 4.17 Alternatively, the palladium-catalyzed oxidative carbonylation of N,N-dimethylaniline can offer a method to generate 3 from the parent arene and carbon monoxide (Scheme 2). While 3 is generated in moderate yield (41% together with its de-methylated isomer), it represents the product of three concurrent arene C–H bond functionalization steps. This reaction presumably proceeds via the initial generation of 4 that undergoes a spontaneous Friedel–Crafts reaction to liberate H2O, which can also be trapped by the in situ generated aroyl electrophiles. As far as we are aware, this is the first synthesis of such an advanced structure directly from arenes and CO.
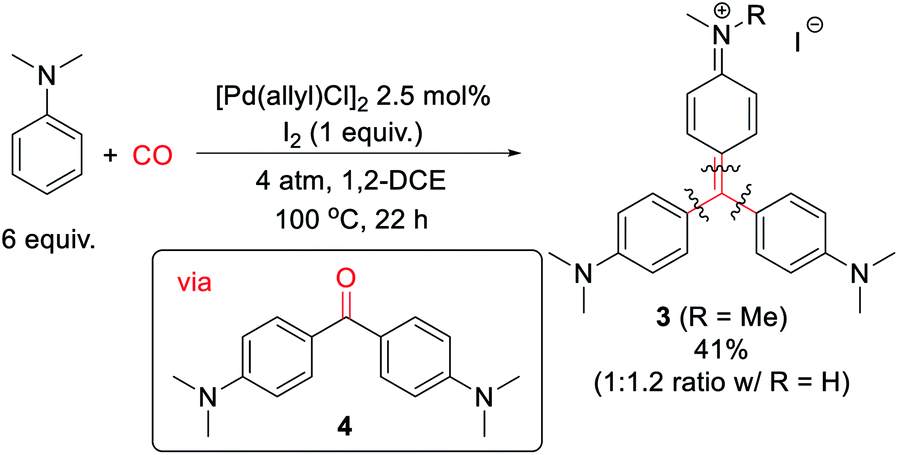 | ||
| Scheme 2 Synthesis of crystal violet from dimethylaniline and carbon monoxide. | ||
Conclusions
In conclusion, we have described herein the first catalytic route to prepare ketones from arenes and CO. The reaction generates either symmetrical or unsymmetrical aryl ketones in high yields does so from combinations of stable, commercially available, and easily modulated reagents, and without the need for synthetic and often expensive aryl iodide reagents. Mechanistic analysis suggests this transformation proceeds via the palladium-catalyzed, in situ generation of potent acylating electrophiles (acyl triflates) from arenes. Considering the broad utility of acylating electrophiles, we anticipate that the ability to access these intermediates from arenes could prove relevant to the design of various new approaches to carbonylative C–H bond functionalization chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NSERC, the Canadian Foundation for Innovation (CFI), the McGill Sustainability Systems Initiative (MSSI) through the Ideas Fund, and the FQRNT supported Centre for Green Chemistry and Catalysis for funding this research.Notes and references
- For reviews: (a) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (d) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (e) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed; (f) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (g) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (h) P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS PubMed.
- (a) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (b) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (d) Y.-F. Zhang and Z.-J. Shi, Acc. Chem. Res., 2019, 52, 161–169 CrossRef CAS PubMed; (e) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC; (f) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- For representative examples: (a) R. Li, L. Jiang and W. Lu, Organometallics, 2006, 25, 5973–5975 CrossRef CAS; (b) X. Wang, D. Leow and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 13864–13867 CrossRef CAS PubMed; (c) D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172–1175 CrossRef CAS PubMed; (d) T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455–4464 CrossRef CAS PubMed; (e) K. Kawasumi, K. Mochida, T. Kajino, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 418–421 CrossRef CAS PubMed; (f) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS PubMed; (g) G. Brasche, J. García-Fortanet and S. L. Buchwald, Org. Lett., 2008, 10, 2207–2210 CrossRef CAS PubMed; (h) K. Matsumoto, S. Takeda, T. Hirokane and M. Yoshida, Org. Lett., 2019, 21, 7279–7283 CrossRef CAS PubMed; (i) D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 5704–5707 CrossRef CAS PubMed; (j) B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115–1118 CrossRef CAS PubMed; (k) Y. Wei and W. Su, J. Am. Chem. Soc., 2010, 132, 16377–16379 CrossRef CAS PubMed; (l) C.-Y. He, S. Fan and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12850–12852 CrossRef CAS PubMed.
- (a) M. Beller and X.-F. Wu, Transition Metal Catalyzed Carbonylations-Carbonylative Activation of C-X Bonds, Springer, Berlin, 2013 CrossRef; (b) Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC; (c) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (d) I. Omae, Coord. Chem. Rev., 2011, 255, 139–160 CrossRef CAS.
- For reviews see: (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed. For recent examples: (b) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 CrossRef CAS PubMed; (c) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS PubMed; (d) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686–693 CrossRef CAS PubMed; (e) Z.-H. Guan, M. Chen and Z.-H. Ren, J. Am. Chem. Soc., 2012, 134, 17490–17493 CrossRef CAS PubMed; (f) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598–10601 CrossRef CAS PubMed; (g) D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Guant, Science, 2016, 354, 851–857 CrossRef CAS PubMed.
- Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
- (a) M. A. Campo and R. C. Larock, J. Org. Chem., 2002, 67, 5616–5620 CrossRef CAS PubMed; (b) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 7316–7319 CrossRef CAS PubMed; (c) Z. Lian, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 1870–1873 RSC; (d) J. Liu, Z. Wei, H. Jiao, R. Jackstell and M. Beller, ACS Cent. Sci., 2018, 4, 30–38 CrossRef CAS PubMed; (e) T. A. Cernak and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 3124–3125 CrossRef CAS PubMed; (f) F. Zeng and H. Alper, Org. Lett., 2013, 15, 2034–2037 CrossRef CAS PubMed.
- (a) H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5204–5207 CrossRef CAS PubMed; (b) J. Wen, S. Tang, F. Zhang, R. Shi and A. Lei, Org. Lett., 2017, 19, 94–97 CrossRef CAS PubMed.
- (a) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC; (b) M.-N. Zhao, L. Ran, M. Chen, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, ACS Catal., 2015, 5, 1210–1213 CrossRef CAS.
- For an alternative approach from aldehydes: (a) S. Tetsuya, I. Tomoaki, M. Masahiro and N. Masakatsu, Chem. Lett., 1996, 25, 823–824 CrossRef; (b) S. Ko, B. Kang and S. Chang, Angew. Chem., Int. Ed., 2005, 44, 455–457 CrossRef CAS PubMed; (c) J. Ruan, O. Saidi, J. A. Iggo and J. Xiao, J. Am. Chem. Soc., 2008, 130, 10510–10511 CrossRef CAS PubMed; (d) X. Jia, S. Zhang, W. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120–3123 CrossRef CAS PubMed; (e) P. Álvarez-Bercedo, A. Flores-Gaspar, A. Correa and R. Martin, J. Am. Chem. Soc., 2010, 132, 466–467 CrossRef PubMed; (f) B. Suchand and G. Satyanarayana, J. Org. Chem., 2016, 81, 6409–6423 CrossRef CAS PubMed; (g) T. Wakaki, T. Togo, D. Yoshidome, Y. Kuninobu and M. Kanai, ACS Catal., 2018, 8, 3123–3128 CrossRef CAS.
- (a) J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841–16844 CrossRef CAS PubMed; (b) J. S. Quesnel, L. V. Kayser, A. Fabrikant and B. A. Arndtsen, Chem.–Eur. J., 2015, 21, 9550–9555 CrossRef CAS PubMed; (c) J. Tjutrins and B. A. Arndtsen, J. Am. Chem. Soc., 2015, 137, 12050–12054 CrossRef CAS PubMed; (d) R. G. Kinney, J. Tjutrins, G. M. Torres, N. J. Liu, O. Kulkarni and B. A. Arndtsen, Nat. Chem., 2018, 10, 193–199 CrossRef PubMed; (e) P.-L. Lagueux-Tremblay, A. Fabrikant and B. A. Arndtsen, ACS Catal., 2018, 8, 5350–5354 CrossRef CAS.
- (a) J. Tsuji, Acc. Chem. Res., 1969, 2, 144–152 CrossRef CAS; (b) J. Tsuji, Palladium Reagents and Catalysts – New Perspectives for the 21st Century, John Wiley & Sons Ltd., West Sussex, 2004 CrossRef; (c) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (d) X.-F. Wu, H. Neumann, A. Spannenberg, T. Schulz, H. Jiao and M. Beller, J. Am. Chem. Soc., 2010, 132, 14596–14602 CrossRef CAS PubMed.
- PdCl2 has been suggested to react with CO to generate phosgene derivatives. (a) T. A. Stromnova, M. N. Vargaftik and I. I. Moiseev, J. Organomet. Chem., 1983, 252, 113–120 CrossRef CAS; (b) D. B. Dell'Amico, F. Calderazzo and N. Zandona, Inorg. Chem., 1984, 23, 137–140 CrossRef; (c) S. Baig, B. Richard, P. Serp, C. Mijoule, K. Hussein, N. Guihéry, J.-C. Barthelat and P. Kalck, Inorg. Chem., 2006, 45, 1935–1944 CrossRef CAS PubMed; (d) F. Ragaini, H. Larici, M. Rimoldi, A. Caselli, F. Ferretti, P. Macchi and N. Casati, Organometallics, 2011, 30, 2385–2393 CrossRef CAS.
- The reaction proceeds well with a near stoichiometric amount of arene (2.1:1 arene to I2 ratio, entry 10), but we observed slightly elevated yields with 1.5 equiv. of arene (3:1 ratio of arene to I2).
- For mechanistic studies with related transition metal acetates: (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (b) S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS; (c) L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821–6836 CrossRef CAS; (d) D. Whitaker, J. Burés and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS PubMed; (e) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS PubMed; (f) M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Organometallics, 2017, 36, 165–171 CrossRef CAS; (g) C. Colletto, A. Panigrahi, J. Fernández-Casado and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS PubMed.
- (a) G. K. Mulholland and Q.-H. Zheng, Synth. Commun., 2001, 31, 3059–3068 CrossRef CAS; (b) S. Wan, S. R. Wang and W. Lu, J. Org. Chem., 2006, 71, 4349–4352 CrossRef CAS PubMed.
- T. Gessner and U. Mayer, Triarylmethane and Diarylmethane Dyes, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00085j |
| This journal is © The Royal Society of Chemistry 2020 |