
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrathin tin monosulfide nanosheets with the exposed (001) plane for efficient electrocatalytic conversion of CO2 into formate†
Hanlin
Chen‡
a,
Junxiang
Chen‡
d,
Jincheng
Si
a,
Yang
Hou
 *ab,
Qiang
Zheng
f,
Bin
Yang
a,
Zhongjian
Li
a,
Liguo
Gao
c,
Lecheng
Lei
a,
Zhenhai
Wen
*d and
Xinliang
Feng
*e
*ab,
Qiang
Zheng
f,
Bin
Yang
a,
Zhongjian
Li
a,
Liguo
Gao
c,
Lecheng
Lei
a,
Zhenhai
Wen
*d and
Xinliang
Feng
*e
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yhou@zju.edu.cn
bNingbo Research Institute, Zhejiang University, Ningbo 315100, China. E-mail: yhou@zju.edu.cn
cSchool Petroleum and Chemical Engineering, Dalian University of Technology, Panjin, 124221, China
dCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: wen@fjirsm.ac.cn
eCenter for Advancing Electronics Dresden (cfaed) and Department of Chemistry and Food Chemistry, Technische Universität Dresden, Mommsenstraße 4, 01062 Dresden, Germany. E-mail: xinliang.feng@tu-dresden.de
fCorrelated Quantum Materials Group, Materials Science and Technology, Division Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN37831, USA
First published on 23rd March 2020
Abstract
Current Sn-based materials are ideal catalysts developed to drive the electrochemical conversion of CO2 to formate, but competing proton reduction to hydrogen is an ever-present drain on catalytic selectivity. Herein, we report a reliable electrochemical exfoliation route, with the assistance of alternating voltage, for large-scale preparation of two-dimensional (2D) ultrathin tin monosulfide nanosheets (SnS NSs), which feature a large lateral size of 1.0 μm with a thickness of ∼5.0 nm. Systematic electrochemical studies demonstrated that the achieved SnS NSs exhibited an outstanding electrocatalytic activity towards the CO2 reduction reaction (CO2RR) to the formate product, as evidenced by a considerable faradaic efficiency (F.E.) of 82.1%, a high partial current density of 18.9 mA cm−2 at −1.1 V, and a low Tafel slope of 180 mV dec−1. Further, using an electrode prepared from the resulting SnS NSs by the particle transfer method, a remarkably high formate F.E. over 91% was achieved in a wide potential window. Such high performance renders the SnS NSs among the best reported tin sulfide-based CO2RR electrocatalysts. Theoretical calculations coupled with comprehensive experimental studies demonstrated that the synergistic effect between the ultrathin layered architecture and dominantly exposed (001) plane of SnS NSs accounted for the uniquely efficient catalytic activity for the CO2RR.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) has been recognized as a promising strategy to convert problematic CO2 into value-added feedstocks, thanks to the multiple advantages of the electrochemical route, including mild conditions (i.e., ambient pressure and room temperature), potential synergy with renewable energy, and product tunability.1–7 However, the electrochemical CO2RR occurs at a potential close to that of the hydrogen evolution reaction (HER), inevitably leading to a rather low selectivity toward the CO2RR since the HER is thermodynamically more favorable in most electrocatalysts. Moreover, the development of advanced CO2RR electrocatalysts still faces issues of high overpotential, low efficiency, slow kinetics, and poor stability.8–10 Thus, exploration and development of high-performance electrocatalysts, i.e., exhibiting high activity, high selectivity, and robust stability, are of great importance and understandably become the necessary prerequisites to fulfill high-efficiency electrochemical CO2 conversion. Among the various CO2RR products, formate (HCOOH) is one of the most appealing products as a liquid fuel with high energy density and is convenient to handle and transport. Although a variety of metal-based materials, such as Pb, Hg, and Cd, have been reported as CO2RR electrocatalysts to produce the HCOOH product, their high toxicity greatly limits their widespread application.11–14 In contrast, as environmentally friendly, relative low cost, non-noble metal catalysts, copper-based, bismuth-based, and tin-based materials have recently received much attention for selective conversion of CO2 into HCOOH due to their intrinsic electrochemical properties, that is, the larger hydrogen evolution potential, the smaller CO adsorption energy, and the stronger stabilization of HCOOH.15–18 For instance, tin sulfides have recently attracted considerable attention for selective conversion of CO2 into HCOOH.11,14 However, the overall electrocatalytic activity and selectivity of tin sulfide-based materials for HCOOH production are not satisfactory due to the limited exposed active sites/planes and low charge transfer rate in bulk materials.19–21 Moreover, it remains unclear which is the major active site for selective catalysis toward the HCOOH product. In this respect, ultrathin nanosheets with a layered crystallographic structure and a specific exposed facet deserve extensive investigation, as they hold promise in further improving the associated electrocatalytic properties and provide a chance to study the catalytically active site, thanks to the dominantly exposed crystalline facet and abundant active sites.20,22–25Herein, we report the efficient synthesis of 2D ultrathin tin monosulfide nanosheets (SnS NSs) through an electrochemical exfoliation method with the assistance of alternating voltage. The thickness of the exfoliated SnS NSs was ∼5.0 nm with an apparent exfoliation yield of 75%. Benefitting from the advantageous ultrathin layer features as well as the highly exposed facets, the achieved SnS NSs exhibited a small Tafel slope of 180 mV dec−1, a high faradaic efficiency (F.E.) of 82.1%, and a large partial current density of 18.9 mA cm−2 at −1.1 V for HCOOH production. In addition, the electrode of the resulting SnS NSs fabricated by a particle transfer method delivered a remarkably high HCOOH F.E. over 91% in a wide potential window, outperforming the bulk SnS and almost all previously reported tin sulfide-based CO2RR electrocatalysts. The experimental results and density functional theory (DFT) calculations shed light on the fact that the tightly stacked (001) high-index facet in SnS NSs is more favorable for producing HCOOH as compared with the (100) plane.
Results and discussion
The electrochemical exfoliation to obtain ultrathin SnS NSs is illustrated in Fig. 1. The bulk SnS with a layered structure was first prepared by a solid–state reaction of Sn and S powders at 500 °C for 24 h. Next, a two-electrode system was set up with the bulk SnS electrode as the working electrode and Pt mesh as the counter electrode for the electrochemical exfoliation in 0.5 M Na2SO4 under alternating voltage. During the cathodic process with a negative voltage of 5.0 V applied at the working electrode, the hydrogen evolution reaction (HER) occurs within SnS interlayer edges, as evidenced by H2 bubbles around the electrode, which induces the electrochemical exfoliation of bulk SnS (Fig. 1a). When tuning to an anodic process by applying a positive voltage of 3.0 V on bulk SnS, the SO42− ions are inserted into SnS crystals (for details see the ESI†), and the interlayer distance of bulk SnS is continuously expanded due to the generation of a large number of H2 and O2 gas molecules between the SnS interlayers (Fig. 1b). As a result, the exfoliated SnS with yellow/black color is removed from the bulk SnS (Fig. 1c).26 The exfoliated SnS powder can be readily collected and dispersed in N,N-dimethylformamide (DMF) to provide a homogeneous and stable dispersion with a concentration of 3.2 mg mL−1 (exfoliation yield of 75%, Fig. S1†). The crystal structure of SnS is shown in Fig. S2,† and more details of the synthesis process and exfoliation mechanism of ultrathin SnS NSs are presented in the ESI.†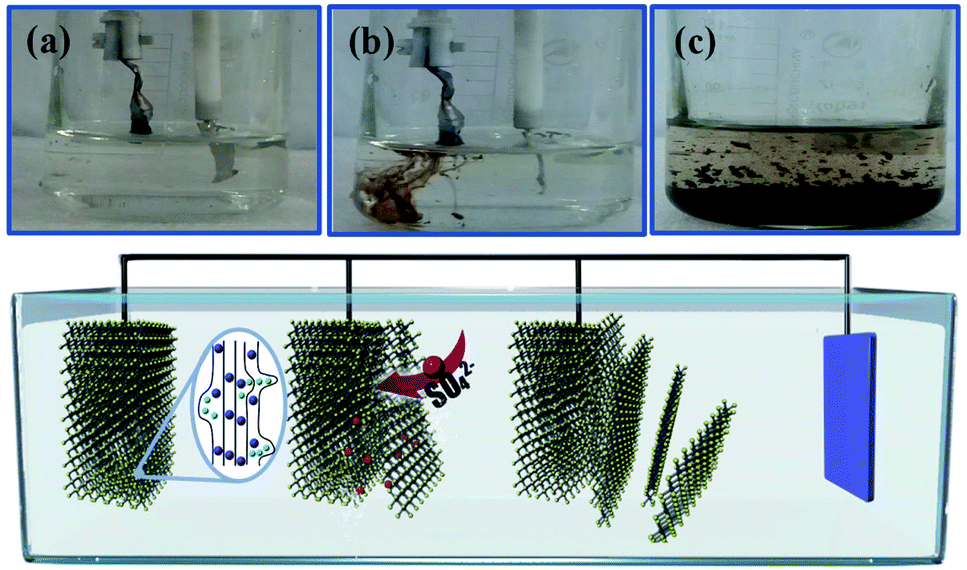 | ||
| Fig. 1 (a) Pictures of the starting bulk SnS with a negative voltage and (b) the exfoliated SnS removed from bulk SnS when changing to a positive voltage. (c) Exfoliated SnS materials dispersed in the solution. | ||
The X-ray photoelectron spectroscopy (XPS) survey spectrum of ultrathin SnS NSs reveals the presence of Sn, S, and O elements, besides the C element (Fig. 2a). In the high-resolution Sn 3d XPS spectrum (Fig. 2b), two Sn 3d5/2 and Sn 3d3/2 peaks of ultrathin SnS NSs can be deconvoluted into four peaks, which are located at 486.0, 486.9, 494.6, and 495.5 eV, respectively.27–29 The peaks with the lower binding energies of 486.0 and 494.6 eV can be assigned to Sn2+.29 The peaks with higher binding energies of 486.9 and 495.5 eV can be attributed to the surface adsorbed oxygen upon exposure to air, which has a higher electronegativity than that of sulfur, thus increasing the Sn binding energy. Fig. 2c presents the high-resolution S 2p XPS spectrum of ultrathin SnS NSs, which is deconvoluted into S 2p3/2 and S 2p1/2 peaks located at 161.2 and 162.2 eV, respectively, ascribed to S2−.29 The Raman spectrum (Fig. S3†) of ultrathin SnS NSs exhibits two typical peaks located at 93 and224 cm−1, corresponding to the Ag modes of the orthorhombic SnS phase.30,31 An obvious decrease in the peak intensity of the Raman spectrum and a blue shift of the Ag band located at 224 cm−1 are observed for ultrathin SnS NSs as compared with bulk SnS, demonstrating the reduced numbers of SnS layers after electrochemical exfoliation.20,27,28 The efficient exfoliation of bulk SnS was further supported by the X-ray diffraction (XRD) patterns, in which the diffraction peaks of ultrathin SnS NSs are broader and weaker than those of the bulk SnS (Fig. S3†), indicating the decreased thickness.28
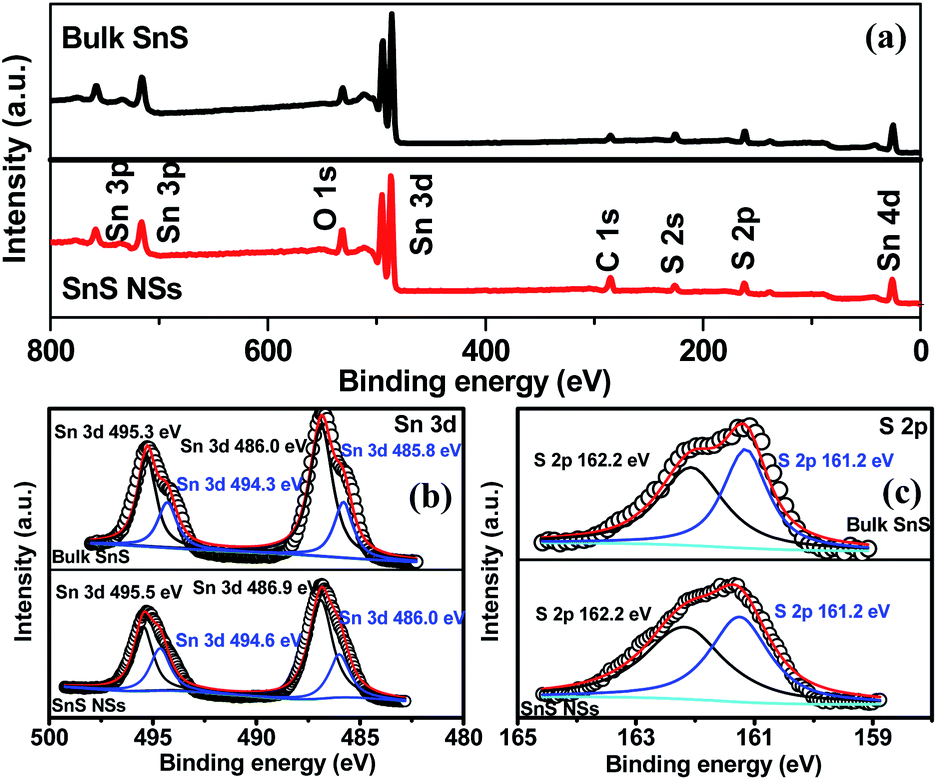 | ||
| Fig. 2 (a) XPS survey spectra and (b and c) high-resolution Sn 3d and S 2p XPS spectra of bulk SnS and SnS NSs. | ||
Different from the bulk SnS (Fig. S4†), the field-emission scanning electron microscopy (FESEM) image of ultrathin SnS NSs exhibited a layered structure with several wrinkles on the surface of the NSs (Fig. 3a). An obvious Tyndall effect for the SnS NS dispersion further demonstrated that the bulk SnS was successfully exfoliated into thin SnS layers (inset of Fig. 3a). The ultrathin SnS NSs have a relatively large lateral size of 1.0 μm and a thickness of ∼5.0 nm (∼6 layers), as illustrated in the atomic force microscopy (AFM) image (Fig. 3b). The high-resolution transmission electron microscopy (HRTEM) image of ultrathin SnS NSs (inset of Fig. 3c) exhibited the (101) plane of SnS with a lattice fringe distance of 0.29 nm, which has a 45° included angle with the (100) plane (Fig. S5†). Fig. 3d displays a typical high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image for ultrathin SnS NSs along the (100) direction. The arrangement of atomic columns in these images was well consistent with the crystal structure of SnS (space group Pbnm) in this projection, as depicted in the inset of Fig. 3d. The corresponding HAADF image simulation was further carried out using the thickness of 2 unit cells, and the results demonstrate the well-defined crystalline structure of ultrathin SnS NSs (Fig. 3e). Moreover, the presence of the (100) plane also supported the highly exposed (001) plane in the vertical direction (Fig. 3f),32 which was separated and exposed during the exfoliation process (Fig. S5†). In the selected area electron diffraction (SEAD) pattern of ultrathin SnS NSs (Fig. S6†), the interplanar spacings of 0.40, 0.28, and 0.20 nm, which correspond to the (110), (040), and (141) planes of SnS, can be observed. The corresponding energy-dispersive X-ray (EDX) elemental mapping images of S, Sn–K, and Sn–L illustrated that both S and Sn are uniformly dispersed throughout the ultrathin SnS NSs (Fig. 3g).
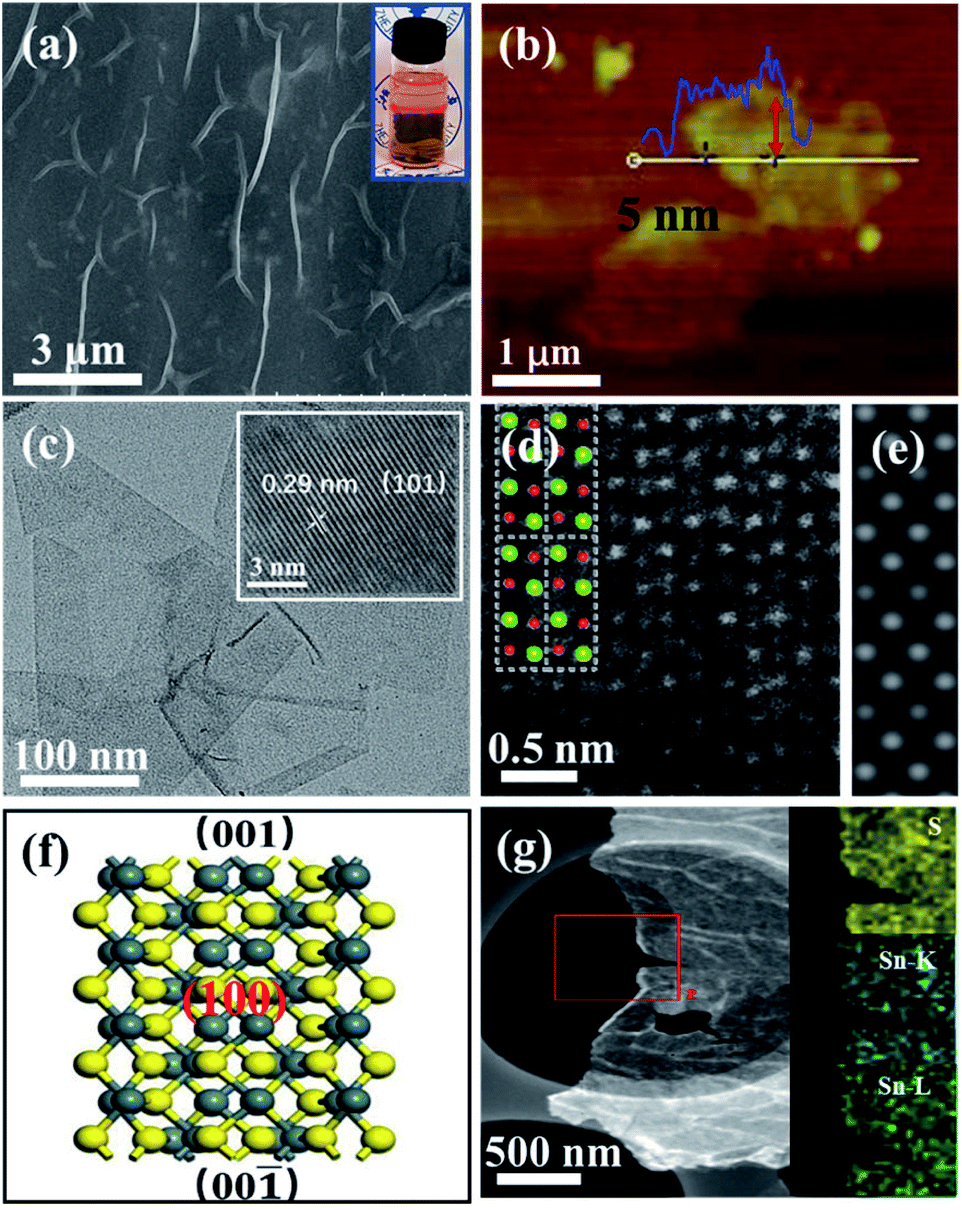 | ||
| Fig. 3 (a) FESEM image of SnS NSs and the corresponding Tyndall effect (inset). (b and c) AFM, TEM, and HRTEM images (inset) of SnS NSs. (d) HAADF image of SnS along the (100) direction. The inset shows the crystal structure of SnS in the same projection, where Sn and S atoms are depicted in green and red, respectively. (e) A simulated HAADF image along (100), using the thickness of 2 unit cells, consistent with that in (d). (f) Crystal structure of the SnS (100) plane where Sn and S atoms are depicted in grey and yellow, respectively. (g) TEM image and corresponding element maps (S, Sn–K, and Sn–L) of SnS NSs. | ||
Taking into consideration the essential role of the ultrathin structure and highly exposed facet of NSs in catalysis, the electrocatalytic properties of the SnS NSs for the CO2RR were investigated in a 0.5 M KHCO3 electrolyte using a three-electrode system. Obviously, the SnS NSs exhibit a significantly enhanced current density in a CO2-saturated electrolyte as compared with that in an Ar-saturated electrolyte (Fig. 4a). The increased current density can be rationally attributed to the electrochemical CO2RR, signifying the more favourable catalytic activity of SnS NSs toward the CO2RR than toward the HER. Gas chromatography (GC) and nuclear magnetic resonance (NMR) were used to determine the gas and liquid products, respectively. The total F.E. of gas and liquid products for SnS NSs is calculated to be nearly 100%. The main products of the CO2RR mainly include HCOOH (Fig. 4b) and a small amount of the CO product. To be specific, the SnS NSs present a maximum F.E. value of 82.1% for HCOOH production at a potential of −1.1 V, which is about 2.78 times higher than that of bulk SnS (29.5%) at the same potential. In addition, the partial current density of ultrathin SnS NSs for HCOOH production could reach 18.9 mA cm−2 at −1.1 V (Fig. 4c), which is over 4.0 times that of bulk SnS (4.1 mA cm−2), further demonstrating an excellent HCOOH selectivity of the SnS NSs. Also, the partial current density of SnS NSs for C1 products (CO and HCOOH) is approximately 4.4 times higher than that of the bulk SnS (4.8 mA cm−2), indicating the superior CO2RR activity of ultrathin SnS NSs (Fig. S7†). Moreover, we further loaded and tested the SnS NSs on a Ti substrate via a particle transfer approach, which is capable of attaining a HCOOH F.E. over 91% in a wide potential window. Notably, this 91% selectivity for HCOOH production for the resulting SnS NSs is comparable to or even higher than those of state-of-the-art tin sulfide-based CO2RR electrocatalysts (Table S1†).
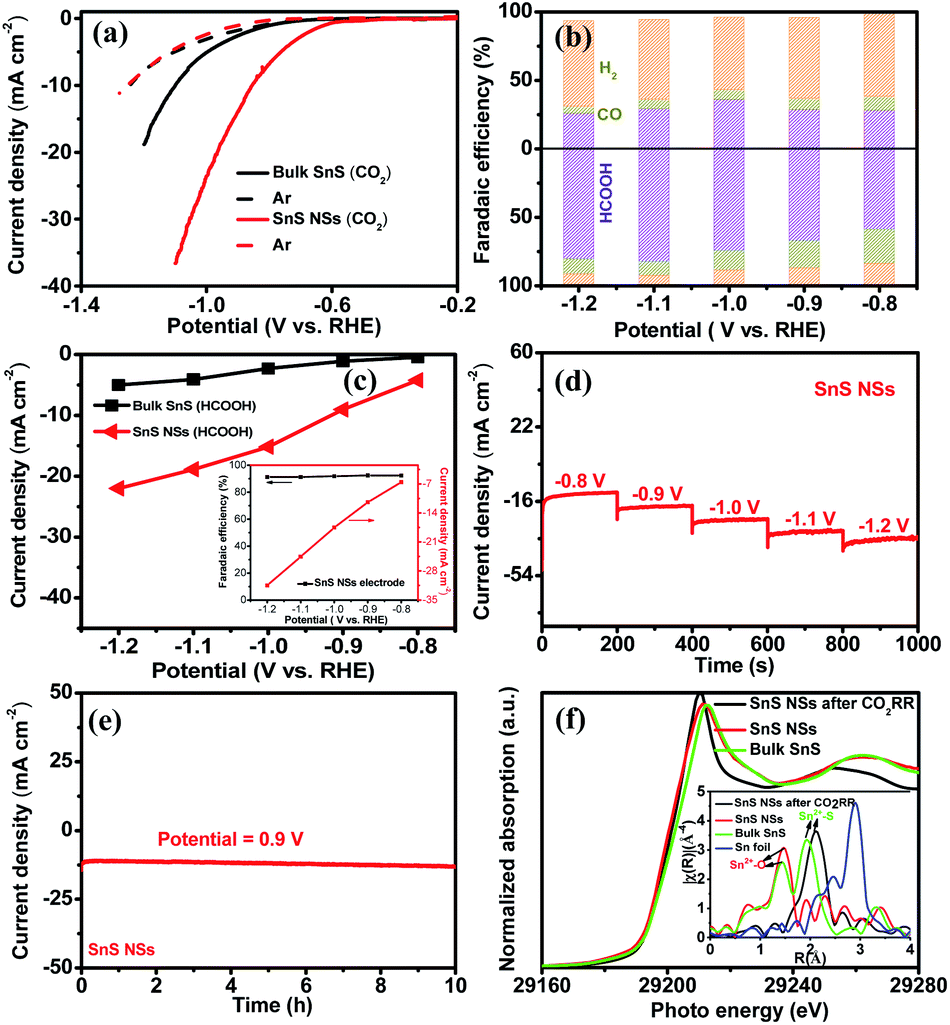 | ||
| Fig. 4 (a) Polarization curves of bulk SnS and SnS NSs in Ar-saturated (dash lines) and CO2-saturated (solid lines) electrolytes. (b) F.E. of bulk SnS (upper) and SnS NSs (lower) for HCOOH, CO, and H2 products at different applied potentials. (c) Partial current densities (HCOOH) for the CO2RR of bulk SnS and SnS NSs. Inset: partial current density and HCOOH F.E. of SnS NSs. (d) Multistep-potential curve of SnS NSs. (e) Chronoamperometry measurements of SnS NSs at 0.9 V. (f) Sn K-edge XANES and EXAFS (inset) spectra for bulk SnS and SnS NSs before and after continuous CO2RR tests. | ||
In order to further study the CO2RR activity of ultrathin SnS NSs, the Tafel slope, electrochemically active surface area (ECSA), and electrochemical impedance spectroscopy (EIS) results of the SnS NSs were investigated. As shown in Fig. S8a,† the Tafel slope of SnS NSs was as low as 180 mV dec−1, much smaller than that of the bulk SnS (357 mV dec−1), indicating favourable reaction kinetics on SnS NSs.20,33 The Tafel slope of 180 mV dec−1 was close to the theoretical value of 200 mV dec−1, which indicates that the rate-determining step of CO2RR catalysis by SnS NSs was CO2 adsorption on the active sites to form 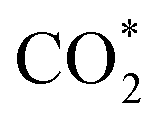 .34–36 Thus the CO2RR process of SnS NSs was controlled by mass transfer. In addition, the cyclic voltammetry curve was plotted to calculate the double-layer capacitance (Cdl), which is supposed to be linearly proportional to the actual active area of the catalyst (Fig. S9†). The value of Cdl was calculated to be 12.0 mF cm−2 for ultrathin SnS NSs (Fig. S8b†), which is much higher than that of bulk SnS (2.28 mF cm−2). The increased ECSA of the ultrathin SnS NSs could be attributed to more exposed edge sites and improved ion exchange between the electrolyte and catalytically active sites. Moreover, as compared with the bulk SnS, an obvious decrease in charge transfer resistance was observed for ultrathin SnS NSs (Fig. S8c†), suggesting faster electron transfer upon electrocatalysis of the CO2RR at the SnS NSs.37 These results reveal that the SnS NSs possess a much larger electro-active surface area and faster electron transfer than the bulk SnS, which majorly contribute to the excellent activity for the CO2RR. Fig. 4d shows the multistep–potential curve of SnS NSs in the potential range from −0.8 V to −1.2 V; the rather stable current densities for each step indicate the excellent mechanical robustness and mass transport properties of SnS NSs for the CO2RR.
.34–36 Thus the CO2RR process of SnS NSs was controlled by mass transfer. In addition, the cyclic voltammetry curve was plotted to calculate the double-layer capacitance (Cdl), which is supposed to be linearly proportional to the actual active area of the catalyst (Fig. S9†). The value of Cdl was calculated to be 12.0 mF cm−2 for ultrathin SnS NSs (Fig. S8b†), which is much higher than that of bulk SnS (2.28 mF cm−2). The increased ECSA of the ultrathin SnS NSs could be attributed to more exposed edge sites and improved ion exchange between the electrolyte and catalytically active sites. Moreover, as compared with the bulk SnS, an obvious decrease in charge transfer resistance was observed for ultrathin SnS NSs (Fig. S8c†), suggesting faster electron transfer upon electrocatalysis of the CO2RR at the SnS NSs.37 These results reveal that the SnS NSs possess a much larger electro-active surface area and faster electron transfer than the bulk SnS, which majorly contribute to the excellent activity for the CO2RR. Fig. 4d shows the multistep–potential curve of SnS NSs in the potential range from −0.8 V to −1.2 V; the rather stable current densities for each step indicate the excellent mechanical robustness and mass transport properties of SnS NSs for the CO2RR.
The long-term stability of the SnS NSs for the CO2RR was evaluated by performing chronoamperometry measurements at −0.9 V (Fig. 4e), in which the current density remained at 13 mA cm−2 upon 10 hours of continuous running, demonstrating a high stability of SnS NSs. In order to reveal the structural changes of ultrathin SnS NSs before and after CO2RR tests, the post Sn K-edge X-ray absorption near-edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS) characterization were performed. As shown in Fig. 4f, the Sn absorption edge positions of SnS NSs before and after CO2RR processes are almost the same as those of bulk SnS, which indicated the same oxidation state of Sn2+ for all investigated samples. Moreover, from Fourier transform (FT) EXAFS results (inset of Fig. 4f), it was found that there are lots of oxygen groups adsorbed on the surface of SnS NSs to form the Sn2+–O coordination. After CO2RR electrocatalysis, the disappearance of the Sn2+–O peak and increased intensity of the Sn–S peak of SnS NSs revealed that the electrochemical process allows the desorption of adsorbed oxygen groups on the Sn surface, which resulted in more exposed Sn–S active sites.38,39
Density functional theory (DFT) calculations were performed to gain a theoretical understanding of the high activity and selectivity of ultrathin SnS NSs for HCOOH production. Four possible models, including the intact and defect SnS (100) and SnS (001) surfaces, were built to represent the possible exposed surfaces of the bulk SnS and SnS NSs. It is noteworthy that our models included the defect sites because they were generally accepted as potential active sites during the electrocatalytic reaction.40,41 The computational hydrogen electrode (CHE) method proposed by Nørskov et al. was used42–44 (Fig. S10†) to study the relative activity of the HER, CO, and HCOOH pathways, with the associated elementary steps of each pathway listed in (RS1)-(RS5). As a result, four free energy diagrams (FEDs) for the four possible exposed sites are obtained and plotted in Fig. 5, which illustrates the following three points: (i) the general catalytic activity. At −1.0 V of URHE, besides that on SnS (100), the CO2RR catalysis on the other three sites are all exothermic reactions, suggesting good activities for these three sites, which explains the experimentally notable current for both bulk SnS and SnS NSs (Fig. 4a); (ii) for the CO pathway, all three active sites, namely, the SnS001, SnS100-def, and SnS001-def, generally exhibit smaller limiting potentials (Ul) (−0.46, −0.87, and −0.59 V in Fig. 5) than the HCOOH (−0.46, −0.87, and −0.11 V) and H2 pathways (−0.55, −0.45, and −0.03 V), suggesting a poorer catalytic selectivity for CO (g) on these sites. This result is well consistent with the smaller CO (g) F.E. for both SnS bulk and SnS NSs shown in Fig. 4b, and further explains the experimentally notable generation of both H2 and HCOOH; (iii) also indicated by the Ul value, as compared with that for the HER, the catalytic selectivity for HCOOH generation is much lower on SnS100-def (−0.87 vs. −0.45 V), slightly lower on SnS001-def (−0.11 vs. −0.03 V), and slightly higher on SnS001 (−0.46 vs. −0.55 V). So relatively, we have a higher selectivity for SnS001-def (−0.11 vs. −0.03 V) and slightly higher selectivity for SnS001 (−0.46 vs. −0.55 V). That is to say, the SnS (001) has a higher selectivity for HCOOH production than SnS (100). On the other hand, for the ratio of the (100) and (001) surfaces exposed practically (R100/001) on SnS NSs and bulk, the SnS NSs have a higher ratio of signal (100) to (001) than the bulk SnS, i.e., they contain a higher ratio of (100) than (001) layers in the volume phase. Since the possible exposed surfaces are only the (100) and (001) planes for SnS, this will contrarily suggest a smaller R100/001 for SnS NSs than for bulk SnS. As such, the smaller R100/001 on SnS NSs, along with the higher selectivity for HCOOH on SnS (001), explains the higher F.E. of HCOOH on SnS NSs in Fig. 4b.
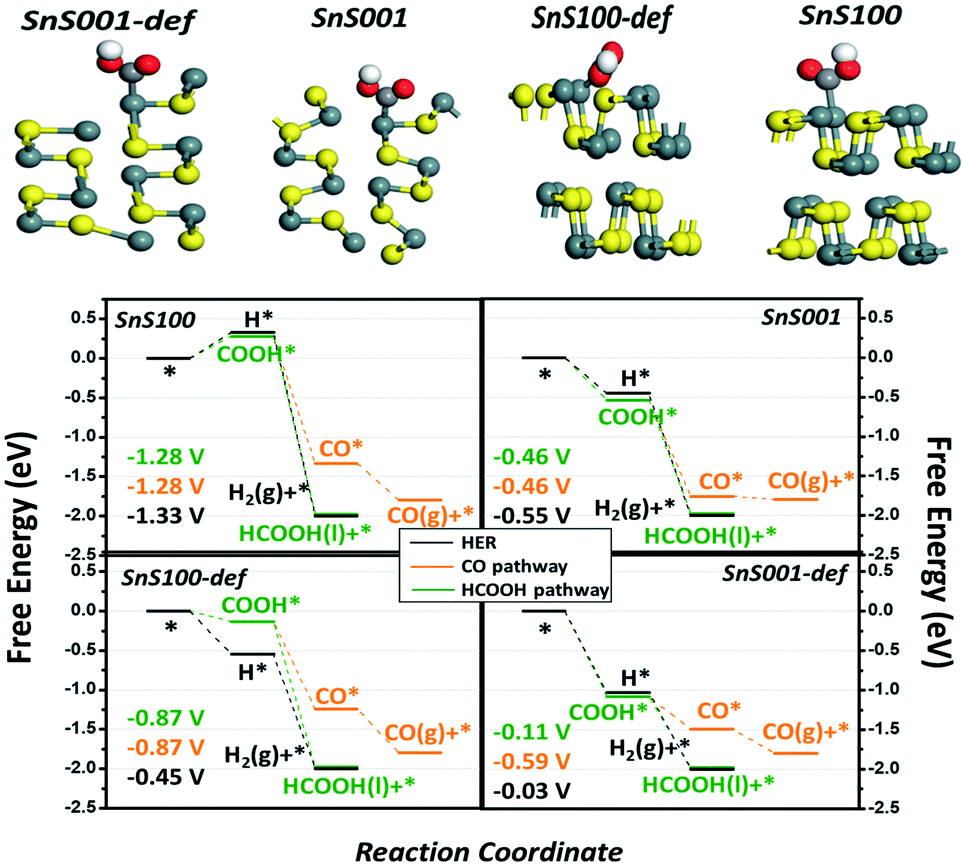 | ||
| Fig. 5 The reaction free energy diagram of possible CO2RR pathways for H2 (black), CO (orange), and HCOOH (green) generation. The associated DFT models with one COOH adsorbed are shown separately. The gray, yellow, red, and white balls stand for tin, sulphur, oxygen, and hydrogen. The coloured numbers are the Ul values for the corresponding pathway. That is, black for H2, orange for CO, and green for HCOOH. | ||
Finally, Fig. 5 also shows that, as compared with the intact sites, the defect sites for both the (100) and (001) planes likely bind the adsorbates more strongly, hence resulting in an improved activity (the attached electronic structure analysis is shown in Fig. S11†). This result indicates that the SnS NSs generated by electrochemical exfoliation will expose more edge sites, thus resulting in higher catalytic activities. In fact, we speculate that lots of defect sites have already been generated by the electrochemical exfoliation approach in the current work, since the electrochemical exfoliation creates an ungentle environment, which exfoliates the bulk form into NSs and exposes more edge active sites. This unique structure could also possibly be another important advantage of the electrochemically exfoliated electrocatalyst.
Inspired by the excellent CO2RR activity, a two-electrode electrolyzer was further assembled with the ultrathin SnS NS cathode for the CO2RR and an Ir/C anode for the oxygen evolution reaction (OER).45–52 As shown in Fig. S12a,† the polarization curve of the CO2RR-OER cell displayed electrocatalytic properties, in which the current density could reach 6.54 mA cm−2 at 3.0 V. Notably, the current density was slightly decreased by only 10% after 10 hours of continuous reaction (inset of Fig. S12a†). Furthermore, two 1.5 V AA batteries were used to drive the electrocatalytic CO2RR with the ultrathin SnS NSs. As shown in Fig. S12b,† a current density of 5.75 mA cm−2 for ultrathin SnS NSs can be obtained using two 1.5 V AA batteries, and current density was decreased by merely 8.5% after 10 hours of continuous running, suggesting a good stability.
Conclusions
In summary, ultrathin SnS NSs with a thickness of ∼5.0 nm and lateral size of 1.0 μm were successfully prepared by an alternating voltage assisted electrochemical exfoliation strategy. The resulting SnS NSs exhibited a high electrochemical activity and selectivity toward the CO2RR with a low Tafel slope, and a high F.E. for HCOOH production, representing one of the most active tin sulfide-based CO2RR electrocatalysts. The excellent CO2RR performance of the as-prepared SnS NSs was attributed primarily to the synergistic catalytic effects produced by their ultrathin layer features and high-index (001) facet. We further demonstrated that the practical application of the SnS NSs for CO2RR electrocatalysis can be achieved using two AA batteries connected in series. Our catalyst design introduced in this work may open new avenues toward development of transition metal dichalcogenide nanosheets with high-index facets for a variety of electrochemical applications, including oxygen evolution, nitrogen reduction, and hydrogen evolution reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51702284, 21922811, and 21878270), the Zhejiang Provincial Natural Science Foundation of China (LR19B060002), the Fundamental Research Funds for the Central Universities, and the Startup Foundation for the Hundred-Talent Program of Zhejiang University. We thank Jianyang Pan (Pharmaceutical Informatics Institute, Zhejiang University) for performing NMR spectrometry for the determination of HCOOH. We also acknowledge the beam time and help from the staff of beamline 1W1B of the Shanghai Synchrotron Radiation Facility BL14W1 and the Beijing Synchrotron Radiation Facility (BSRF, Beijing, China).References
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong and J. A. Reimer, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef PubMed.
- Z. Li, G. Li, X. Chen, Z. Xia, J. Yao, B. Yang, L. Lei and Y. Hou, ChemSusChem, 2018, 11, 2382–2387 CrossRef PubMed.
- L. Liang, F. Lei, S. Gao, Y. Sun, X. Jiao, J. Wu, S. Qamar and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 13971–13974 CrossRef PubMed.
- C. Lu, J. Yang, S. Wei, S. Bi, Y. Xia, M. Chen, Y. Hou, M. Qiu, C. Yuan and Y. Su, Adv. Funct. Mater., 2019, 29, 1806884 CrossRef.
- T. Wang, Q. Zhao, Y. Fu, C. Lei, B. Yang, Z. Li, L. Lei, G. Wu and Y. Hou, Small Methods, 2019, 3, 1900210 CrossRef CAS.
- Z. Xia, M. Freeman, D. Zhang, B. Yang, L. Lei, Z. Li and Y. Hou, ChemElectroChem, 2018, 5, 253–259 CrossRef CAS.
- W. Zheng, C. Guo, J. Yang, F. He, B. Yang, Z. Li, L. Lei, J. Xiao, G. Wu and Y. Hou, Carbon, 2019, 150, 52–59 CrossRef.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- S. Zhang, B. Meyer, G. H. Moh and F. Scholz, Electroanalysis, 1995, 7, 319–328 CrossRef CAS.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan and W. Yan, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- J. He, X. Liu, H. Liu, Z. Zhao, Y. Ding and J. Luo, J. Catal., 2018, 364, 125–130 CrossRef.
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma and J. Liu, Nano Energy, 2018, 53, 808–816 CrossRef CAS.
- H. Zhong, Y. Qiu, T. Zhang, X. Li, H. Zhang and X. Chen, J. Mater. Chem., A., 2016, 4, 13746–13753 RSC.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu and J. Zhu, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef PubMed.
- Y. Zhang, X. Zhang, Y. Ling, F. Li, A. Bond and J. Zhang, Angew. Chem., Int. Ed., 2018, 130, 13467–13471 CrossRef.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, Nano Lett., 2017, 17, 384–391 CrossRef PubMed.
- F. Li, M. Xue, J. Li, X. Ma, L. Chen, X. Zhang, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 129, 14910–14914 CrossRef.
- G. Babu, N. Masurkar, H. Al Salem and L. M. R. Arava, J. Am. Chem. Soc., 2017, 139, 171–178 CrossRef PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung and E. Tutuc, Science, 2009, 324, 1312–1314 CrossRef PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim and Y. I. Song, Nat. Nanotechnol., 2010, 5, 574 CrossRef PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De and R. J. Smith, Science, 2011, 331, 568–571 CrossRef PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 10443–10445 CrossRef PubMed.
- S. Yang, K. Zhang, A. G. Ricciardulli, P. Zhang, Z. Liao, M. R. Lohe, E. Zschech, P. W. Blom, W. Pisula and K. Müllen, Angew. Chem., Int. Ed., 2018, 130, 4767–4771 CrossRef.
- D. H. Youn, S. K. Stauffer, P. Xiao, H. Park, Y. Nam, A. Dolocan, G. Henkelman, A. Heller and C. B. Mullins, ACS Nano, 2016, 10, 10778–10788 CrossRef CAS PubMed.
- X. Xiong, C. Yang, G. Wang, Y. Lin, X. Ou, J.-H. Wang, B. Zhao, M. Liu, Z. Lin and K. Huang, Energy Environ. Sci., 2017, 10, 1757–1763 RSC.
- T. J. Whittles, L. A. Burton, J. M. Skelton, A. Walsh, T. D. Veal and V. R. Dhanak, Chem. Mater., 2016, 28, 3718–3726 CrossRef.
- Q. Li, A. Wei, J. Lu, L. Tao, Y. Yang, D. Luo, J. Liu, Y. Xiao, Y. Zhao and J. Li, Adv. Elect. Mater., 2018, 4, 1800154 CrossRef.
- J. Xia, D. Zhu, X. Li, L. Wang, L. Tian, J. Li, J. Wang, X. Huang and X. M. Meng, Adv. Funct. Mater., 2016, 26, 4673–4679 CrossRef.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- S. Gao, X. Jiao, Z. Sun, W. Zhang, Y. Sun, C. Wang, Q. Hu, X. Zu, F. Yang and S. Yang, Angew. Chem., Int. Ed., 2016, 55, 698–702 CrossRef PubMed.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2019, 10, 358–364 CrossRef.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef PubMed.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang and S. Wei, Nat. Commun., 2017, 8, 14503 CrossRef PubMed.
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H. G. Steinrück, K. Lim, Y. Li, M. F. Toney and K. Chan, Nat. Catal., 2019, 2, 55–61 CrossRef.
- R. Bacewicz, J. Antonowicz, S. Podsiadło and S. Schorr, Solid State Commun., 2014, 177, 54–56 CrossRef.
- Y. Han, S. Axnanda, E. J. Crumlin, R. Chang, B. Mao, Z. Hussain, P. N. Ross, Y. Li and Z. Liu, J. Phys. Chem. B, 2018, 122, 666–671 CrossRef PubMed.
- M. Favaro, C. Valero-Vidal, J. Eichhorn, F. Toma, P. Ross, J. Yano, Z. Liu and E. Crumlin, J. Mater. Chem. A, 2017, 5, 11634–11643 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- C. Lei, H. Chen, J. Cao, J. Yang, M. Qiu, Y. Xia, C. Yuan, B. Yang, Z. Li and X. Zhang, Adv. Energy Mater., 2018, 8, 1801912 CrossRef.
- C. Lei, Y. Wang, Y. Hou, P. Liu, J. Yang, T. Zhang, X. Zhuang, M. Chen, B. Yang and L. Lei, Energy Environ. Sci., 2019, 12, 149–156 RSC.
- J. Cao, C. Lei, J. Yang, X. Cheng, Z. Li, B. Yang, X. Zhang, L. Lei, Y. Hou and K. K. Ostrikov, J. Mater. Chem., A., 2018, 6, 18877–18883 RSC.
- X. Cheng, C. Lei, J. Yang, B. Yang, Z. Li, J. Lu, X. Zhang, L. Lei, Y. Hou and K. Ostrikov, ChemElectroChem, 2018, 5, 3866–3872 CrossRef CAS.
- Y. Hou, M. Qiu, G. Nam, M. G. Kim, T. Zhang, K. Liu, X. Zhuang, J. Cho, C. Yuan and X. Feng, Nano Lett., 2017, 17, 4202–4209 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, T. Zhang, J. Ma, S. Liu, X. Zhuang, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1604480 CrossRef PubMed.
- Y. He, X. Zhuang, C. Lei, L. Lei, Y. Hou, Y. Mai and X. Feng, Nano Today, 2019, 24, 103–119 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06548b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |