
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTesting the limits of radical-anionic CH-amination: a 10-million-fold decrease in basicity opens a new path to hydroxyisoindolines via a mixed C–N/C–O-forming cascade†
Quintin
Elliott
 ,
Gabriel
dos Passos Gomes‡
,
Christopher J.
Evoniuk
and
Igor V.
Alabugin
*
,
Gabriel
dos Passos Gomes‡
,
Christopher J.
Evoniuk
and
Igor V.
Alabugin
*
Department of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, USA. E-mail: alabugin@chem.fsu.edu
First published on 21st February 2020
Abstract
An intramolecular C(sp3)–H amidation proceeds in the presence of t-BuOK, molecular oxygen, and DMF. This transformation is initiated by the deprotonation of an acidic N–H bond and selective radical activation of a benzylic C–H bond towards hydrogen atom transfer (HAT). Cyclization of this radical–anion intermediate en route to a two-centered/three-electron (2c,3e) C–N bond removes electron density from nitrogen. As this electronegative element resists such an “oxidation”, making nitrogen more electron rich is key to overcoming this problem. This work dramatically expands the range of N-anions that can participate in this process by using amides instead of anilines. The resulting 107-fold decrease in the N-component basicity (and nucleophilicity) doubles the activation barrier for C–N bond formation and makes this process nearly thermoneutral. Remarkably, this reaction also converts a weak reductant into a much stronger reductant. Such “reductant upconversion” allows mild oxidants like molecular oxygen to complete the first part of the cascade. In contrast, the second stage of NH/CH activation forms a highly stabilized radical–anion intermediate incapable of undergoing electron transfer to oxygen. Because the oxidation is unfavored, an alternative reaction path opens via coupling between the radical anion intermediate and either superoxide or hydroperoxide radical. The hydroperoxide intermediate transforms into the final hydroxyisoindoline products under basic conditions. The use of TEMPO as an additive was found to activate less reactive amides. The combination of experimental and computational data outlines a conceptually new mechanism for conversion of unprotected amides into hydroxyisoindolines proceeding as a sequence of C–H amidation and C–H oxidation.
Introduction
Due to their abundance, C(sp3)–H bonds offer an excellent starting point for the functionalization of organic compounds.1 However, the productive use of C–H bonds is complicated due to the difficulties associated with their selective activation.2 These challenges prompt chemists to search for new approaches for utilizing C–H bonds as a reactive organic functionality.3C–H amination couples C–H activation with the concomitant formation of a C–N bond and opens new avenues for the synthesis of nitrogen-containing organic compounds.4 This approach has evolved into a versatile group of reactions that overcome the challenges with C–H activation and C–N bond formation. Depending upon nitrogen's participation in C–H activation and C–N bond formation, one can broadly classify C–H amination reactions within the following four approaches: (a) C–H activation directly coupled with C–N bond formation (direct activation), (b) C–H activation with delayed C–N bond formation, (c) N-assisted C–H activation, and (d) independent C–H/N–H activation (Scheme 1).
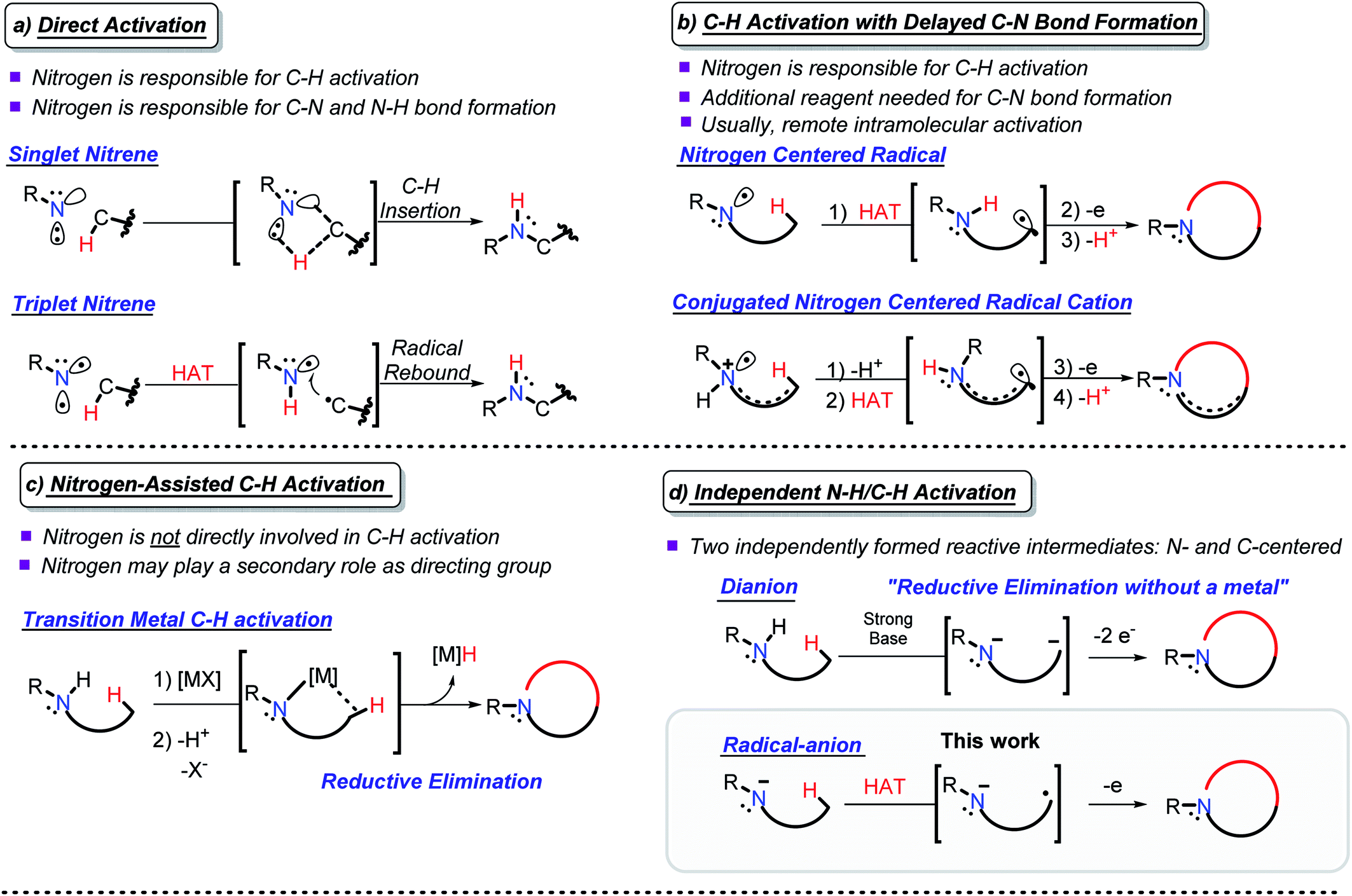 | ||
| Scheme 1 Selected examples of the four approaches to C–N bond formation via C–H activation. All carbons are tetravalent, the non-participating C–H bonds are omitted for clarity. | ||
In direct activation (Scheme 1a), nitrogen is responsible for the activation of C–H bonds and the formation of both the C–N and the N–H bonds. This mode of activation works with highly coordinatively unsaturated species, e.g., nitrenes and nitrenium ions, where nitrogen can insert into a C–H bond in either a stepwise or concerted manner.5
In C–H activation with delayed C–N bond formation (Scheme 1b), nitrogen assists in C–H activation by breaking the C–H bond, but the C–N bond forming step needs an additional participant (e.g., an external oxidant). This process also requires an electron-deficient nitrogen-centered intermediate, i.e., a radical6 or a radical–cation.7 If placed in proximity to a C–H bond, a nitrogen-centered radical can abstract a hydrogen atom, forming a carbon centered radical. A C–N bond is formed by trapping this radical. Alternatively, activation of a C–H bond can be achieved by deprotonation of a nitrogen radical–cation, prepared by oxidation of an amine. Although such deprotonation usually proceeds at the α-position where it can provide a stabilized C-centered α-radical,8 activation of a remote C–H bond becomes possible in conjugated radical–cations, even when the activating NH2 moiety and the activated CH2 group are five bonds away.9,10
In N-assisted C–H activation reactions (Scheme 1c), nitrogen does not directly participate in the C–H activation step but can trap the C-centered reactive species once they are formed. An external reagent, such as an appropriately chosen transition metal, is used for C–H activation. An appealing strategy is to use nitrogen as a directing group, guiding the transition metal to the targeted C–H bond and facilitating C–N bond formation afterwards.11,12 A new C–N bond can be formed via reductive elimination at the transition metal.13
The fourth, conceptually different approach is to activate both the N–H and the C–H bonds by independently converting them into reactive intermediates (Scheme 1d). By decoupling the two steps, this approach potentially becomes the most flexible, but the conditions for selective and independent N–H/C–H activation are not always easy to achieve. The possible situations here involve: (i) formation of N- and C-centered radicals, (ii) formation of a radical and an anion (usually, C-radical and N-anion), and (iii) formation of an N-anion and a C-anion. The latter two paths must be terminated by 1e and 2e oxidations, respectively, to yield the “normal” two-centered/two-electron (2c,2e) C–N bond.
Productive combination of two reactive intermediates is efficient only when one of these intermediates is relatively persistent.14 From this perspective, formation of stable N-anions (approaches ii and iii) is attractive (Scheme 1d).
Sarpong and coworkers illustrated that a C, N dianion, formed in the presence of strong base can form a C–N bond upon two-electron oxidation with I2 (iii).15 This approach, which can be conceptually considered “reductive elimination without a metal”, allows C–N bond formation without the need for preoxidized coupling partners via the formal loss of H2. Formation of five-, six-, and seven-membered rings was found to proceed even in conformationally unbiased substrates.
In our work, we explore the advantage of a three-electron approach (ii).10 Because the three electron radical/anion interactions are potentially stabilizing, they can lead to favorable C/N precoordination which leads to instantaneous C–N bond formation upon oxidation. In contrast, the 4e anion/anion interactions are repulsive, and hence, the C,N-dianion may adopt a conformation that is not conducive to bond formation.
We have recently illustrated the value of the radical-anionic approach in an intramolecular C(sp3)–H amination.10 This method relies on a sequence of N–H deprotonations and selective H-atom transfers (HAT) from C–H bonds to generate a radical–anion intermediate (Scheme 3a). This intermediate forms a thermodynamically favored two-center/three-electron (2c,3e) “half bond”, where one of the electrons is forced to occupy a high energy antibonding orbital (“electron upconversion”, Scheme 2).16 The newly formed radical–anion can then be readily oxidized into a “normal” 2c,2e bond by a mild oxidant, such as molecular oxygen. By generating a radical anion in situ, we effectively take a weak reductant and evolve it into a more potent reductant. Such “electron upconversion”16 allows for the effective use of a mild oxidant such as molecular oxygen. By avoiding stronger oxidants, we prevented undesired product oxidation in our cascade reactions that yield expanded N-doped polyaromatic systems.
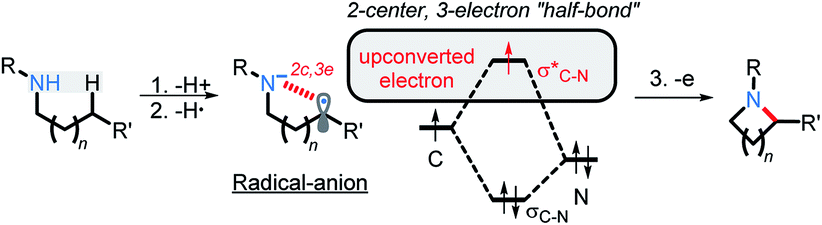 | ||
| Scheme 2 Steps in the proposed C–H/N–H activation and the mechanism of electron upconversion. | ||
In this manuscript, we push the limits of this approach by using amides, a drastically less nucleophilic nitrogen source (Scheme 3b). We will show how this structural modification diverts the cascade towards incorporation of a late C–O bond forming step17 and opens a synthetic route to 3-hydroxyisoindolinones. Recently, Xiao et al.18 independently developed a mechanistically different version of 3-hydroxyisoindolinone synthesis from secondary amides (Scheme 3c). An interesting feature of Xiao's work is that the same catalyst is used in two different ways to promote the two cascade stages. In the first ground state stage, eosin Y acts as a radical redox catalyst which is oxidized to a radical–cation by reaction with a stoichiometric amount of an external oxidant (SelectFluor). Without light, the reaction stops at the first step (C–N bond formation). The second step (the formation of the C–O bond) is achieved by using eosin Y as a photocatalyst in the presence of oxygen. The overall transformation had a good scope for the aromatic substituents but was only reported for N-monosubstituted amides (N-methoxy, N-aryl, N-alkyl).
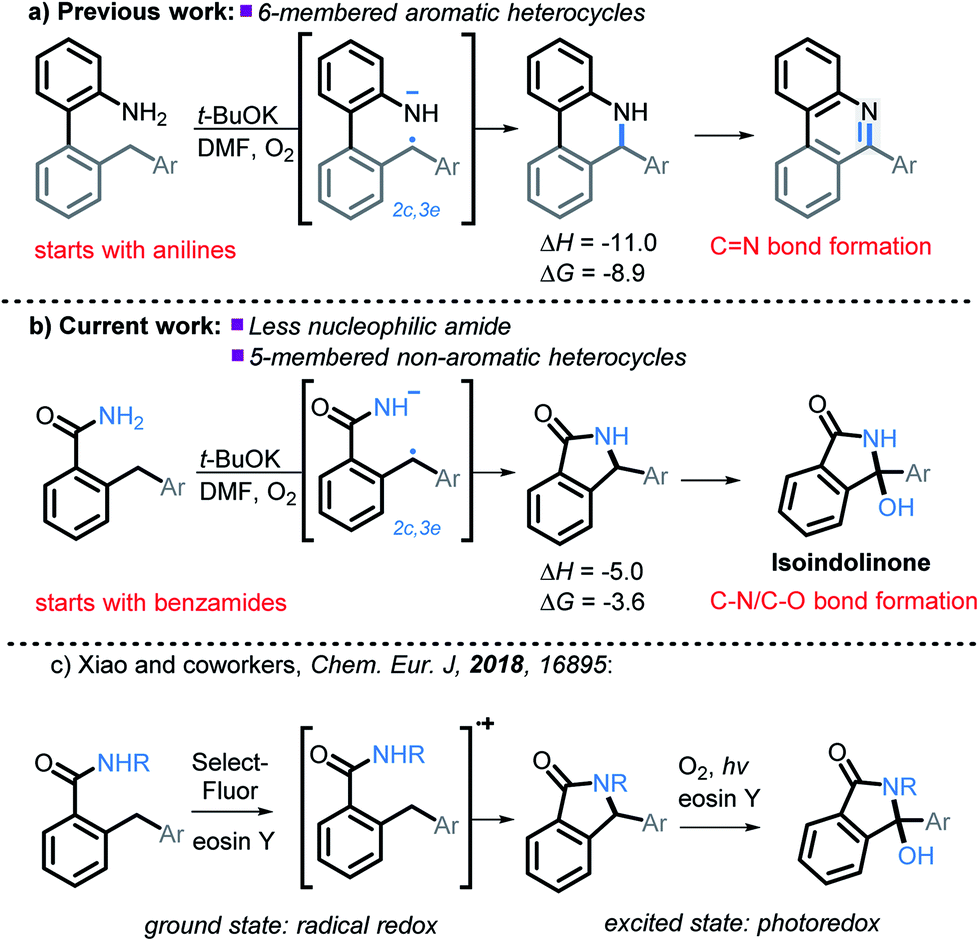 | ||
| Scheme 3 Expanding previous work to utilize less nucleophilic amides, as well as the formation of non-aromatic five membered heterocycles, and C–N/C–O bond formation. All energies are in kcal mol−1. | ||
It is clear that our approach and the approach of Xiao are very different mechanistically (anionic vs. cationic) and, hence, should provide complementary reactivity patterns for future synthetic designs. Our work presents an alternative route that proceeds in the ground state and does not require additional oxidants besides molecular oxygen. In addition, it avoids formation of a cationic species, a feature which can be beneficial to substrates that are sensitive to oxidative conditions. Furthermore, as we will show below, our conditions are applicable for primary amides.
The successful use of amides as nucleophilic partners in a base-promoted oxidative C(sp3)–H amidation yields isoindolinones under transition-metal free conditions. Because the presence of metal impurities should be minimized in pharmaceutics,19 a transition-metal free alternative is an attractive choice for the synthesis of isoindolinones related to the antihypertensive agent chlortalidone,20 inhibitors of MDM2-p53 interactions,21 and selected natural products.22 Furthermore, a variety of post-synthetic modifications of the core hydroxyisoindoline structure are possible (Scheme S5 in ESI†) including transformations mediated by chiral phosphoric acids23,24 and by transition metal catalysts.25
Results and discussion
We began by subjecting amide 1a (0.01 mmol) to the previously optimized conditions, (i.e., DMF, 4 Å-molecular sieves (MS), O2 atmosphere, and 3 equivalents of t-BuOK at room temperature) for 4 hours.9 To our delight, 2a was formed in an excellent yield, 87% (Table 1, entry 1). When DMSO and THF were used in place of DMF, the yields of 2a were lower (36% and 47%, entries 2 and 5) along with the formation of undesired side products. Furthermore, when DMF was replaced with acetonitrile, toluene, or DCM, 2a was not observed (entry 3, 4 and 6). These results prompted us to perform the rest of our studies in DMF.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Eq. | Atm | Time | Yield% |
| a Reaction conditions: all reactions performed in a 20 mL scintillation via, 1a (0.025 M), 2.5 mL of solvent, 4 Å-molecular sieves (MS), and room temperature (22 °C). All yields determined by 1H NMR using internal standard. | ||||||
| 1 | DMF | t-BuOK | 3 | O2 | 4 h | 87% |
| 2 | DMSO | t-BuOK | 3 | O2 | 4 h | 36% |
| 3 | MeCN | t-BuOK | 3 | O2 | 4 h | <1% |
| 4 | Toluene | t-BuOK | 3 | O2 | 4 h | <1% |
| 5 | THF | t-BuOK | 3 | O2 | 4 h | 47% |
| 6 | DCM | t-BuOK | 3 | O2 | 4 h | <1% |
| 7 | DMF | t-BuONa | 3 | O2 | 4 h | 79% |
| 8 | DMF | KOH | 3 | O2 | 4 h | 55% |
| 9 | DMF | NaOH | 3 | O2 | 4 h | 31% |
| 10 | DMF | LiOH | 3 | O2 | 4 h | <1% |
| 11 | DMF | K2CO3 | 3 | O2 | 4 h | <1% |
| 12 | DMF | t-BuOK | 1 | O2 | 4 h | 55% |
| 13 | DMF | t-BuOK | 2 | O2 | 4 h | 68% |
| 14 | DMF | t-BuOK | 4 | O2 | 4 h | 85% |
| 15 | DMF | t-BuOK | 5 | O2 | 4 h | 88% |
| 16 | DMF | t-BuOK | 3 | O2 | 3 h | 85% |
| 17 | DMF | t-BuOK | 3 | O2 | 2 h | 82% |
| 18 | DMF | t-BuOK | 3 | O2 | 1 h | 84% |
| 19 | DMF | t-BuOK | 3 | Air | 1 h | 75% |

We then tested five additional bases (entry 7–11). The tert-butoxide salts produced 2a in the highest yields. Interestingly, the size of the counterion impacts the conversion of 1a into 2a. While the difference was small for the sodium and potassium tert-butoxide salts (87% vs. 79% respectively), a noticeable difference in yield was observed between the K, Na, and Li hydroxides (55%, 31%, <1% respectively). Fewer than 3 equivalents of base (entry 12 and 13) were insufficient for the full consumption of the starting material. More than 3 equivalents of base (entry 14 and 15) were found to be unnecessary as there was no improvement in yield.
We then varied the reaction time (entry 16–18) and observed full consumption of starting material within one hour. Finally, when the reaction vial was charged with air instead of oxygen, 2a was produced in 75% yield (entry 19). However, the rate of the reaction was slower and not all of 1a was consumed.
We then explored the scope of substituents that are compatible with the reaction conditions. Variation in the pendant aryl ring revealed that heterocyclic substrates, as well as the substrates with ortho, meta, and para electron withdrawing groups produced the target products in good to excellent yields. On the other hand, electron donating groups were only tolerated in the meta position. Lower conversions to the corresponding isoindolinone were observed (39% and 48% respectively) for the reactions of 4-methoxy and 4-methyl substrates. However, substrate 2c with a para tert-butyl group underwent the desired transformation in high yield (84%) (Table 2).
| a Reaction conditions: benzamide (0.025 M), t-BuOK (3 eq.), 4 Å MS, DMF, O2 balloon and the reactions were allowed to stir for 4 hours at rt. Unless stated otherwise, the yield is of the isolated product. |
|---|
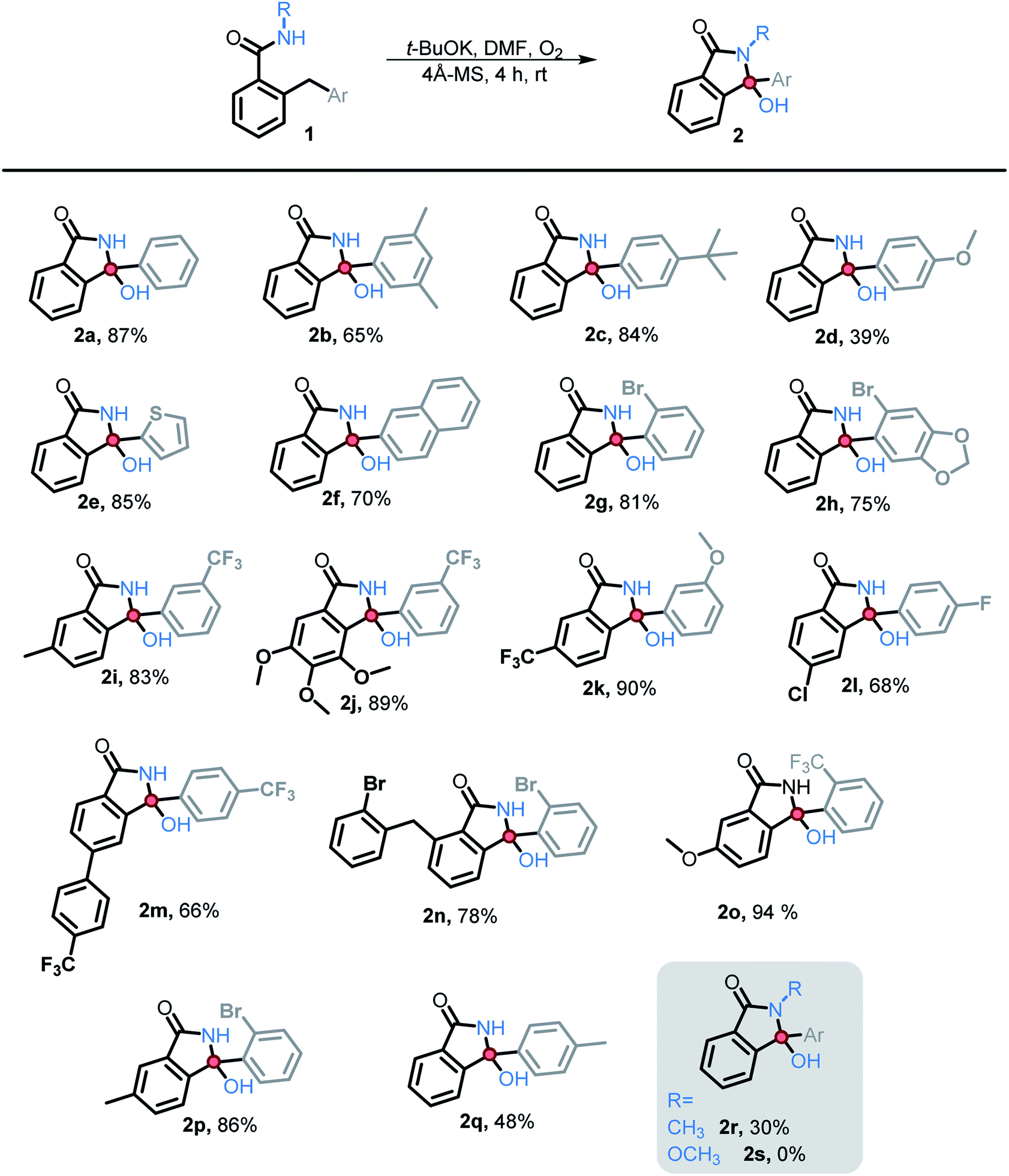
|
Substitution in the amide ring had a smaller effect – the reaction tolerated both electron donating and withdrawing groups, ortho, meta, and para to the amide. The overall transformation remained unaffected with halogens (i.e., Br, Cl, and F) on either the amide ring or pendant aryl ring, allowing for the introduction of a useful synthetic handle for future modifications.
We also investigated the possibility of introducing additional substituents at the amide nitrogen. The N-methyl amide 2r underwent the desired transformation sluggishly (30% yield, 74% based on reclaimed starting material) under the standard conditions. On the other hand, a N-methoxy amide remained unreactive under our standard conditions (vide infra).
Mechanistic studies
A series of studies designed to gain insight into the reaction mechanisms were performed and are listed below (Scheme 4). First, the C–H bond strength is critical for the reaction success. A BDE ≤ 85 kcal mol−1 was needed and simple benzylic or alkylbenzylic C–H bonds remained unreactive under our conditions (eqn (1)).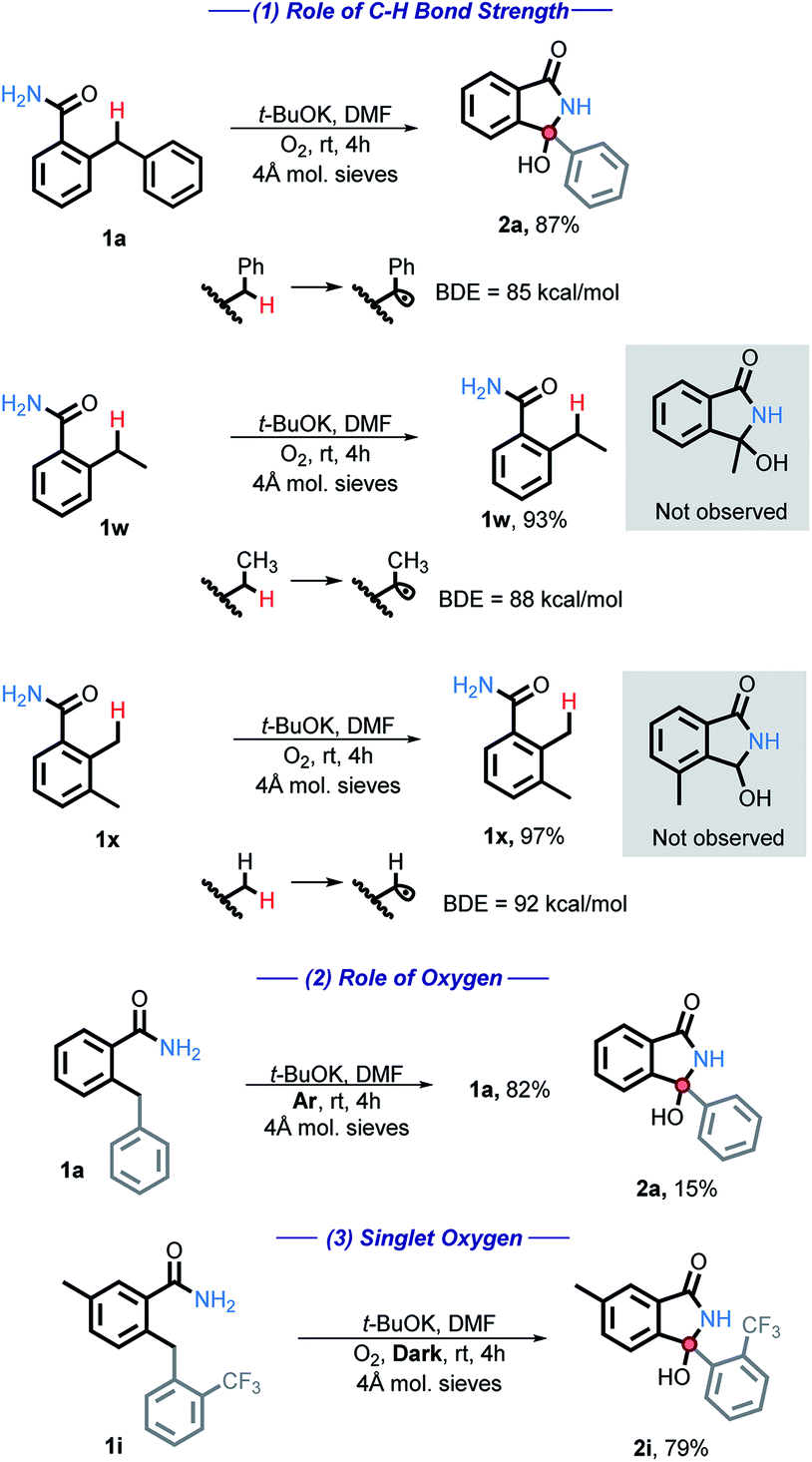 | ||
| Scheme 4 Mechanistic tests for the C–H bond strength and for the participation of oxygen. All energies are reported in kcal mol−1. | ||
When the reaction is performed under an inert atmosphere (Ar), the consumption of 1a is greatly diminished (eqn (2)), confirming the importance of O2 as the oxidant.
We have also considered the possible involvement of singlet oxygen. Singlet oxygen has been shown to be synthetically useful in the oxidation of heteroatoms, cyclization reactions, and the synthesis of hydroperoxides.26 However, 1i was fully consumed even in the absence of light, producing 2i in 79% yield (eqn (3)). Reactivity in the dark indicates that singlet oxygen is not involved in the main path of this reaction.
Under these oxidative conditions, one could suggest that the reaction occurs via an oxidation of the CH2 moiety to a carbonyl intermediate.27 This intermediate could then close the cycle via nucleophilic attack of the amide onto the carbonyl under basic conditions. We have eliminated benzylic C–H oxidation in our early work by using N-Me substituted anilines, which yield products that clearly could not originate from the ketone.10 Obtaining such direct evidence in the present case is problematic because, unlike the aniline cascade, the present amide cascade is terminated by C–O bond formation that renders the C/N radical–anion coupling and the carbonyl pathway products identical.
To test for the possible formation of a carbonyl intermediate, we wanted to use an unreactive amide in order to avoid the cyclization, while still allowing for the benzylic carbon to remain reactive enough to be oxidized into a ketone. However, no reaction was observed when amide 1s was exposed to the standard conditions. Calculations show that if a ketone intermediate were formed, its cyclization would be thermodynamically favorable (ΔG = −5.1 kcal mol−1). Assuming that this substitution at nitrogen does not directly change reactivity at the remote benzylic position, the lack of benzylic oxidation suggests that the carbonyl intermediate is not formed under our conditions10 (Scheme 5). We have also found that an isoindolinone is readily converted into the respective 3-hydroxyisolindolinone under these conditions, a process that is unlikely to proceed through a carbonyl intermediate.
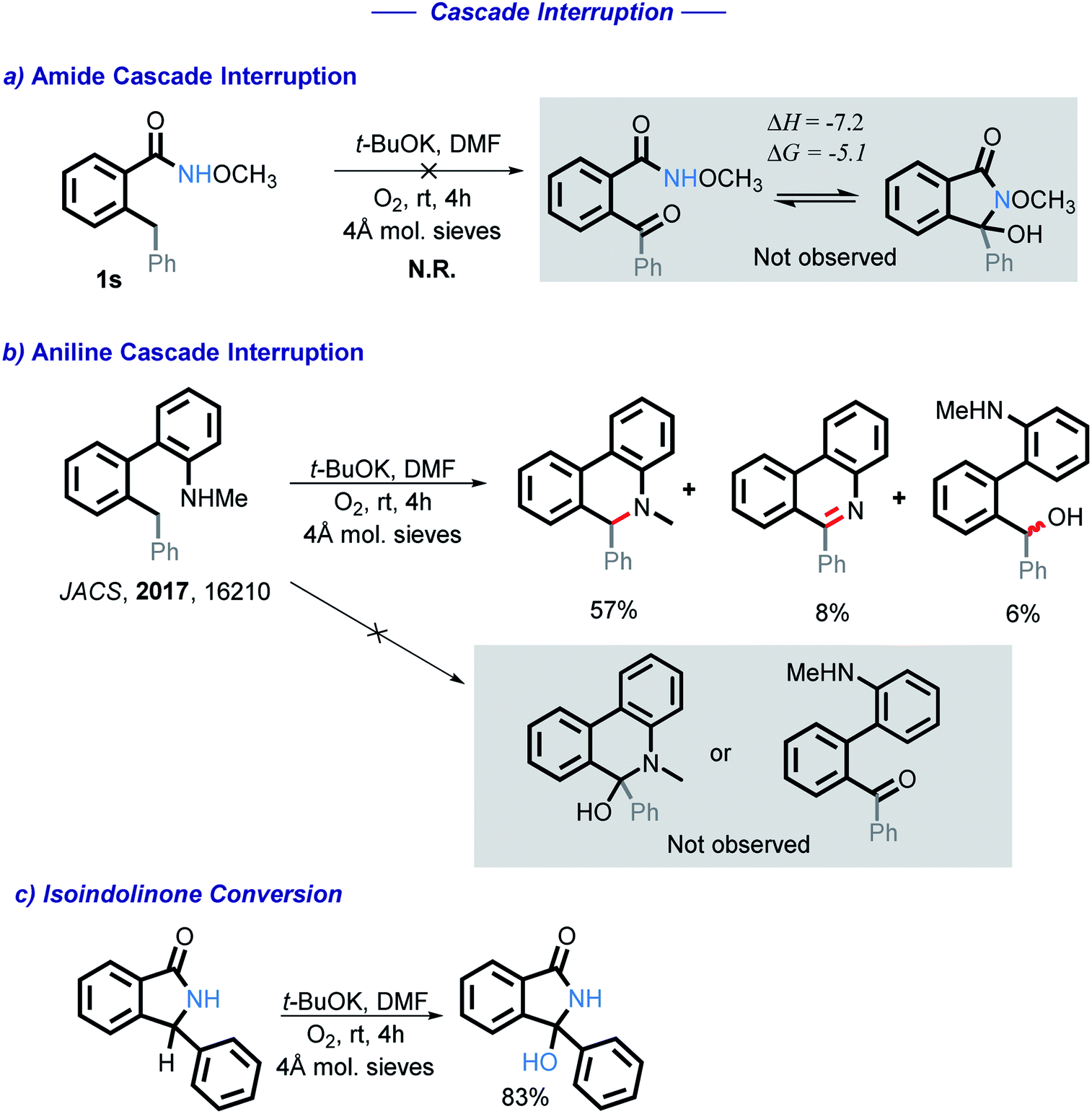 | ||
| Scheme 5 Interruption of the amide and aniline cascades. | ||
Computational data
Calculations were carried using the global-hybrid meta-GGA (U)M06-2X functional28 and the 6-31+G(d,p) basis set for all atoms, with an ultrafine integration grid (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590 points). A broken-spin approach was applied when necessary. The implicit SMD29 solvation model was used to simulate the effects of N,N-dimethyl-formamide (DMF) throughout the calculated structures. Grimme's D3 version (zero damping) for empirical dispersion30 was also included. Unless otherwise noted, all results presented are at the (SMD=DMF)/(U)M06-2X(D3)/6-31+G(d,p)/int=ufine level of theory. Frequency calculations were carried out for all structures to confirm them as either a minimum or a TS. All calculations were performed with the Gaussian 09 software package.31 Three-dimensional depictions and orbital plots were produced with CYLView 1.0.132 and Chemcraft 1.8.33
590 points). A broken-spin approach was applied when necessary. The implicit SMD29 solvation model was used to simulate the effects of N,N-dimethyl-formamide (DMF) throughout the calculated structures. Grimme's D3 version (zero damping) for empirical dispersion30 was also included. Unless otherwise noted, all results presented are at the (SMD=DMF)/(U)M06-2X(D3)/6-31+G(d,p)/int=ufine level of theory. Frequency calculations were carried out for all structures to confirm them as either a minimum or a TS. All calculations were performed with the Gaussian 09 software package.31 Three-dimensional depictions and orbital plots were produced with CYLView 1.0.132 and Chemcraft 1.8.33
Radical cascade mechanism
Guided by these experimental results, we turned to computations for identifying the key intermediates of this transformation. A full thermodynamic landscape for the proposed reaction cascade is shown in Scheme 6. Each step in this cascade is thermodynamically favorable. In subsequent individual sections, we will discuss the individual steps along with their activation barriers and the respective experimental evidence. | ||
| Scheme 6 Proposed mechanism and calculated reaction thermodynamics for the individual steps in the N–H/C–H amidation. All energies are in kcal mol−1. | ||
Generation of DMF radical
We suggest that the chemical species responsible for the formation of the bis-benzylic C-centered radical is DMF radical (C(O)N(CH3)2). The formation of this radical under these conditions is well-documented. Yan et al. used them in a variety of interesting transformations that combined C–H activation and C–C bond formation.34 However, the mechanism by which the DMF radical is generated is not fully established. A commonly suggested path that involves oxidation of DMF anion by DMF is highly thermodynamically unfavorable (ΔG = +54.0 kcal mol−1, Scheme 7).10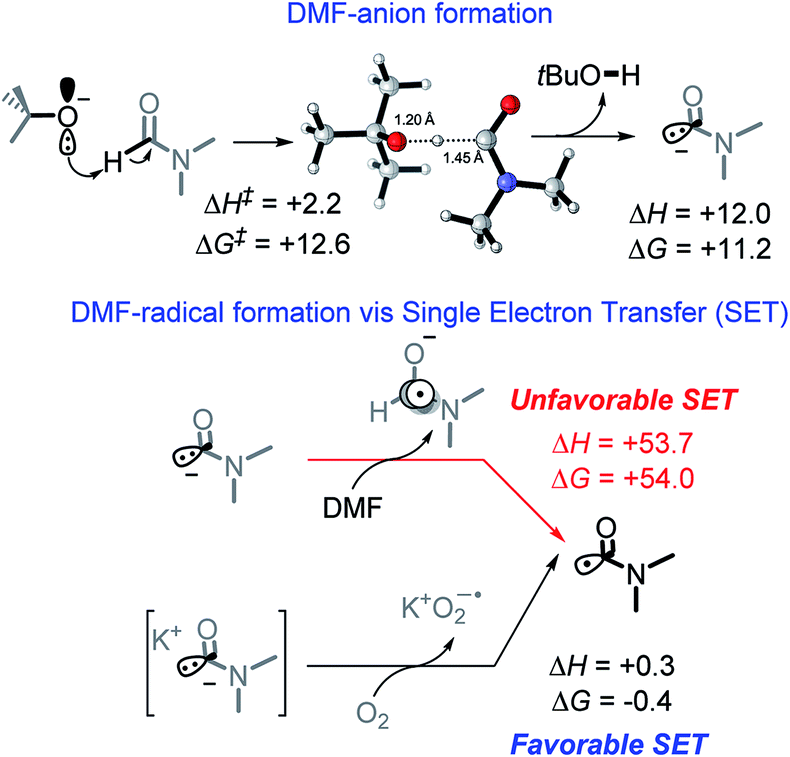 | ||
| Scheme 7 Formation of DMF radical. All energies reported in kcal mol−1. | ||
While we were unable to find the pKa of t-BuOH in DMF, literature reports35 the pKa of 1-butanol in DMF to be 33.3. It is reasonable to estimate the pKa of t-BuOH in DMF to be ca. 34–35. The pKa of DMF in DMF (38),35 makes it approximately 3–4 pKa units less acidic than t-BuOH. Although it makes the initial deprotonation of H–C(O)NMe2 uphill, a small amount of DMF anion is still formed at equilibrium. Calculations show that although this deprotonation is endergonic by 11.2 kcal mol−1, the activation barrier for the deprotonation of DMF is relatively low (12.6 kcal mol−1, Scheme 7), and equilibrium should be reached quickly.
Indeed, Drapeau and co-workers noted a formamide proton shift, via1H and 13C NMR, when t-BuOK was added to wet DMF-d7.36 Their computational work additionally highlighted two interesting points regarding the impact of counter ions. First, the cation stabilizes the DMF anion, assisting in deprotonation. Second, a cation of a certain size (e.g., Li) can stabilize the DMF anion and increase the barrier for its oxidation to DMF radical.36 Our experimental results also suggest that reaction is slow in the presence of Na and, to a greater extent, Li counterions (Table 1, entry 1, 7–10).
Our calculations show that the oxidation of DMF anion by oxygen is slightly exergonic (with the inclusion of K+, Scheme 7). This makes the combined deprotonation and oxidation of DMF endergonic by ∼10 kcal mol−1, which would allow only small quantities of DMF radical to be present at a given time. If the propagation step of the cascade is fast, then the initial penalty of forming the DMF radical is a small price to pay when all sequential steps in the cascade reactions are favorable and efficient. Overall, the generation of DMF radical imposes a ∼10 kcal mol−1 penalty needed to initiate an otherwise exergonic sequence. Interestingly, calculations suggest that the activation barrier for proton abstraction from DMF is almost entirely entropic (enthalpy of activation is only 2.2 kcal mol−1). The TS Gibbs energy (12.6 kcal mol−1) is only 1.4 kcal mol−1 higher than the Gibbs energy of the product, suggesting that the deprotonation should be rapidly reversible.
Deprotonation/H-atom transfer
The unusual nature of our intramolecular C–H amination stems from the generation of a radical and an anion in situ. To achieve this, two criteria must be met. First, an acidic proton is needed for a deprotonation that produces a persistent N-anion. Second, the substrates should have a sufficiently weak C–H bond that undergoes HAT with the formation of a C-radical.The cascade mechanism begins with the exergonic deprotonation of the mildly acidic benzamide by tert-butoxide (ΔG = −17.2 kcal mol−1, Scheme 6, A1). Once a persistent nitrogen anion is quickly generated, the slowly produced DMF radical (see the ESI† for discussion of DMF radical formation) can engage the sufficiently weak bis-benzylic C–H bond (BDE = 85 kcal mol−1) in a HAT step (ΔG = −10.0 kcal mol−1, Scheme 6, A2). This step yields the initial acyclic radical–anion intermediate. While the HAT ΔG‡ and ΔG are slightly lower for the neutral amide than the deprotonated amide (ΔG‡ = 17.6 vs. 19.1, ΔG = −10.9 vs. −10.0 kcal mol−1), the high concentration of base and the highly favorable thermodynamics for the deprotonation step should result in the majority of HAT occurring after deprotonation (Scheme 8).
 | ||
| Scheme 8 Reaction energy profiles and twisted transition states for C–H activation in neutral and deprotonated substrates. All energies are in kcal mol−1. | ||
While tert-butoxide can deprotonate the benzylic C–H bond of diphenylmethane37 (pKa = 32.2 in DMSO), the benzamide's N–H bond is far more acidic (pKa = 23.3 in DMSO). The nine orders of magnitude difference in acidity leaves no doubt that the N–H bond should be deprotonated preferentially. In our previous work, we explored the possibility of t-BuOK deprotonating the benzylic C–H bond and found that ΔG for this process is also favorable (−3 kcal mol−1) suggesting that C–H deprotonation is feasible as well. However, once the initial nitrogen anion is formed, the second deprotonation would form a dianion. Although the possibility of a dianionic process cannot be completely eliminated at the present stage, we favor the radical HAT path for the C–H activation because of the coulombic penalty associated with the formation of multiply charged species.
The transition state for the initial HAT is interesting. At the stage where the benzylic C–H bond begins to break to form the C-centered radical, the benzylic C–H is misaligned with the aromatic π-system. Hence, the forming radical finds itself in alignment only with the pendant aromatic ring. One might expect that as this radical would prefer to be in a close alignment with both aromatic rings to take advantage of stereoelectronic stabilization. This finding explains why the pendant aryl group is necessary – although the CH2 groups in substrates 1w and 1x are formally “benzylic” (Scheme 4), the core aryl group does not directly contribute to their C–H activation. Under these intramolecular stereoelectronic constraints, their relatively low BDE values are somewhat misleading.
Furthermore, the amide is rotated out of conjugation the aromatic π-system in a nearly orthogonal geometry.38 The twisted geometry may provide an explanation to why amide deprotonation does not activate the ortho-benzylic C–H bond towards H-abstraction. Steric hindrance between the two relatively large ortho-substituents contributes to this geometric preference. On the other hand, the amide π-system may engage in through-space interactions with the back lobe of the anti-bonding orbital of the breaking C–H bond. In this scenario, the amide may be assisting in the C–H activation process by helping to stabilize the newly forming radical via a through-space interaction.
Radical–anionic cyclization and C–N bond formation
In the key step of the overall cascade, the N-anion and the C-centered radical in A2 react to form a 2c,3e “half” bond, also making a cyclic radical–anion in the process. Interestingly, this process occurs without the apparent loss of amide resonance since it is the 2nd “in-plane” lone pair of nitrogen that is involved in the N–C coupling. On the other hand, the radical center rotates out of conjugation with the central aryl ring as well. The latter stereolectronic penalty is likely to contribute to a relatively high, 17.2 kcal mol−1, Gibbs barrier and the low thermodynamic driving force, ΔG = −3.6 kcal mol−1, for this seemingly trivial step (Scheme 9). These values are significantly less favorable than the analogous values for the C–N bond formation from deprotonated anilines (8.5 and −27.8 kcal mol−1, respectively) reported in our earlier work. This dramatic difference highlights the importance of N-partner basicity/donor ability for this process. We will discuss this factor further in the conclusions section of this manuscript.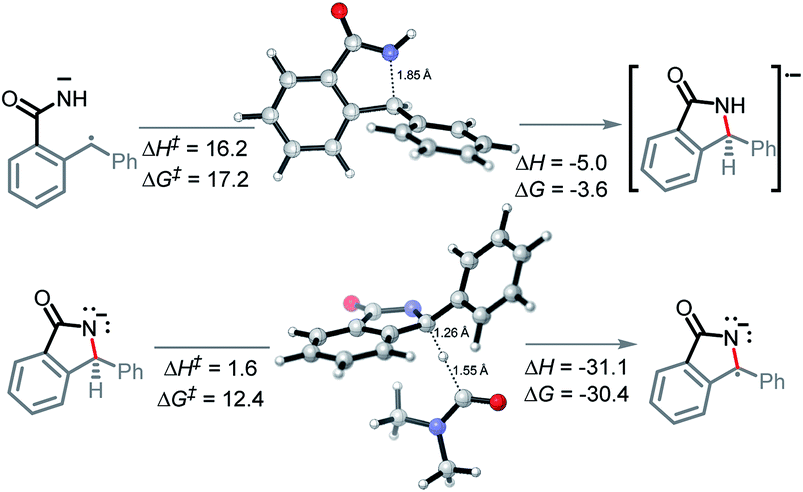 | ||
| Scheme 9 Transition states for the C–N bond formation and second HAT. | ||
Additionally, the three non-bonding electrons of the radical and anion reacting partners have to find “a new home” in this step. Two of these electrons are accommodated in the newly formed σ orbital of the C–N bond. The third electron avoids its apparent destiny of ending up at the high energy 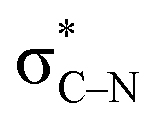 orbital by “hopping” to a lower energy π* orbital of the aromatic ring (Scheme 10). This state crossing, not unusual for radical-anionic reactions,39 stabilizes the product by creating a delocalized π-type radical–anion.
orbital by “hopping” to a lower energy π* orbital of the aromatic ring (Scheme 10). This state crossing, not unusual for radical-anionic reactions,39 stabilizes the product by creating a delocalized π-type radical–anion.
 | ||
Scheme 10 State crossing avoids populating the high energy  orbital. orbital. | ||
Nevertheless, the reacting system evolved from a mild reductant (electron in a non-bonding orbital) into a more potent reductant (electron is an antibonding orbital). The high reducing power of the cyclic product allows its reaction with a mild oxidant, such as molecular oxygen. This reaction (i.e., single electron transfer) removes the antibonding electron, converting a 2c,3e-bond into a normal 2c,2e C–N bond. This step simultaneously forms superoxide in a thermodynamically favorable way with ΔG = −29.1 kcal mol−1 (Scheme 6, A3).
After the benzamide is cyclized into an isoindolinone, the cyclic intermediate is deprotonated to give anion A4 (Scheme 6, ΔG = −17.7 kcal mol−1). The α-C–H bond in this anion is sufficiently weakened to form a stabilized radical–anion A5 (ΔG = −30.4 kcal mol−1 after a HAT to DMF radical).
Diverting from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N to C–O bond formation
N to C–O bond formation
From the stabilized radical intermediate A5, one can envision two possible mechanisms for the formation of the C–O bond (Scheme 11). The first is similar to what we reported in our previous work,10 in which the intermediate is oxidized a second time by molecular oxygen to form the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety of an imine intermediate. This intermediate may then be intercepted by a suitable nucleophile generated in situ, such as superoxide or hydroxide. In particular, superoxide is known to undergo disproportionation to give O2 and hydroxide under aqueous conditions.40 It is possible that a similar process to generate hydroxide could occur under the conditions of our C–H amination reaction, whereas t-BuOH would serve as the proton source instead of H2O. The second possibility, is that a radical coupling partner such as superoxide or hydroperoxyl radical couples to the C-centered radical making a hydroperoxide intermediate.
N moiety of an imine intermediate. This intermediate may then be intercepted by a suitable nucleophile generated in situ, such as superoxide or hydroxide. In particular, superoxide is known to undergo disproportionation to give O2 and hydroxide under aqueous conditions.40 It is possible that a similar process to generate hydroxide could occur under the conditions of our C–H amination reaction, whereas t-BuOH would serve as the proton source instead of H2O. The second possibility, is that a radical coupling partner such as superoxide or hydroperoxyl radical couples to the C-centered radical making a hydroperoxide intermediate.
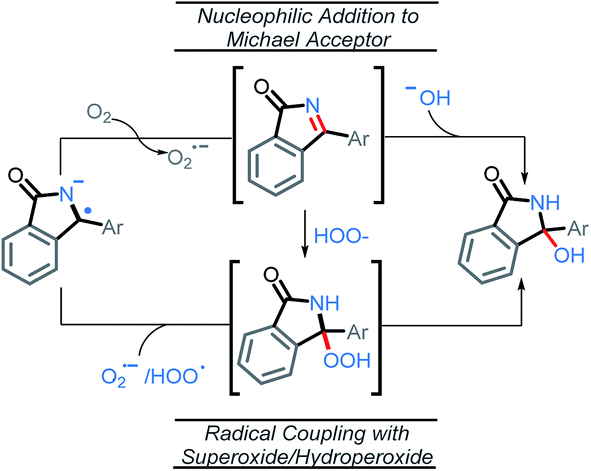 | ||
| Scheme 11 Possible routes to product from stabilized radical–anion intermediate. | ||
We initially tested to see if the addition of an external nucleophile could be used to trap the plausible imine intermediate. The addition of either sodium or potassium methoxide hinders the reaction, with no methoxy product observed in these experiments (Scheme 12).
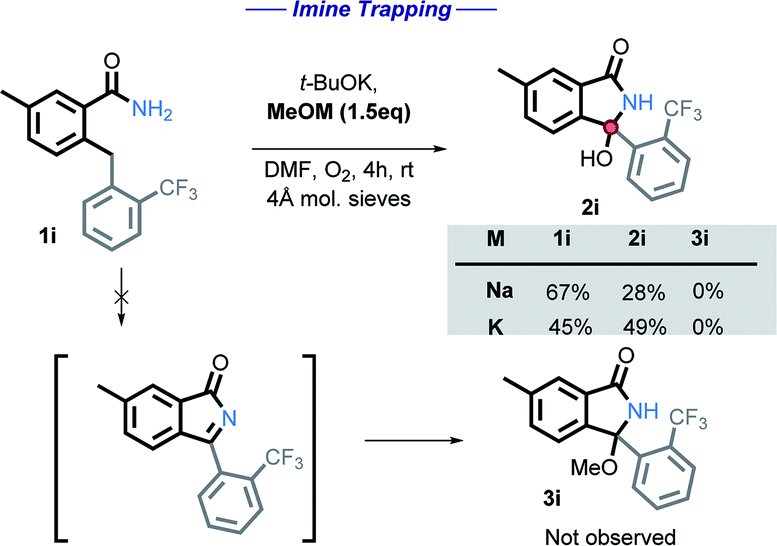 | ||
| Scheme 12 Attempting to trap the hypothetical imine intermediate. | ||
Our calculations show the oxidation of radical–anion A5 by O2 is thermodynamically uphill (ΔG = +10.0 kcal mol−1, Scheme 13). While this is not a prohibitive price to pay, it is worth noting that this oxidation step is 39.1 kcal mol−1 more endergonic than the initial oxidation step. This substantial increase stems from several factors, including lack of aromatic stabilization in the N-heterocyclic part of the product as well as the additional radical and charge stabilization in the reactant, not only through the bis-phenyl groups, but additionally through the nitrogen and carbonyl.
 | ||
| Scheme 13 Contrasting thermodynamics (kcal mol−1) for the CN forming oxidations in the amide and aniline cascades. | ||
This finding contrasts our previous work with anilines where the second oxidation step was favorable (ΔG = −16.6 kcal mol−1) due to the formation of an aromatic system.10 In the present case, the situation is different, and this differences manifests itself in a divergent reaction pathway (formation of a C–O bond instead of CN moiety). To understand this step better, we explored the possibility of radical coupling being responsible for the final C–O formation.
A series of studies designed to gain insight into the possible radical coupling of A5 were performed and are listed below (Scheme 14).
 | ||
| Scheme 14 Radical coupling mechanistic studies. | ||
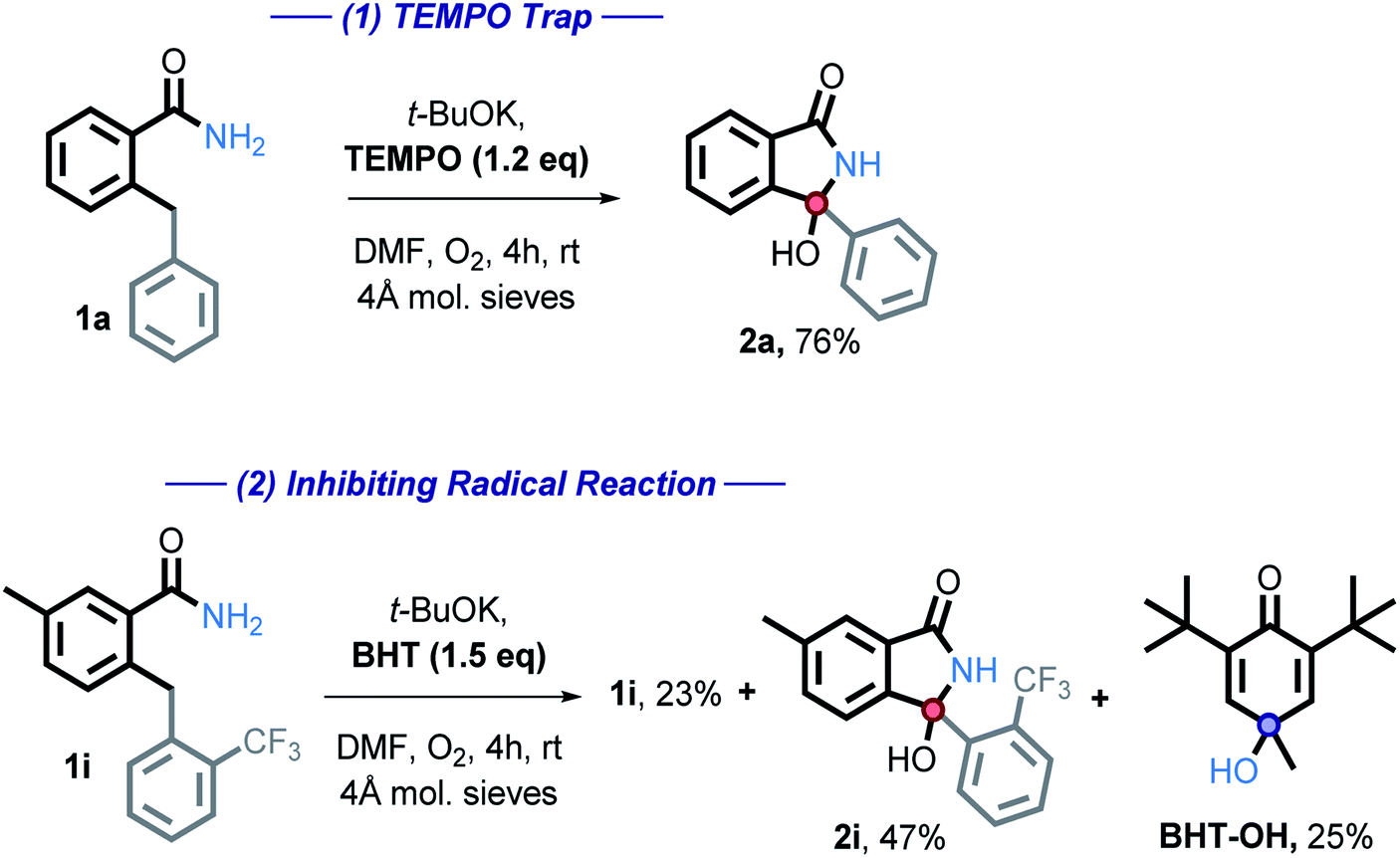
We then employed two common radical trapping agents, attempting to either trap a radical intermediate or to inhibit the reaction. TEMPO, caused a slight decrease in isolated yield (87% vs. 76%) but no TEMPO trapped product or recovered starting material was observed (eqn (1)). This finding is consistent with the radical mechanism if intramolecular radical trapping (i.e., the cyclization) is faster than the intermolecular trapping, or if TEMPO can play an alternative role by promoting the C–H activation step. More detailed discussion on the possible role of TEMPO will be given in a subsequent section.
We also tested the effect of a common radical and peroxide trapping agent, 3,5-di-tert-4-butylhydroxytoluene (BHT), at the standard reaction conditions (eqn (2)). The phenol moiety of BHT is often used to halt the autoxidation of organic molecules with oxygen, analogous to that of vitamin E. BHT can deactivate two (usually peroxy) radicals – the first one by a hydrogen atom transfer and the second one by reaction at the cycle.41
We found that the reaction is partially inhibited – 2i was isolated in a 47% yield, starting material recovered in 27% isolated yield. Furthermore, BHT-OH was additionally isolated in 25% yield. Low yields are a result of isolation difficulties. Formation of this product further suggests that a radical pathway is involved.
Addition of superoxide/hydroperoxyl radical to the radical–anion A5
It is unlikely that the OH group in the final product is a result of radical coupling with hydroxide radical, as it is highly reactive and will likely abstract a hydrogen atom from the solvent or t-BuOH before reaching the reactant.42 This consideration led us to explore the possibility of radical coupling with superoxide or with its conjugate acid, the hydroperoxyl radical (HOO˙).43Our calculations show this to be a favorable path for forming the C–O bond (Scheme 15). In particular, radical addition to intermediate A5via HOO˙ was found to be highly exergonic, ΔG = −23.6 kcal mol−1. Addition of superoxide (the precursor of HOO˙) to A5 was favorable, ΔG = −11.0 kcal mol−1. On the other hand, the addition of molecular oxygen was found to be endergonic, ΔG = +7.5 kcal mol−1. It is worth noting that all three of these proposed pathways are thermodynamically more favorable than an oxidation of A5 by O2 (ΔG = +10.0 kcal mol−1).
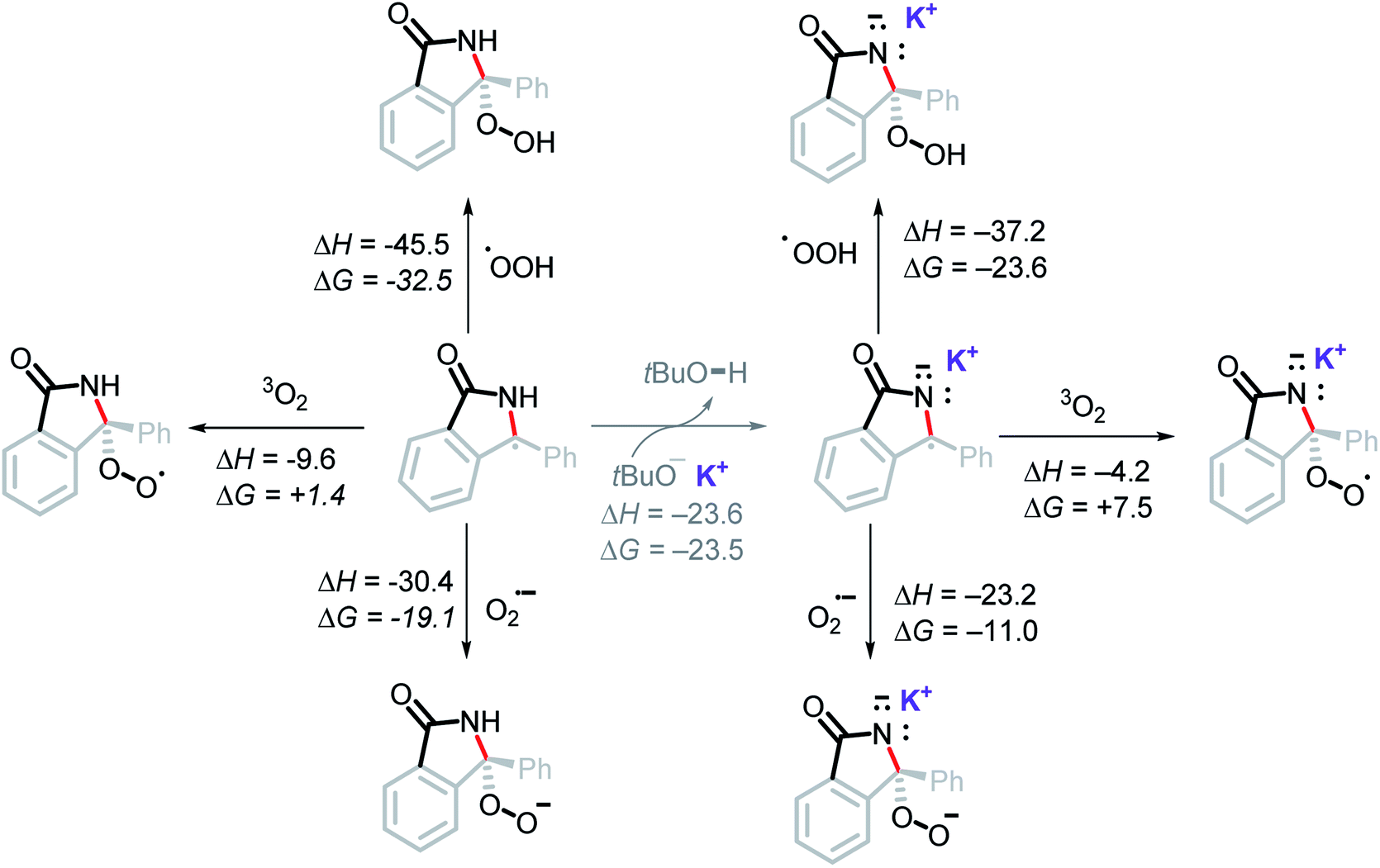 | ||
| Scheme 15 Computational thermodynamic data for the addition of molecular oxygen, superoxide, and hydroperoxyl radical to the stabilized radical intermediate. Energies in kcal mol−1. | ||
Superoxide has been shown to act as a Brønsted base in aprotic media and deprotonate weak acids such as 1-butanol in DMF (pKa = 33.3 in DMF).44 It is therefore possible, that superoxide may deprotonate t-BuOH (estimated pKa ≈ 34–35 in DMF) generated in situ, forming HOO˙. Hence, both HOO˙ and superoxide may couple with A5.
The evidence that we have presented up to this point suggests that formation of a hydroperoxide intermediate45 should be considered (Scheme 6, A6). To explore this possibility, we prepared this intermediate and tested its properties as described in the following section.
Hydroperoxide intermediate
The hydroxy group of 2p was converted into hydroperoxy group of 3p (Scheme 16, eqn (1)) by acid-catalyzed reaction with H2O2. The reference spectra contained the characteristic broad downshifted peak of a hydroperoxide that readily underwent deuterium exchange.46 After subjecting 3p to our standard conditions, the formation of 2p was observed by TLC within five minutes. In half an hour, 2p was formed in 88% yield (Scheme 16, eqn (2)). The lack of 3p reactivity in the absence of t-BuOK (Scheme 16, eqn (2)) indicates the important role of t-BuOK in the reduction of the hydroperoxide intermediate into the final product. | ||
| Scheme 16 Preparation and instability of the suggested hydroperoxide intermediate. | ||
Because of the rapid consumption of hydroperoxide in the presence of base, we anticipate its existence in situ to be fleeting. The existence of transient hydroperoxide intermediates, that convert to OH-bearing final products, has previously been reported in O2-mediated oxidations of C(sp3)–H bonds.47 Indeed, in several experiments, 1H NMR of the reaction mixtures showed a broad downshifted singlet, potentially indicative of the hydroperoxide, (see ESI for additional details†). Although this intermediate was too unstable to persist and to be reliably detected under the reaction conditions, these findings suggest that a hydroperoxide intermediate (Scheme 6, A6) is formed transiently and reduced by t-BuOK/t-BuOH into the final isoindolinone product. The hydroperoxide may also be involved in the radical chain propagation by serving as a possible source of O-centered radicals that can assist in the C–H activation step.
Secondary amides
We were intrigued by the sluggish reactivity of the secondary amides. Since we isolate unreacted starting material, the reaction is interrupted in the initial stages. Yet our calculations find that each of the three initiation steps, i.e., the deprotonation (ΔG = −16.0), HAT (ΔG = −9.3), and the radical–anion cyclization (ΔG = −5.9), for 1r are thermodynamically favorable (Scheme 17).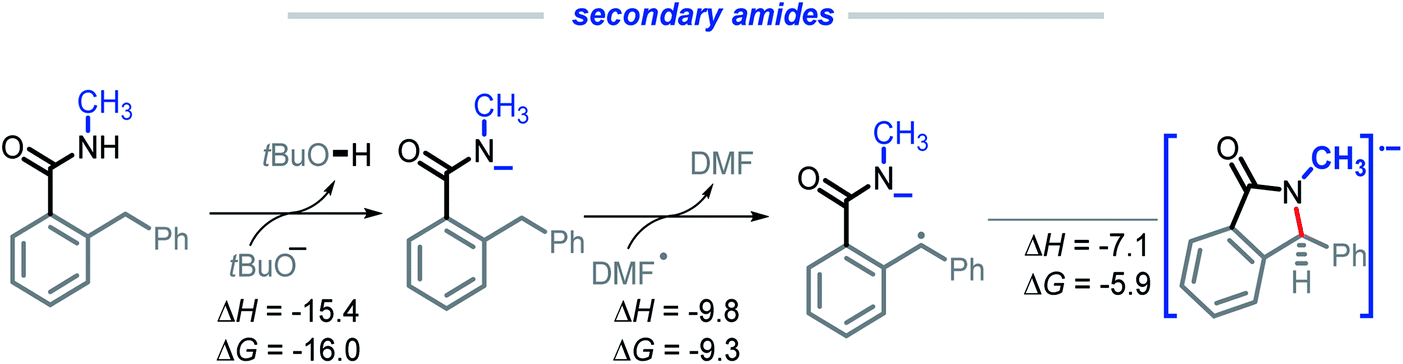 | ||
| Scheme 17 Calculations for the deprotonation, HAT, and radical–anion cyclization of 1r. All energies are reported in kcal mol−1. | ||
We used computed activation barriers to understand why the secondary amides are unreactive (see ESI†). Our analysis suggests that the barrier for the H-abstraction is about 1.2 kcal mol−1 higher for the secondary amides than it is for the primary amides. This difference should lead to a ca. 10-fold rate decrease for this step, providing a possible explanation for the low reactivity of the secondary amide substrates under the reaction conditions.
Conversely, the calculated barrier for the C–N bond formation is lower for the secondary amides, indicating that this step is unlikely to be the cascade bottleneck (Scheme 18). Aimed by these observation, we have concentrated our attention on the H-transfer step.
 | ||
| Scheme 18 Computed activation and reaction enthalpies and Gibbs energies for the cyclization of secondary amide 1r. Numbers in parentheses are for primary amide 1a. All numbers reported in kcal mol−1. | ||
Indeed, focus on C–H activation allowed us to solve the problem of secondary amides as described below. Recalling that TEMPO had no detrimental effect on our radical reaction, we considered the possibility of TEMPO assisting in C–H activation.
Indeed, literature suggests that TEMPO can be a useful additive to the oxidation of benzylic C(sp3)–H bonds. For example, TEMPO can be oxidized into an oxoammonium salt (TEMPO+) in the presence of hydroperoxyl radical, peroxyl radicals, or a secondary oxidant (e.g. NaOCl) as shown in Scheme 19a.48 The latter has been shown to act as an oxidant that may facilitate the forward progression of redox reactions.49 In particular, TEMPO+, generated in situ, from TEMPO was used as a cocatalyst in the aerobic oxidation of benzylic C(sp3)–H into carbonyls.50 Even if the carbonyl intermediate is formed in this case, it would, as we previously discussed in Scheme 5, readily cyclize under basic conditions into our isoindolinone product.
 | ||
| Scheme 19 Potential pathways for TEMPO assistance in C–H activation. | ||
Additionally, TEMPO+ has been suggested to facilitate benzylic hydride transfers.51 It is plausible that in our case, a concerted C–N bond formation and benzylic hydride transfer to TEMPO+ occurs generating TEMPOH and our cyclized product (Scheme 19b). Alternatively, one can consider a hydride transfer to TEMPO+ forming a benzylic carbocation that is immediately trapped intramolecularly via cyclization (Scheme 19c).
Although the exact mechanistic path for C–H activation in our system is so far unknown, the combination of possible attractive scenarios motivated us to test the effect of TEMPO on the reaction. To our delight, we observed the nearly full consumption when secondary amide 1r was subjected to the optimized conditions along with TEMPO. Interestingly, no TEMPO-trapping product was present in the reaction mixture. Instead, 2r was obtained in 74% isolated yield a dramatic improvement over the standard conditions (30% without TEMPO vs. 74% with TEMPO, Scheme 20). Motivated by this finding, we tested reactivity of 1s in the presence of TEMPO. Gratifyingly, the reaction affords 2s, albeit in a moderate yield of 31% (the majority of reaction mixture is unreacted 1s). Furthermore, substrates 1t, 1u, and 1v also showed a substantial increase in reactivity with the addition of TEMPO and the products 2t, 2u, and 2v were isolated in 87%, 54%, and 55% respectively.
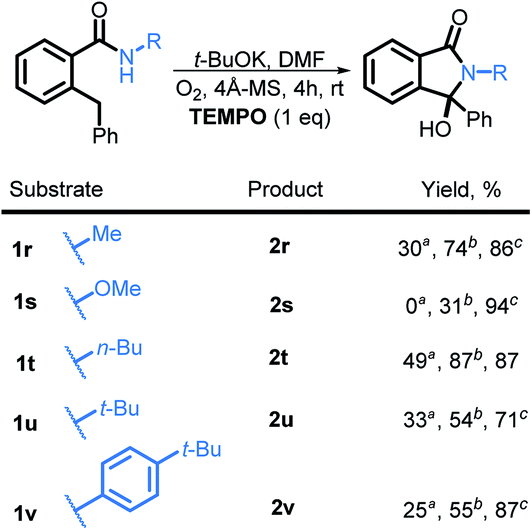 | ||
| Scheme 20 Improved C–H activation with TEMPO as an additive. Reaction conditions: secondary benzamide (0.025 M), t-BuOK (3 eq.), DMF (2 mL), 4 Å MS, O2 balloon, reactions stirred at r.t. overnight. aIsolated yields for reaction performed under standard conditions. bIsolated yields for reactions with the addition of TEMPO. cYields based of reclaimed starting materials with the addition TEMPO. | ||
Nitriles
As we explored the reactivity of amides under our oxidative C–H amination conditions, we decided to take advantage of the electrophilic nature of benzonitriles, a common precursor in our amide synthesis. In the presence of a suitable nucleophile, anionic addition to the electrophilic carbon on nitriles will result in formation of a nitrogen anion. This anion may then trap a carbon radical, forming a C–N bond upon oxidation.Indeed, nitrile 4a was found to transform into 2a in good yield (72%) in the presence of t-BuOK and O2 in DMF (Scheme 21). Two interesting side products were isolated as well. The rearranged product 5a is a result of C–H and C–C activation. Because product 6a was not reported previously, we have confirmed its structure by single crystal X-ray crystallography (see ESI†). Although the mechanism and scope of these transformations will have to be explored further in the future work, this finding does illustrate that the scope of the present approach to C–H activation extends beyond amide and aniline couplings.
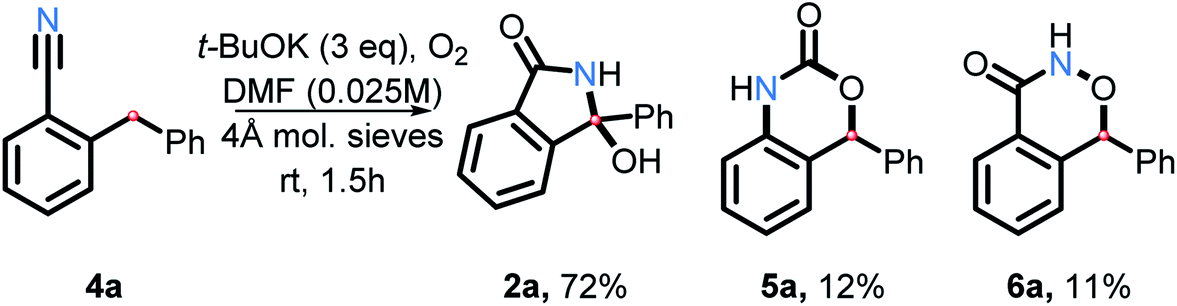 | ||
| Scheme 21 C–H activation and cyclization of a nitrile substrate. | ||
Conclusion
We have developed a direct non-photochemical method for converting C(sp3)–H bonds into C–N and C–O bonds under mild conditions with the aid of base, molecular oxygen, and DMF. Each component of the overall reaction plays a pivotal role in a coordinated sequence of deprotonation, H-atom transfer, and electron transfer that forges the C–N bond. The base has three main functions: (1) to deprotonate the N–H bond, (2) to provide an adequate concentration of DMF carbamoyl anion to be converted to DMF radical, and (3) to convert the hydroperoxide intermediate into the final hydroxyl product. The DMF radical performs selective HAT at the di-benzylic C(sp3)–H to form a C-centered radical. The C–N bond is formed initially through a 2c,3e interaction between the N-anion and C-radical. Oxidation of the radical–anion intermediate by oxygen completes the C–N bond forming sequence. Computational data suggest that the stabilized radical anion formed after the second HAT could not be oxidized by molecular oxygen. Instead, the radical couples with either superoxide or hydroperoxide species to generate a hydroperoxide intermediate that ultimately decomposes to form the hydroxide product. Addition of TEMPO opens the door for the use of secondary amides and improves the performance of some insufficiently reactive primary amides. This process allows for the formation of functionalized N-heterocycles in an operationally simple and robust fashion.Importantly, this work dramatically expands the range of N-anions that can participate in the three-electron approach to C–N bond formation. In general, 2c,3e-bonds are weaker than their classic 2c,2e-counterparts. Hence, limitations for their formation are much more severe. For example, the C–N bond formation in the radical cyclization with anilines is uphill by >20 kcal mol−1. One of the reasons why this deceivingly simple transformation is unfavorable is that it leads to the development of cationic character at nitrogen. Trading the lone pair of nitrogen for a 2c,3e-bond removes electron density from the N atom – an “oxidation” process that this electronegative element generally resists. Making nitrogen more electron rich, preferably anionic, is key to overcoming this problem.10
In the present work, we have increased N–H bond acidity by seven orders of magnitude from our earlier studies by switching from anilines10 (pKa ∼ 30 in DMSO) to amides (pKa ∼ 23 in DMSO). Such deactivation of the conjugated nitrogen base increases the activation barrier for C–N bond formation from 8.510 to 17.2 kcal mol−1 and decreased reaction exergonicity from 2810 to 4 kcal mol−1. Thus, the present use of a stabilized N-anion provided important “bracketing” information on the thermodynamic limits of three-electron C–N bond formation. Although the initial correlations presented in Scheme 22 are crude, considering the small number of points and differences in the formed ring size, they should provide the first approximate guidelines for the design of such reactions.
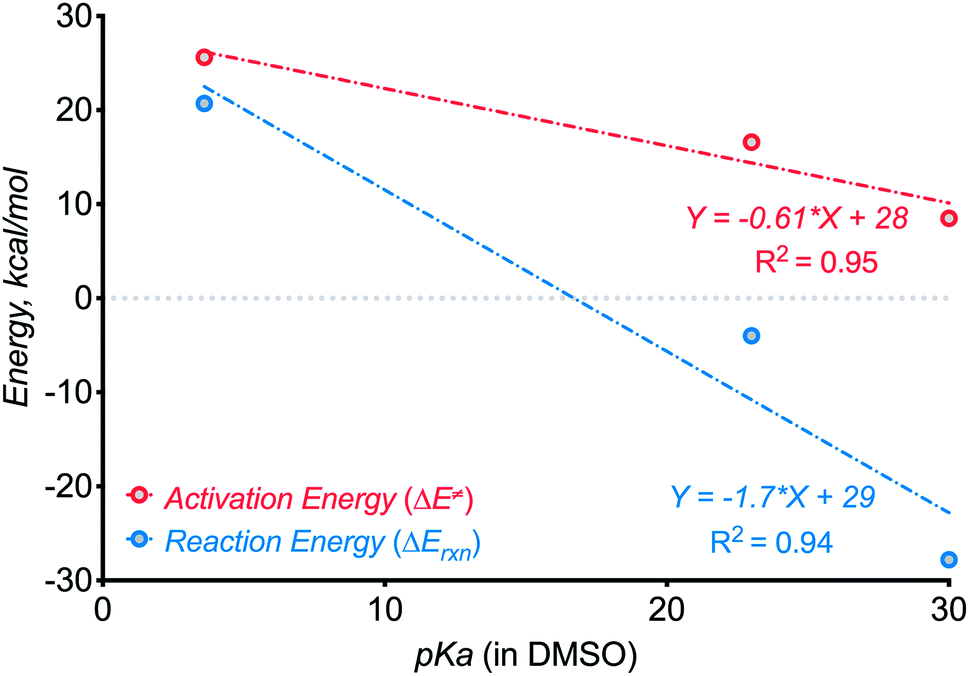 | ||
| Scheme 22 Testing for the effect of N–H component acidity on the kinetics and thermodynamics of three-electron C–N bond formation. | ||
Furthermore, even though the C–N bond formation from deprotonated benzamides is still efficient despite the additional kinetic and thermodynamic penalties, the next step, i.e., the CN moiety formation, is not favorable anymore! Here, the cyclized radical–anion is so stable that it does not undergo one-electron oxidation under the reaction conditions. Instead, a new reactivity pattern becomes available and the reaction sequence culminates in the formation of a C–O bond instead of a CN moiety.
These observations can guide the choice of N–H components in future reactions that form 2c,3e bonds. There is a broad range (pKa ∼ 20–32 in DMSO) of N–H bonds that can be deprotonated by potassium tert-butoxide but do not form an overstabilized and unreactive conjugate base. For such N–H partners, a variety of possible C–H bonds may be used for generating a suitable C-radical for the three-electron C–H aminations. One can sufficiently weaken the C–H bond (BDEs in the range of 80–90 kcal mol−1) by placing heteroatoms or non-aromatic π-systems next to the methylene carbon.
The N-deprotonated benzamides presented in this work are close to the “pKa limit” for three-electron bond formation from benzylic radicals. In order to expand this limit to less reactive, more stabilized nitrogen bases, one has to use more reactive, less stabilized radical partners. We hope that our report will encourage broad investigation of new strategies for unconventional C–N bond formation.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The fundamental and synthetic aspects of this study were supported by donors of the ACS Petroleum Research Fund (PRF#57377-ND4) and by the National Science Foundation (CHE-1465142). We appreciate the allocation of computational resources from FSU RCC and the NSF XSEDE (TG-CHE160006) and assistance in acquiring 1H and 13C NMR spectra from the NMR Facility of FSU Department of Chemistry and Biochemistry. G. P. G. is grateful to Professor Alán Aspuru-Guzik (University of Toronto) for his support.References
- (a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (b) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49–51 CrossRef CAS PubMed; (c) G. Choi, Q. Zhu, D. C. Miller, C. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed; (d) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed; (e) M. S. Burg, M. Gicquel, S. Breitenlechner and A. Pöthig, Angew. Chem., Int. Ed., 2018, 57, 2953–2957 CrossRef PubMed; (f) J. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- (a) B. Peng and N. Maulide, Chem.–Eur. J., 2013, 19, 13274–13287 CrossRef CAS PubMed; (b) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (d) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874–5883 CrossRef CAS PubMed.
- (a) M. S. Chen and C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed; (b) S. Murahuashi, N. Komiya and T. Nake, J. Am. Chem. Soc., 2003, 125, 15312–15313 CrossRef PubMed; (c) B. Dangel, K. Godula, S. Won Youn, B. Sezen and D. Sames, J. Am. Chem. Soc., 2002, 124, 11856–11857 CrossRef CAS PubMed; (d) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (e) O. V. Zatolochnaya and V. Gevorgyan, Nat. Chem., 2014, 6, 661–663 CrossRef CAS PubMed; (f) Y. N. Timsina, B. F. Gupton and K. C. Ellis, ACS Catal., 2018, 8, 5732–5776 CrossRef CAS; (g) A. Dhakshinamoorthy, A. Asiri and H. Garcia, ACS Catal., 2019, 9, 1081–1102 CrossRef CAS; (h) Z. Zhou and L. Kürti, Synlett, 2019, 30, 1525–1535 CrossRef CAS.
- (a) D. Hazelard, P. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500–2521 RSC; (b) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 34, 5061–5074 RSC; (c) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 921–942 RSC; (d) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (e) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- (a) A. Arai, Y. Ueda, K. Morisaki, T. Furuta, T. Sasamori, N. Tokitoh and T. Kawabata, Chem. Commun., 2018, 54, 2264–2267 RSC; (b) M. Kono, S. Harada and T. Nemoto, Chem.–Eur. J., 2017, 23, 7428–7432 CrossRef CAS PubMed; (c) M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570–11574 CrossRef CAS PubMed; (d) P. Starkov, T. F. Jamison and I. Marek, Chem.–Eur. J., 2015, 21, 5278–5300 CrossRef CAS PubMed; (e) T. A. Ramierez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC; (f) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (g) J. D. Bois, Org. Process Res. Dev., 2011, 15, 758–762 CrossRef PubMed; (h) J. B. Aguila, Y. M. Badiei and T. H. Warren, J. Am. Chem. Soc., 2013, 135, 9399–9406 CrossRef PubMed; (i) X. Zhang, H. Xu, X. Liu, D. L. Phillips and C. Zhao, Chem.–Eur. J., 2016, 22, 7288–7297 CrossRef CAS PubMed.
- (a) X. Hu, X. Qi, J. Chen, Q. Zhao, Q. Wei, Y. Lan and W. Xia, Nat. Commun., 2016, 7, 11188–11200 CrossRef PubMed; (b) K. C. Nicolaou, P. S. Baran, Y.-L. Zhong, S. Barluenga, K. W. Hunt, R. Kranich and J. A. Vega, Iodine, J. Am. Chem. Soc., 2002, 124, 2233–2244 CrossRef CAS PubMed; (c) Y. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980–11983 CrossRef CAS PubMed; (d) G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed; (e) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed; (f) K. T. Tranatino, D. C. Miller, T. A. Callon and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 6440–6443 CrossRef PubMed.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1879, 12, 984–990 CrossRef; (b) M. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef CAS.
- (a) J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed; (b) K. A. Margrey, A. Leven and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 15644–15648 CrossRef CAS PubMed; (c) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed; (d) J. L. Jeffrey, F. R. Petronijević and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404–8407 CrossRef CAS PubMed; (e) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC; (f) H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860–11871 CrossRef CAS PubMed.
- C. J. Evoniuk, S. P. Hill, K. Hanson and I. V. Alabugin, Chem. Commun., 2016, 52, 7138–7141 RSC.
- C. J. Evoniuk, G. P. Gomes, S. Hill, F. Satoshi, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 16210–16221 CrossRef CAS PubMed.
- (a) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y. Zhang, X. Zhang, G. Chen, Y. Cheng, Z. Cao, Q. Guo, L. Wang and Z. Shi, Nat. Commun., 2014, 5, 4707–4713 CrossRef CAS PubMed; (b) Y. Timsina, F. Gupton and K. Ellis, ACS Catal., 2018, 8, 5732–5776 CrossRef CAS; (c) Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496–3499 CrossRef CAS PubMed; (d) G. He, S. Zhang, W. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef CAS PubMed; (e) X. Wu, Y. Zhao and H. Ge, Chem.–Eur. J., 2014, 20, 9530–9533 CrossRef CAS PubMed; (f) J. J. Neumann, S. Rakshi, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892–6895 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- (a) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS PubMed; (b) S. W. Youn, J. H. Bihn and B. S. Kim, Org. Lett., 2011, 13, 3738–3741 CrossRef CAS PubMed; (c) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed; (d) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed.
- A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS.
- (a) S. P. West, A. Bisai, A. D. Lim, R. R. Narayan and R. Sarpong, J. Am. Chem. Soc., 2009, 131, 11187–11194 CrossRef CAS PubMed; (b) J. M. Gruver, S. P. West, D. B. Collum and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 13212–13213 CrossRef CAS PubMed; (c) J. L. Jeffrey, E. B. Bartlett and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 2194–2197 CrossRef CAS PubMed.
- (a) M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532–5550 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- For the general review of dehydrogenative C–O bond formation, see: I. B. Krylov, V. A. Vil' and A. O. Terent'ev, Cross-dehydrogenative Coupling for the Intermolecular C-O Bond Formation, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef PubMed.
- D.-M. Yan, Q.-Q. Zhao, L. Rao, J.-R. Chen and W.-J. Xiao, Chem.–Eur. J., 2018, 24, 16895–16901 CrossRef CAS PubMed.
- U. S. Department of Health and Human Services Food and Drug Administration, Q3D Elemental Impurities Guidance for Industry, 2015 Search PubMed.
- J. G. Topliss, L. M. Konzelman, N. Sperber and E. R. Franklin, J. Med. Chem., 1964, 7, 453–456 CrossRef CAS PubMed.
- (a) I. R. Hardcastle, S. U. Ahmed, H. Atkins, G. Farnie, B. T. Golding, R. J. Griffin, S. Guyenne, C. Hutton, P. Källblad, S. J. Kemp, M. S. Kitching, D. R. Newell, S. Norbedo, J. Northen, R. J. Reid, K. Saravanan, H. M. Willems and J. Lunec, J. Med. Chem., 2006, 49, 6209–6221 CrossRef CAS PubMed; (b) C. Riedinger, J. A. Endicott, S. J. Kemp, L. A. Smyth, A. Watson, E. Valeur, B. T. Golding, R. J. Griffin, I. R. Hardcastle, M. E. Noble and J. M. McDonnell, J. Am. Chem. Soc., 2008, 130, 16038–16044 CrossRef CAS PubMed; (c) I. R. Hardcastle, J. Liu, E. Valeur, A. Watson, S. U. Ahmed, T. J. Blackburn, K. Bennaceur, W. Clegg, C. Drummond, J. A. Endicott, B. T. Golding, R. J. Griffin, J. Gruber, K. Haggerty, R. W. Harrington, C. Hutton, S. Kemp, X. Lu, J. M. McDonnell, D. R. Newell, M. E. Noble, S. L. Payne, C. H. Revill, C. Riedinger, Q. Xu and J. Lunec, J. Med. Chem., 2011, 54, 1233–1243 CrossRef CAS PubMed; (d) A. F. Watson, J. Liu, K. Bennaceur, C. J. Drummond, J. A. Endicott, B. T. Golding, R. J. Griffin, K. Haggerty, X. Lu, J. M. McDonnell, D. R. Newell, M. E. M. Noble, C. H. Revill, C. Riedinger, Q. Xu, Y. Zhao, J. Lunec and I. R. Hardcastle, Bioorg. Med. Chem. Lett., 2011, 21, 5916–5919 CAS; (e) T. A. Grigoreva, D. S. Novikova, A. V. Petukhov, M. A. Gureev, A. V. Garabadzhiu, G. Melino, N. A. Barlevv and V. G. Tribulovich, Bioorg. Med. Chem. Lett., 2017, 27, 5197–5202 CrossRef CAS PubMed.
- (a) V. Fajardo, V. Elango, B. K. Cassels and M. Shamma, Tetrahedron Lett., 1982, 23, 39–42 CrossRef CAS; (b) H. Kamauchi, Y. Shiraishi, A. Kojima, N. Kawazoe, L. Kinoshita and K. Koyama, J. Nat. Prod., 2018, 81, 1290–1294 CrossRef CAS PubMed.
- M.-W. Chen, Q.-A. Chen, Y. Duan, Z.-S. Ye and Y.-G. Zhou, Chem. Commun., 2012, 48, 1698–1700 RSC.
- Z. Kang, D. Zhang, J. Shou and W. Hu, Org. Lett., 2018, 20, 983–986 CrossRef CAS PubMed.
- (a) T. Nishimura, M. Nagamoto, Y. Ebe and T. Hayashi, Chem. Sci., 2013, 4, 4499–4504 RSC; (b) S. Sharma, Y. Oh, N. K. Mishra, U. De, H. Jo, R. Sachan, H. S. Kim, Y. H. Jung and I. S. Kim, J. Org. Chem., 2017, 82, 3359–3367 CrossRef CAS PubMed; (c) M. Nagamoto and T. Nishimura, Chem. Commun., 2014, 50, 6274–6277 RSC; (d) T. Nishimura, A. Noishiki, Y. Ebe and T. Hayashi, Angew. Chem., Int. Ed., 2013, 52, 1777–1780 CrossRef CAS PubMed.
- (a) H. H. Wasserman and J. l. Ives, Tetrahedron, 1981, 37, 1825–1852 CrossRef CAS; (b) P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC; (c) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- (a) Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. Su and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 CrossRef CAS PubMed; (b) G. Urgoita, R. SanMartin, M. T. Herrero and E. Domínguez, Chem. Commun., 2015, 51, 4799–4802 RSC; (c) J. Ma, Z. Hu, M. Li, W. Zhao, X. Hu, W. Mo, B. Hu, N. Sun and Z. Shen, Tetrahedron, 2015, 71, 6733–6739 CrossRef CAS; (d) H. Wang, Z. Wang, H. Huang, J. Tan and K. Xu, Org. Lett., 2016, 18, 5680–5683 CrossRef CAS PubMed; (e) C. Guo, Y. Zhang, Y. Zhang and J. Wang, Chem. Commun., 2018, 54, 3701–3704 RSC; (f) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed.
- The M06-2X functional has demonstrated to provide good thermodynamic data for organic reactions. For more details, see: (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- ChemCraft 1.8, http://www.chemcraftprog.com, accessed in February 2016 Search PubMed.
- (a) Y.-Y. Chen, X.-J. Zhang, H.-M. Yuan, W.-T. Wei and M. Yan, Chem. Commun., 2013, 49, 10974–10976 RSC; (b) W.-J. Wang, X. Zhao, L. Tong, J.-H. Chen, X.-J. Zhang and M. Yan, J. Org. Chem., 2014, 79, 8557–8565 CrossRef CAS PubMed; (c) Y.-Y. Chen, N.-N. Zhang, L.-M. Ye, J.-H. Chen, X. Sun, X.-J. Zhang and M. Yan, RSC Adv., 2015, 5, 48046–48049 RSC; (d) W.-T. Wei, Y.-J. Cheng, Y. Hu, Y.-Y. Chen, X.-J. Zhang, Y. Zou and M. Yan, Adv. Synth. Catal., 2015, 357, 3474–3478 CrossRef CAS; (e) Y.-Y. Chen, Z.-Y. Chen, N.-N. Zhang, J.-H. Chen, X.-J. Zhang and M. Yan, Eur. J. Org. Chem., 2016, 3, 599–606 CrossRef.
- E. J. Nanni Jr, M. D. Stallings and D. T. Sawyer, J. Am. Chem. Soc., 1980, 102, 4481–4485 CrossRef.
- M. Pichette Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587–10591 CrossRef CAS PubMed.
- J.-S. Li, F. Yang, Q. Yang, Z.-L. Li, G.-Q. Chen, Y.-D. Da, P.-M. Huang, C. Chen, Y. Zhang and L.-Z. Huang, Synlett, 2017, 28, 994–998 CrossRef CAS.
- For the effect of such geometric changes on stability and reactivity, see: I. V. Komarov, S. Yanik, A. Y. Ishchenko, J. E. Davies, J. M. Goodman and A. J. Kirby, J. Am. Chem. Soc., 2015, 137, 926–930 CrossRef CAS PubMed; M. Hutchby, C. E. Houlden, M. F. Haddow, S. N. G. Tyler, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., 2012, 124, 563–566 CrossRef; M. Szostak and J. Aube, Chem. Rev., 2013, 113, 5701–5765 CrossRef PubMed; S. Z. Vatsadze, Y. D. Loginova, G. Gomes and I. V. Alabugin, Chem.–Eur. J., 2017, 23, 3225–3245 CrossRef PubMed.
- P. Peterson, N. Shevchenko, B. Breiner, M. Manoharan, F. Lufti, J. Delaune, M. Kingsley, K. Kovnir and I. V. Alabugin, J. Am. Chem. Soc., 2016, 138, 15617–15628 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999, p. 461 Search PubMed.
- G. W. Burton and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 6472–6477 CrossRef CAS.
- (a) E. Bothe, M. N. Schuchmann, D. Schulte-Frohlinde and C. Sonntag, Z. Naturforsch., B: J. Chem. Sci., 1983, 38, 212–219 Search PubMed; (b) W. Song, T. Xu, W. J. Cooper, D. D. Dionysiou, A. A. d. l. Cruz and K. E. O'Shea, Environ. Sci. Technol., 2009, 43, 1487–1492 CrossRef CAS PubMed; (c) L. M. Dorfman and G. E. Adams, Reactivity of the Hydroxyl Radical in Aqueous Solutions, U.S. National Bureau of Standards, Washington, DC, 1973 Search PubMed; (d) H. Chen, L. Lin, Z. Lin, G. Guo and J. M. Lin, J. Phys. Chem. A, 2010, 114, 10049–10058 CrossRef CAS PubMed.
- (a) E. J. Nanni, D. T. Sawyer, S. S. Ball and T. C. Bruice, J. Am. Chem. Soc., 1981, 103, 2797–2802 CrossRef CAS; (b) D. T. Sawyer, E. J. Nanni and J. L. Roberts Jr, Electrochemical and Spectrochemicla Studies of Biological Redox Components, 1982, pp. 585–600 Search PubMed; (c) M. Hayyan, M. A. Hasim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- (a) E. J. Nanni Jr, M. D. Stallings and D. T. Sawyer, J. Am. Chem. Soc., 1980, 102, 4481–4485 CrossRef; (b) D.-H. Chin, G. Chiericato Jr, E. J. Nanni Jr and D. T. Sawyer, J. Am. Chem. Soc., 1982, 104, 1296–1299 CrossRef CAS.
- Note that the peroxide moiety in this molecule would engage in the anomeric
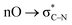 interaction. Such interactions were shown to significantly stabilize peroxides with geminal acceptor groups:
(a) G. P. Gomes, V. Vil', A. Terent'ev and I. V. Alabugin, Chem. Sci., 2015, 6, 6783–6791 RSC;
(b) V. A. Vil', G. P. Gomes, M. V. Ekimova, K. A. Lyssenko, M. A. Syroeshkin, G. I. Nikishin, I. V. Alabugin and A. O. Terent'ev, J. Org. Chem., 2018, 83, 13427–13445 CrossRef PubMed;
(c) E. Juaristi, G. P. Gomes, A. O. Terent'ev, R. Notario and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 10799–10813 CrossRef CAS PubMed;
(d) G. P. Gomes, I. A. Yaremenko, P. S. Radulov, R. A. Novikov, V. V. Chernyshev, A. A. Korlyukov, G. I. Nikishin, I. V. Alabugin and A. O. Terent'ev, Angew. Chem., Int. Ed., 2017, 56, 4955–4959 CrossRef PubMed.
interaction. Such interactions were shown to significantly stabilize peroxides with geminal acceptor groups:
(a) G. P. Gomes, V. Vil', A. Terent'ev and I. V. Alabugin, Chem. Sci., 2015, 6, 6783–6791 RSC;
(b) V. A. Vil', G. P. Gomes, M. V. Ekimova, K. A. Lyssenko, M. A. Syroeshkin, G. I. Nikishin, I. V. Alabugin and A. O. Terent'ev, J. Org. Chem., 2018, 83, 13427–13445 CrossRef PubMed;
(c) E. Juaristi, G. P. Gomes, A. O. Terent'ev, R. Notario and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 10799–10813 CrossRef CAS PubMed;
(d) G. P. Gomes, I. A. Yaremenko, P. S. Radulov, R. A. Novikov, V. V. Chernyshev, A. A. Korlyukov, G. I. Nikishin, I. V. Alabugin and A. O. Terent'ev, Angew. Chem., Int. Ed., 2017, 56, 4955–4959 CrossRef PubMed. - D. Swern, A. H. Clements and T. M. Lung, Anal. Chem., 1969, 41, 412–416 CrossRef CAS.
- (a) D. M. Schultz, F. Levesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS PubMed; (b) A. S.-K. Tsang, A. Kapat and F. Schoenebeck, J. Am. Chem. Soc., 2016, 138, 518–526 CrossRef CAS PubMed; (c) K.-J. Liu, Z.-H. Duan, X.-L. Zeng, M. Sun, Z. Tang, S. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 10293–10298 CrossRef CAS; (d) M. Lesieur, C. Genicot and P. Pasau, Org. Lett., 2018, 20, 1987–1990 CrossRef CAS PubMed.
- (a) M. Griesser, R. Shah, A. T. Van Kessel, O. Zilka, E. A. Haidasz and D. A. Pratt, J. Am. Chem. Soc., 2018, 140, 3798–3808 CrossRef CAS PubMed; (b) A. Samuni, C. M. Krishna, P. Riesz, E. Finkelstein and A. Russo, J. Biol. Chem., 1988, 263, 17921–17924 CAS; (c) J. B. Mitchell, A. Samuni, M. C. Krishna, W. G. DeGraff, M. S. Ahn, U. Samuni and A. Russo, Biochemistry, 1990, 29, 2802–2807 CrossRef CAS PubMed; (d) M. C. Krishna, D. A. Grahame, A. Samuni, J. B. Mitchell and A. Russo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5537–5541 CrossRef CAS PubMed; (e) M. C. Krishna, A. Russo, J. B. Mitchell, S. Goldstein, H. Dafni and A. Samuni, J. Biol. Chem., 1996, 271, 26026–26031 CrossRef CAS PubMed.
- (a) J. M. Bobbit, C. Brückner and N. Merbouh, Org. React., 2009, 74, 103 Search PubMed; (b) H. Richter and O. G. Mancheño, Eur. J. Org. Chem., 2010, 2010, 4460–4467 Search PubMed; (c) A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044–14047 CrossRef CAS PubMed; (d) K. T. Tarantino, D. C. Miller, T. A. Callon and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 6440–6443 CrossRef CAS PubMed.
- Z. Zhang, Y. Gao, Y. Liu, J. Li, H. Xie, H. Li and W. Wang, Org. Lett., 2015, 17, 5492–5495 CrossRef CAS PubMed.
- (a) M. F. Semmelhack, C. R. Schmid and D. A. Cortes, Tetrahedron Lett., 1986, 27, 1119–1122 CrossRef CAS; (b) T. A. Hamlin, C. B. Kelly, J. M. Ovian, R. J. Wiles, L. J. Tilley and N. E. Leadbeater, J. Org. Chem., 2015, 80, 8150–8167 CrossRef CAS PubMed; (c) H. Richter and O. G. Mancheño, Eur. J. Org Chem., 2010, 4460–4467 CAS; (d) J. Dong, Q. Xia, C. Yan, H. Song, Y. Liu and Q. Wang, J. Org. Chem., 2018, 83, 4516–4524 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, and computational details for all calculated structures. CCDC 1973775. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06511c |
| ‡ Current address: Department of Chemistry and Department of Computer Science, University of Toronto, Toronto, Ontario, Canada. |
| This journal is © The Royal Society of Chemistry 2020 |