
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of unsymmetrically substituted triarylamines via acceptorless dehydrogenative aromatization using a Pd/C and p-toluenesulfonic acid hybrid relay catalyst†
Satoshi
Takayama
a,
Takafumi
Yatabe
 a,
Yu
Koizumi
a,
Xiongjie
Jin
b,
Kyoko
Nozaki
b,
Noritaka
Mizuno
a and
Kazuya
Yamaguchi
*a
a,
Yu
Koizumi
a,
Xiongjie
Jin
b,
Kyoko
Nozaki
b,
Noritaka
Mizuno
a and
Kazuya
Yamaguchi
*a
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kyama@appchem.t.u-tokyo.ac.jp; Fax: +81-3-5841-7220
bDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 25th March 2020
Abstract
An efficient and convenient procedure for synthesizing triarylamines based on a dehydrogenative aromatization strategy has been developed. A hybrid relay catalyst comprising carbon-supported Pd (Pd/C) and p-toluenesulfonic acid (TsOH) was found to be effective for synthesizing a variety of triarylamines bearing different aryl groups starting from arylamines (diarylamines or anilines), using cyclohexanones as the arylation sources under acceptorless conditions with the release of gaseous H2. The proposed reaction comprises the following relay steps: condensation of arylamines and cyclohexanones to produce imines or enamines, dehydrogenative aromatization of the imines or enamines over Pd nanoparticles (NPs), and elimination of H2 from the Pd NPs. In this study, an interesting finding was obtained indicating that TsOH may promote the dehydrogenation.
Introduction
Triarylamines are an important class of chemicals that are widely used for synthesizing valuable materials including polymers, pharmaceuticals, dyes, and natural products.1 In addition, they have been utilized as hole transport materials for organic electroluminescent devices and dye-sensitized solar cells.2 In general, triarylamines that are effective as hole transport materials often possess three different aryl groups.2 Such triarylamines have classically been synthesized by a two-step process in which diarylamines are synthesized first and then coupled with iodobenzenes by the Ullmann reaction.3 In recent years, triarylamine synthesis by the Buchwald–Hartwig coupling in the presence of homogeneous Pd catalysts with intricately designed ligands has become mainstream.4 Although various triarylamines can be produced by these synthetic methods, the use of haloarenes and the formation of metal halide byproducts are inevitable. Therefore, it is very important to develop new greener methods that can be used to synthesize triarylamines from various starting materials.Here, we describe a new strategy for synthesizing unsymmetrically substituted triarylamines based on dehydrogenative aromatization. Dehydrogenative aromatization has recently emerged as an attractive method for synthesizing various arenes from ubiquitous saturated six-membered carbocyclic compounds.5 Over the past few years, various kinds of synthetically useful catalytic reactions employing dehydrogenative aromatization have been developed, which can be used to effectively access phenols,6 aryl ethers,7N-substituted anilines,8 biaryls,9 and heterocycles10 (Fig. 1a). Recently, Li and co-workers have developed a dearomatization–rearomatization strategy in which phenol substrates are once dearomatized followed by reacting with nucleophiles, and then rearomatized to produce various arene products.11 This dearomatization–rearomatization strategy has a great potential to be able to utilize biomass-derived phenols for organic synthesis, and is expected to expand in the future.
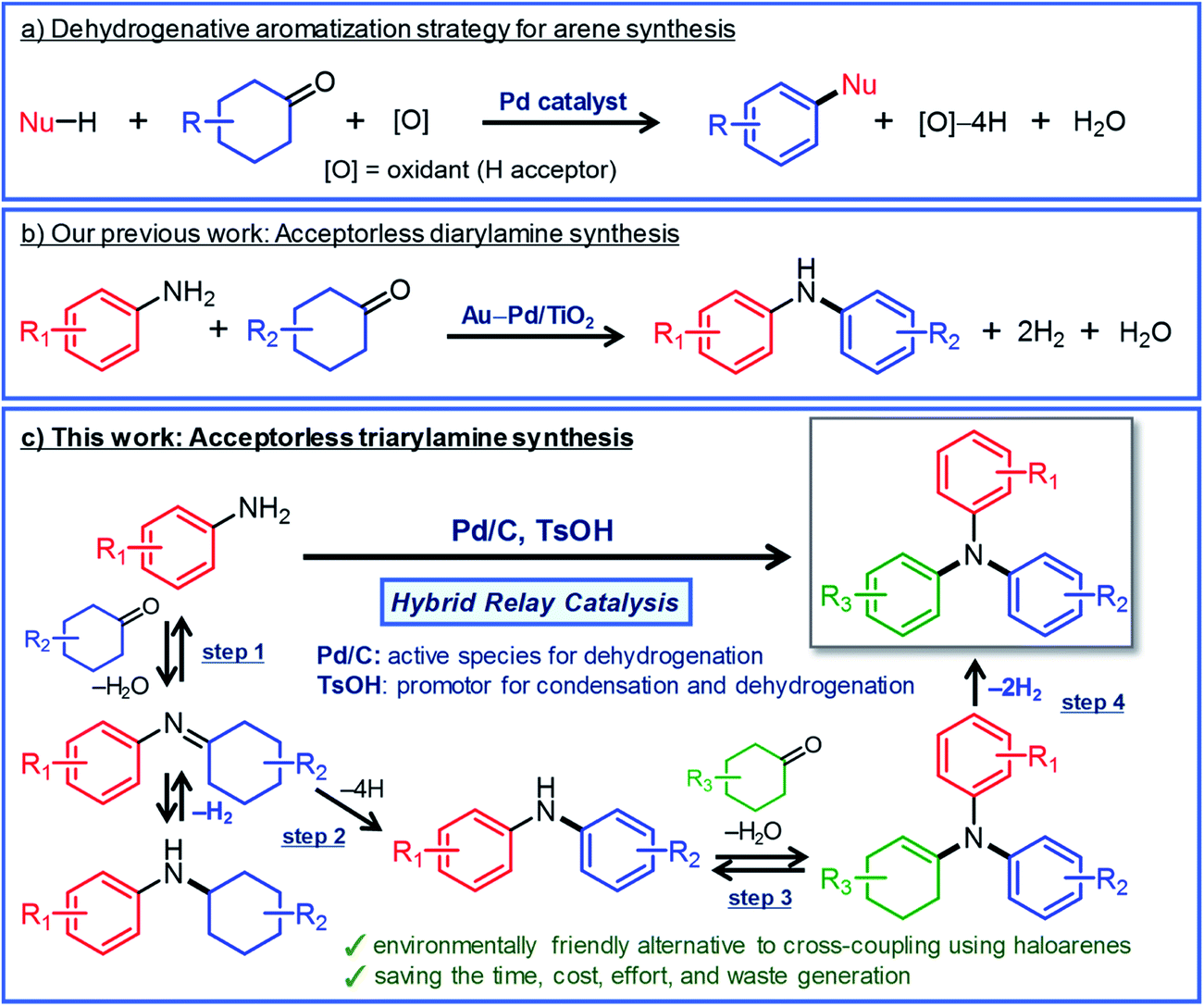 | ||
| Fig. 1 (a) Dehydrogenative aromatization strategy for synthesizing various arenes from six-membered carbocyclic compounds, (b) our recent work on the synthesis of diarylamines from anilines and cyclohexanones via acceptorless dehydrogenative aromatization,12a and (c) the synthesis of unsymmetrically substituted triarylamines via acceptorless dehydrogenative aromatization utilizing hybrid relay catalysis. | ||
For dehydrogenative aromatization reactions, a stoichiometric amount of a hydrogen acceptor is often required. In contrast, acceptorless dehydrogenative aromatization, which generates gaseous H2 as a co-product, represents a more environmentally friendly and economical method for synthesizing arenes. Thus far, we have developed several acceptorless dehydrogenative aromatization reactions using supported Pd-based nanoparticle (NP) catalysts;12 for example, in the presence of supported Au–Pd alloy NP catalysts (for example, Au–Pd/TiO2), unsymmetrically substituted diarylamines with a variety of substituent patterns were synthesized starting from anilines and cyclohexanones (Fig. 1b).12a This method was expanded to other combinations of substrates such as cyclohexylamines/cyclohexanones and nitrobenzenes/cyclohexanols.12a However, in the Au–Pd alloy NP-catalyzed system, diarylamines were obtained as the major products, and triarylamines were only produced in very low yield (<1%) (Fig. 1b).12a
If efficient acceptorless arylation of diarylamines using cyclohexanones can be achieved, then environmentally friendly one-pot sequential synthesis of triarylamines with various substitution patterns and co-production of H2 should be possible by using a variety of substrate combinations. However, to date, an efficient acceptorless dehydrogenative aromatization system to produce triarylamines from diarylamines and cyclohexanones has not been realized.13,14 There are several critical issues facing selective triarylamine production. Because of the low nucleophilicity of diarylamines, condensation with cyclohexanones (step 3 in Fig. 1) is very inefficient. Furthermore, acceptorless dehydrogenation of the corresponding enamines (step 4 in Fig. 1) is unknown. In fact, when the reaction between 3-methyldiphenylamine (1a) and 4-ethylcyclohexanone (2a) was carried out using a commercially available carbon-supported Pd (Pd/C) catalyst, the yield of the desired unsymmetrically substituted triarylamine 3a was quite low (see Table 1, entry 1, and related discussion). To realize the effective triarylamine production, acceleration of the condensation and/or the successive dehydrogenation is very important. We hypothesized that suitable acid co-catalysts promote the reaction. To examine the feasibility of our hypothesis, we investigated the effect of acid co-catalysts thoroughly. As a result, we found that various triarylamines could be synthesized from an equimolar mixture of diarylamines and cyclohexanones using a hybrid relay catalyst comprising Pd/C and p-toluenesulfonic acid (TsOH) (Fig. 1c). Furthermore, one-pot sequential syntheses of unsymmetrically substituted triarylamines from anilines and two different cyclohexanones were also successful (Fig. 1c). In the proposed one-pot triarylamine synthesis, the absence of isolation/purification of the diarylamine intermediates reduces time, cost, effort, and waste generation. Therefore, the present system has the potential to be an environmentally friendly alternative to cross-coupling reactions using haloarenes. We revealed that triarylamines are produced via direct dehydrogenation of enamine intermediates, whereas diarylamines from anilines and cyclohexanones are formed via disproportionation of imine intermediates. It is worth noting that an interesting finding was obtained indicating that TsOH may promote the dehydrogenation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Co-catalysts | Yield (%) | ||
| 3a | 4a | 5a | |||
| a Reaction conditions: catalyst (metal: 2 mol%), additive (10 mol%), 1a (0.5 mmol), 2a (0.5 mmol), mesitylene (2 mL), Ar (1 atm), temperature 160 °C, and time 2 h. Yields were determined by GC and are relative to 1a. | |||||
| 1 | Pd/C | w/o | 6 | <1 | 4 |
| 2 | Pd/C | TsOH | 85 | <1 | <1 |
| 3 | Pd/C | PhCOOH | 7 | <1 | 5 |
| 4 | Pd/C | CF3COOH | 13 | <1 | 4 |
| 5 | Pd/C | CH3SO3H | 20 | <1 | <1 |
| 6 | Pd/C | Ti(OiPr)4 | 8 | <1 | 3 |
| 7 | Pd/C | Al(OiPr)3 | 5 | <1 | 6 |
| 8 | Pd/C | ZnCl2 | <1 | <1 | <1 |
| 9 | Pd/C | B(C6F5)3 | 6 | <1 | <1 |
| 10 | Pd/C | Sc(O3SCF3)3 | 1 | <1 | 1 |
| 11 | Pd/C | Na2CO3 | 2 | <1 | 7 |
| 12 | Pd/C | K2CO3 | 1 | <1 | 69 |
| 13 | Pd/TiO2 | TsOH | 54 | <1 | <1 |
| 14 | Pd/Al2O3 | TsOH | 4 | <1 | <1 |
| 15 | Pd/CeO2 | TsOH | 11 | <1 | 1 |
| 16 | Pd/LDH | TsOH | <1 | <1 | 9 |
| 17 | Ru/C | TsOH | 3 | <1 | <1 |
| 18 | Rh/C | TsOH | 54 | 13 | <1 |
| 19 | Pt/C | TsOH | 39 | 31 | <1 |
Results and discussion
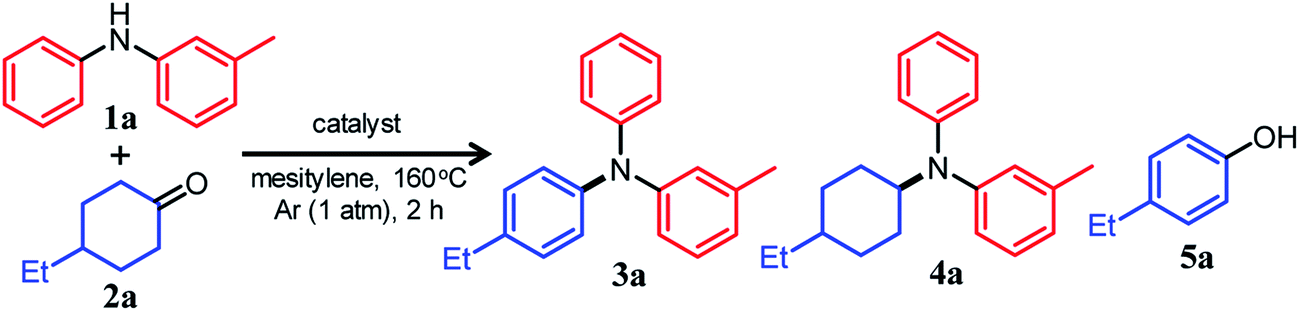
Reaction between 3-methyldiphenylamine and 4-ethylcyclohexanone
We conducted the reaction between 3-methyldiphenylamine (1a) and 4-ethylcyclohexanone (2a) under various conditions. Initially, the reaction between 1a and 2a in mesitylene at 160 °C was carried out in the presence of Pd/C without any co-catalysts. However, the yield of and selectivity to the desired unsymmetrically substituted triarylamine 3a were unsatisfactory (Table 1, entry 1). We concluded that the condensation between 1a and 2a did not proceed well, and in order to solve the problem, we performed the reaction in the presence of various acid or base co-catalysts. As a result, TsOH was identified as a suitable acid co-catalyst. In the presence of Pd/C (2 mol%) and TsOH (10 mol%), the reaction gave the desired triarylamine 3a in 85% yield after 2 h, and side products such as the reductive amination product 4a and 4-ethylphenol (5a) were hardly detected (Table 1, entry 2). It should be noted that the reaction proceeded efficiently with an equimolar mixture of 1a and 2a. It was confirmed by volumetric measurement and mass spectrometry (MS) analysis of the evolved gas that approximately two equivalents of H2 gas with respect to 3a were produced during the reaction. Other Brønsted acid co-catalysts, such as benzoic acid (PhCOOH), trifluoroacetic acid (CF3COOH), and methanesulfonic acid (CH3SO3H), were less effective (Table 1, entries 3–5). The relatively high solubility of TsOH in mesitylene compared with the other Brønsted acid co-catalysts tested is considered to be one reason for the effectiveness of TsOH. For the reaction between 1a and 2a, Lewis acid co-catalysts were ineffective (Table 1, entries 6–10). Conversely, the direct dehydrogenation of 2a was conspicuously promoted in the presence of base co-catalysts (especially K2CO3), resulting in the undesirable formation of phenol 5a as the major product (Table 1, entries 11 and 12).15The choice of support was very significant, and the effect of TsOH was most noticeable when using Pd/C (Table 1, entry 2). We prepared several supported Pd catalysts using various oxide or hydroxide supports (Pd/support; support = TiO2, Al2O3, CeO2, or Mg6Al2(OH)16CO3·4H2O (LDH)) and performed the reaction between 1a and 2a using these catalysts. Although the Pd content and average particle sizes of the Pd NPs in the Pd/support catalysts were almost the same as those in Pd/C (Fig. S1, Table S1, ESI†), the catalytic performance of all other Pd/support catalysts was greatly inferior to that of Pd/C (Table 1, entries 13–16). Thus, it is presumed that the promotion effect of TsOH cannot be fully exhibited when using Pd/support because of its adsorption on the basic sites of the oxide or hydroxide surface. In particular, when a strong basic support (LDH) was used, the desired 3a was not produced at all, and the corresponding phenol 5a was obtained as the major product (Table 1, entry 16).15 Therefore, the use of basic supports is inappropriate for this reaction. Among the solvents examined, mesitylene yielded the best results (Table S2, entry 1, ESI†), and decane and diethyleneglycol dimethyl ether (diglyme) also afforded 3a in good yields (Table S2, entries 2 and 3, ESI†). On the other hand, highly polar solvents such as N,N-dimethylacetamide and N-methylpyrrolidone were not effective (Table S2, entries 4 and 5, ESI†). When the reaction temperature was examined in the range 130–160 °C, it was found that the reaction progressed more efficiently as the temperature increased (Table S3, ESI†).
We also examined the reaction between 1a and 2a using supported metal catalysts other than Pd. In the presence of Ru/C, the reaction hardly proceeded (Table 1, entry 17). Conducting the reaction in the presence of Rh/C or Pt/C afforded 3a in moderate yields along with significant amounts of the reductive amination product 4a (Table 1, entries 18 and 19). Therefore, Pd/C was determined to be the best catalyst for this reaction. Hereafter, we mainly used the most effective Pd/C and TsOH hybrid relay catalyst under the aforementioned optimized conditions for further detailed investigations.
To establish whether the observed catalysis of the synthesis of 3a from 1a and 2a occurred heterogeneously on Pd/C or was a result of the presence of leached Pd species in the reaction solution, the Pd/C catalyst was removed by hot filtration 1 h after the reaction was started, and the reaction was restarted with the filtrate under the same conditions. As shown in Fig. S2, ESI,† the production of 3a immediately ceased upon the removal of Pd/C. Additionally, after the reaction, the filtrate was analyzed using inductively coupled plasma atomic emission spectroscopy (ICP-AES), and it was found that Pd species were barely detectable (less than 0.09% of Pd used for the reaction). Consequently, it was concluded that the observed catalysis of the reaction was truly heterogeneous.16 Furthermore, the Pd/C retrieved after the reaction could be reused for the reaction between 1a and 2a, though the catalytic performance slightly declined; a yield of 72% for 3a was obtained with a reused Pd/C catalyst (Fig. S3, ESI†).
Triarylamine synthesis from diarylamines and cyclohexanones
Under the optimized reaction conditions described above using Pd/C and TsOH, we next examined the scope of suitable substrates for the proposed triarylamine synthesis from diarylamines and cyclohexanones. The substrates used in this study are shown in Fig. 2. As summarized in Fig. 3, various kinds of substrate combinations were converted into the corresponding unsymmetrically substituted triarylamines in moderate-to-high yields. These reactions (except for the synthesis of 3c and 3g) efficiently proceeded using equimolar mixtures of diarylamines and cyclohexanones. The desired triarylamine products were readily isolated by simple column chromatography on silica gel.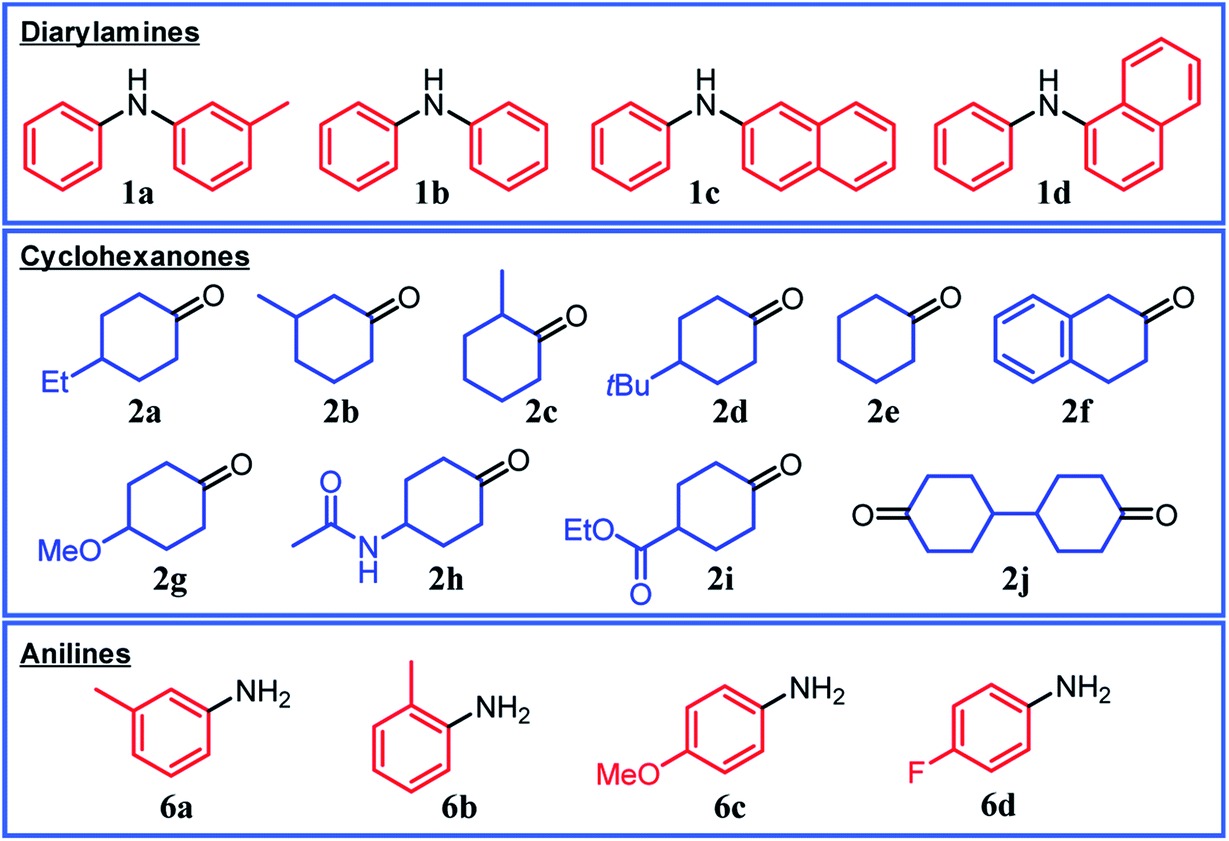 | ||
| Fig. 2 Substrates used in this study. | ||
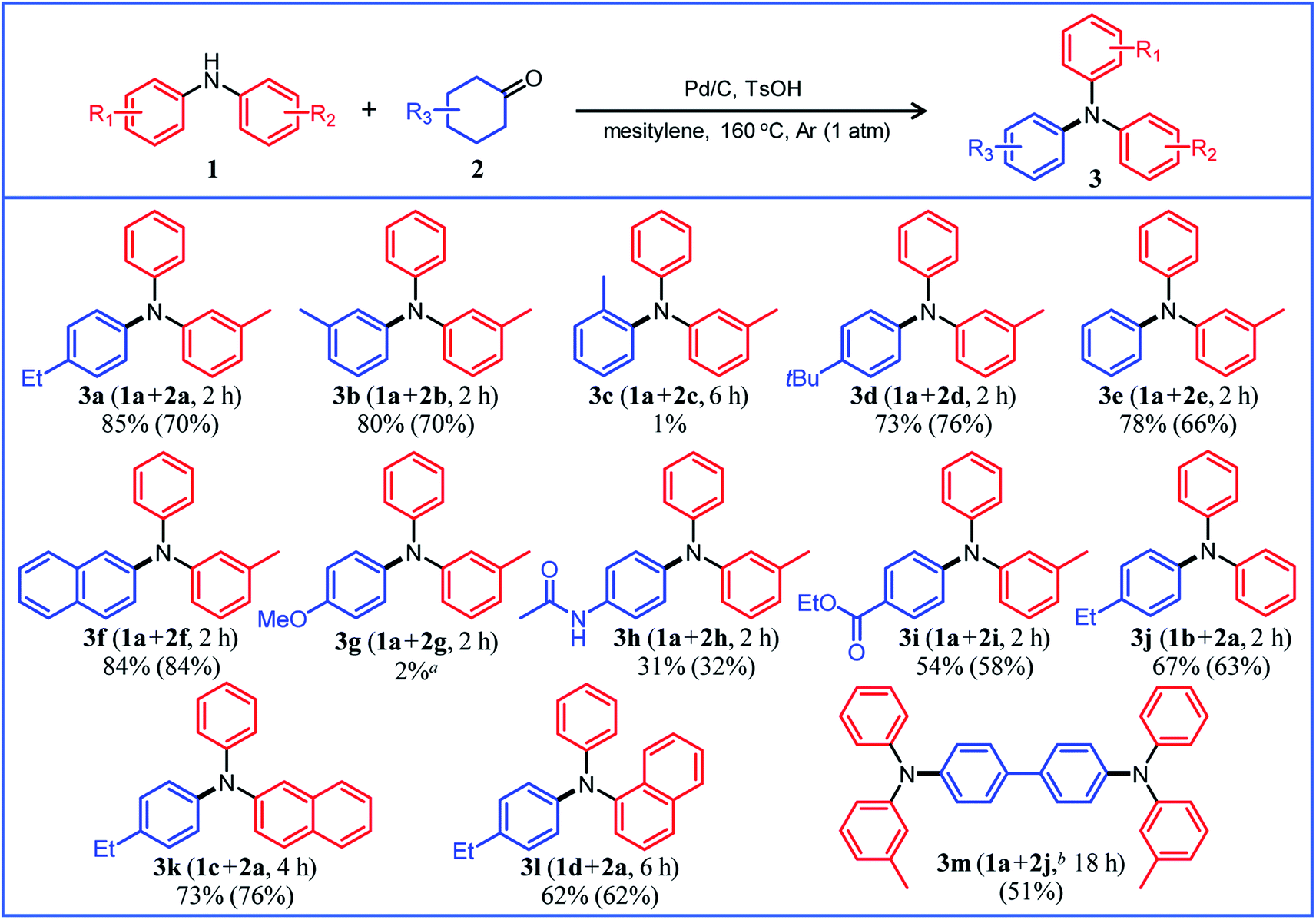 | ||
| Fig. 3 Substrate scope of the proposed triarylamine synthesis from diarylamines and cyclohexanones using a Pd/C and TsOH hybrid relay catalyst. Reaction conditions: Pd/C (2 mol%), TsOH (10 mol%), 1 (0.5 mmol), 2 (0.5 mmol), mesitylene (2 mL), Ar (1 atm), and temperature 160 °C. Yields are relative to 1. GC yields and isolated yields (values in parentheses) are shown. In all cases, phenol side products 5 were hardly detected. aElimination of the methoxy group occurred during the reaction, giving 3e as the major product (35%). b1a (1.0 mmol), 2j (0.5 mmol). | ||
Cyclohexanone (2e) and its derivatives with alkyl groups at the 3- or 4-position (2a, 2b, and 2d) smoothly reacted with diarylamine 1a, affording the corresponding triarylamine derivatives in high yields (3a, 3b, 3d, and 3e). Unfortunately, the use of 2-methylcyclohexanone (2c) as the arylation source was difficult; for the reaction between 1a and 2c, the desired triarylamine 3c was hardly produced. We speculated that if there is a substituent at the 2-position, the steric hindrance of the cyclohexene ring to be dehydrogenated will increase, thus impeding the access of the enamine intermediate to the Pd NP surface. However, if a substituent is introduced at the 2-position of aniline in advance, it is possible to synthesize triarylamines bearing a substituent at the 2-position such as 3c, as discussed in more detail later.
By using tetrahydronaphthalenone (2f), it was possible to introduce a naphthalene skeleton into the diarylamine (3f). When using 4-methoxycyclohexanone (2g) as the arylation source, an undesirable elimination of the methoxy group occurred during the aromatization step, giving 3e as the major product. In order to synthesize triarylamines bearing a methoxy group such as 3g, it is indispensable to introduce the methoxy group into the aniline in advance, as discussed in more detail later. Cyclohexanones bearing an amide (2h) or an ester group (2i) were tested with the proposed system, affording the corresponding triarylamines in moderate yields (3h and 3i). Diphenylamine (1b) and naphthylphenylamines (1c and 1d) also worked well as reaction partners of 2a, affording the corresponding triarylamines in good yields (3j, 3k, and 3l). It is noteworthy that TPD (3m), which is a highly valuable hole transport material for organic electroluminescent devices,2 was readily synthesized from 1a and bis(4-cyclohexanone) 2j (1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2j = 2:1) utilizing the proposed method.
:2j = 2:1) utilizing the proposed method.
One-pot triarylamine synthesis from anilines and two different cyclohexanones
In this section, we describe our attempts to develop a one-pot triarylamine synthesis from anilines and two different cyclohexanones. As discussed in the previous section, it was found that various triarylamines can be synthesized from diarylamines and cyclohexanones using a Pd/C and TsOH hybrid relay catalyst. Therefore, we first investigated whether the hybrid relay catalyst could be applied to selective diarylamine synthesis (1a) from 3-methylaniline (6a) and cyclohexanone (2e) under the optimized conditions for triarylamine synthesis from diarylamines and cyclohexanones. As shown in Fig. 4, in the presence of Pd/C and TsOH, an equimolar mixture of 6a and 2e selectively converted to the desired diarylamine 1a in 82% yield without formation of the reductive amination product N-cyclohexylaniline 7a as well as triarylamine 3e. Under these conditions, 2e (arylation source) was not present during the reaction because the consecutive reaction of condensation between 6a and 2e and disproportionation of the corresponding imine (8e) was completed immediately (within a few minutes) after the reaction started, as mentioned later in detail. Therefore, even if the reactions between anilines and cyclohexanones are carried out under the same conditions as used for triarylamine synthesis, diarylamines are selectively obtained without producing triarylamines because there are no arylation sources. This facilitates a very simple one-pot triarylamine synthesis, i.e., after completion of the first reaction between an aniline and a cyclohexanone, it is only necessary to add a cyclohexanone as the second arylation source without isolation of the diarylamine intermediate or changing any reaction conditions, which reduces time, cost, effort, and waste generation.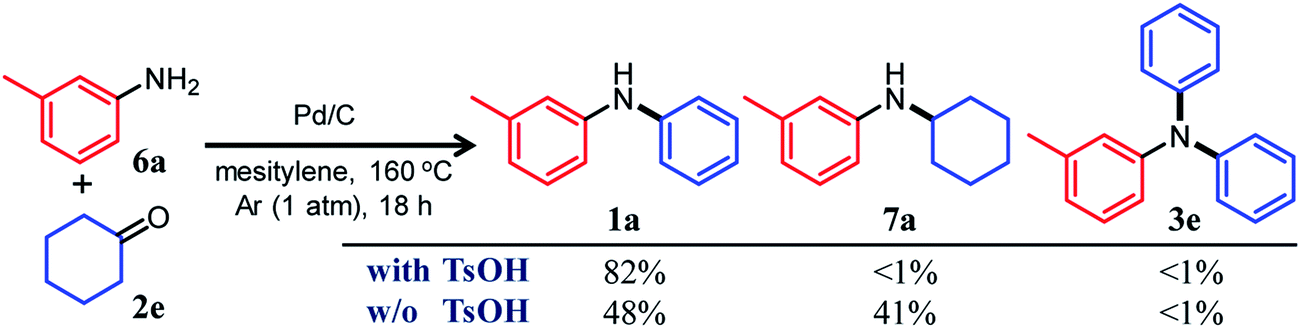 | ||
| Fig. 4 Effect of TsOH on the reaction between 3-methylaniline (6a) and cyclohexanone (2e). Reaction conditions: Pd/C (2 mol%), TsOH (0 or 10 mol%), 6a (0.5 mmol), 2e (0.5 mmol), mesitylene (2 mL), Ar (1 atm), temperature 160 °C, and time 18 h. Yields are relative to 6a and were determined by GC. | ||
The effect of TsOH on the reaction between 6a and 2e was crucial. When using Pd/C and TsOH, the reaction exclusively yielded 1a after 18 h. On the other hand, in the absence of TsOH, the desired diarylamine 1a was only obtained in 48% yield together with the formation of 7a in 41% yield under the same conditions (Fig. 4). Therefore, in the reaction between 6a and 2e, TsOH likely promotes the condensation to some extent but is considered to be more important in promoting the Pd-catalyzed dehydrogenative aromatization of 7a to 1a, as discussed later.
Several results of the proposed one-pot triarylamine synthesis are summarized in Fig. 5. We successfully synthesized triarylamines with alkyl substituents at the 3- or 4-position of the phenyl rings in good yields (3a, 3b, and 3d). These triarylamines could be synthesized even if the order in which the aryl groups are introduced is changed; for example, 3a was obtained in 65% yield using 2a as the first arylation source (18 h) and 2e as the second arylation source (4 h) under the conditions in Fig. 5. Regarding the synthesis of triarylamines bearing substituents at the 2-position of the phenyl rings, which was impossible in the previous section, for example, by starting from 2-methylaniline (6b) and sequentially introducing aryl groups into it, the desired triarylamine 3c was synthesized in high yield using the developed one-pot synthesis. The new one-pot synthesis was also effective at introducing substituents that tend to be eliminated into the aniline substrate in advance; for example, triarylamine 3g, which could not be successfully synthesized using 4-methoxycyclohexanone (2g) as the arylation source, was obtained in high yield by utilizing 4-methoxyaniline (6c) as the starting material and 2e and 2b as the arylation sources. A triarylamine bearing a fluoro group (3n) was also synthesized.
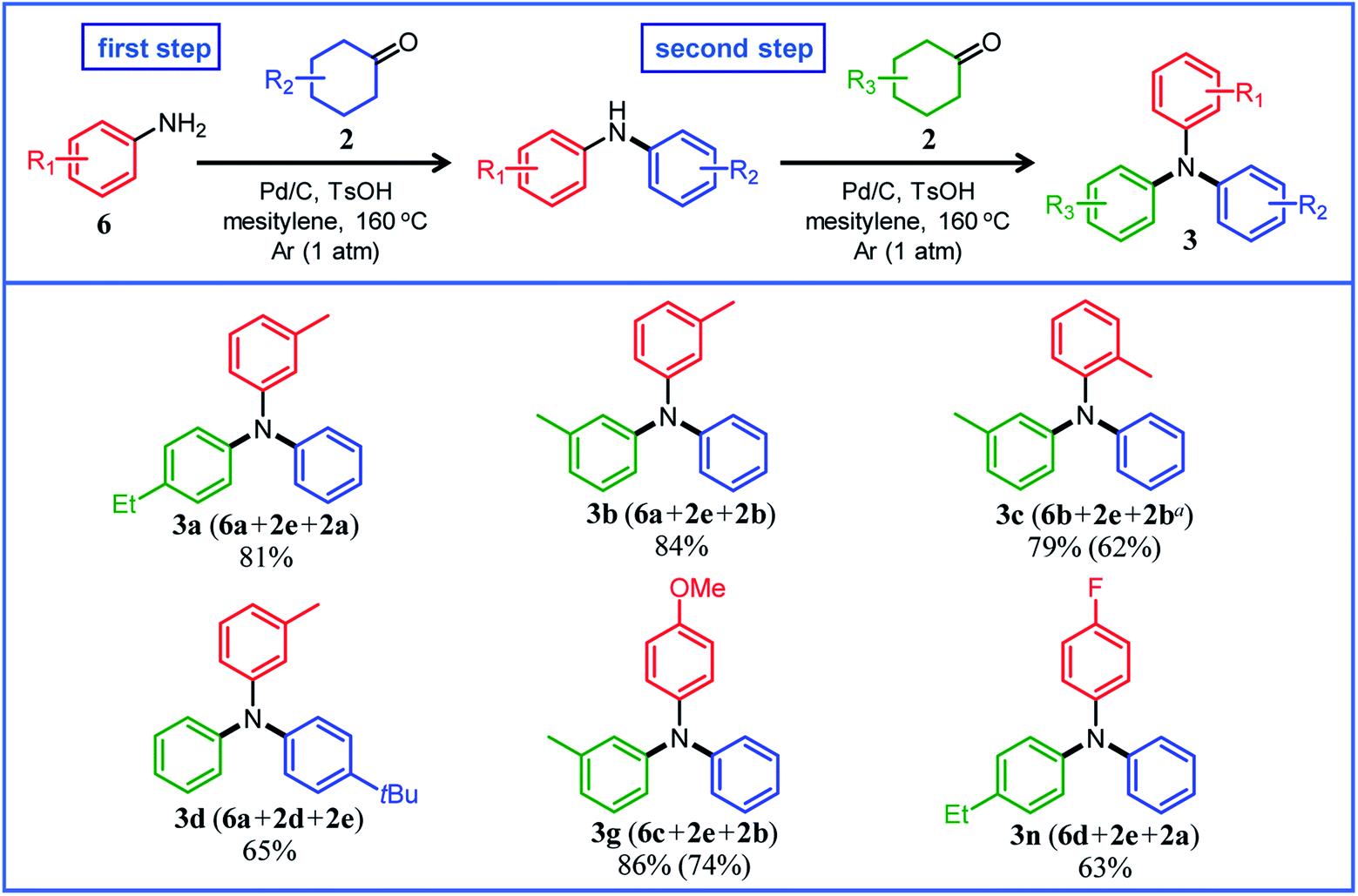 | ||
| Fig. 5 Substrate scope of the proposed one-pot triarylamine synthesis from anilines and two different cyclohexanones using a Pd/C and TsOH hybrid relay catalyst. Reaction conditions: (first step) Pd/C (2 mol%), TsOH (10 mol%), 6 (0.5 mmol), 2 (0.5 mmol), mesitylene (2 mL), Ar (1 atm), temperature 160 °C, and time 18 h. (second step) After the first step, 2 (0.55 mmol) was added to the reaction mixture, and the reaction was continued for further 2 h under the same conditions. Yields are relative to 6. GC yields and isolated yields (values in parentheses) are shown. In all cases, phenol side products 5 were hardly detected. a8 h for the second step. | ||
Reaction pathways
In this section, the reaction pathways of the proposed triarylamine synthesis are discussed in detail. To begin with, the first step involving the reaction between anilines and cyclohexanones (steps 1 and 2 in Fig. 1c) was examined. As can be seen from the time course of the reaction between 6a and 2e in the presence of Pd/C and TsOH (Fig. 6a), these substrates were completely consumed immediately after the start of the reaction and converted to diarylamine 1a and N-cyclohexylaniline 7a. After that, it became clear that 7a gradually converted to 1a. The corresponding imine (8a) that forms via the condensation of 6a and 2e was barely detected throughout the reaction. In a separate experiment, we confirmed that N-cyclohexylaniline (7b) was smoothly converted to diphenylamine (1b) in high yield under the same conditions in the presence of Pd/C and TsOH (Fig. 7a). Previously, we have reported that N-cyclohexylidenebenzenamine (imine 8b) was smoothly converted to a 1:2 mixture of diphenylamine and N-cyclohexylaniline under similar acceptorless conditions in the presence of Pd-based NP catalysts.12a Considering the present experimental results and those of our previous studies, it can be concluded that diarylamine 1a was produced via the disproportionation of the imine intermediate (formed via the condensation of 6a and 2e) and that the disproportionation of 8a was much faster than the dehydrogenation of 7a (Fig. 6b).
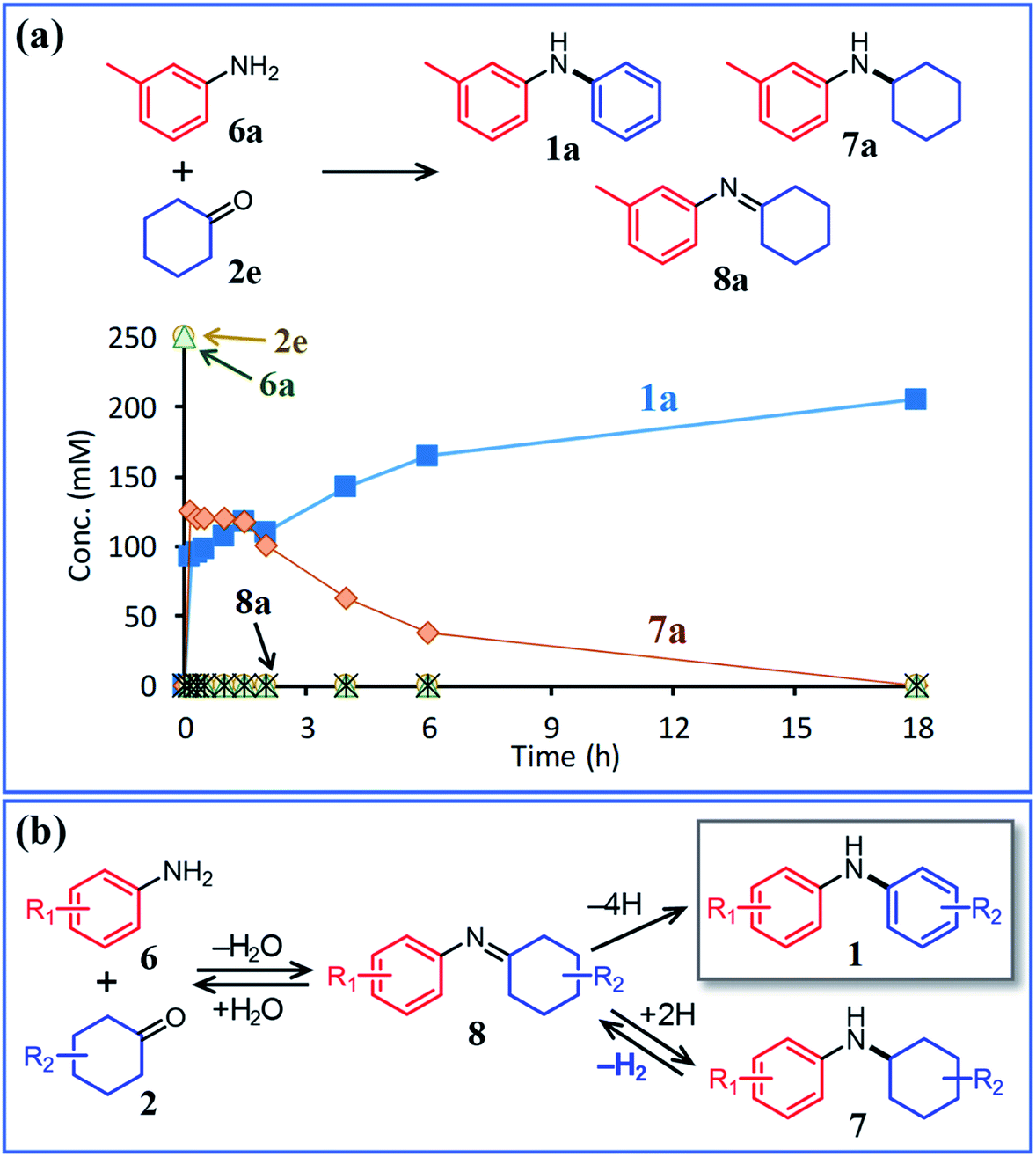 | ||
| Fig. 6 (a) Time course for the reaction between 3-methylaniline (6a) and cyclohexanone (2e) and (b) plausible reaction pathway. Reaction conditions: Pd/C (2 mol%), TsOH (10 mol%), 6a (0.5 mmol), 2e (0.5 mmol), mesitylene (2 mL), Ar (1 atm), and temperature 160 °C. | ||
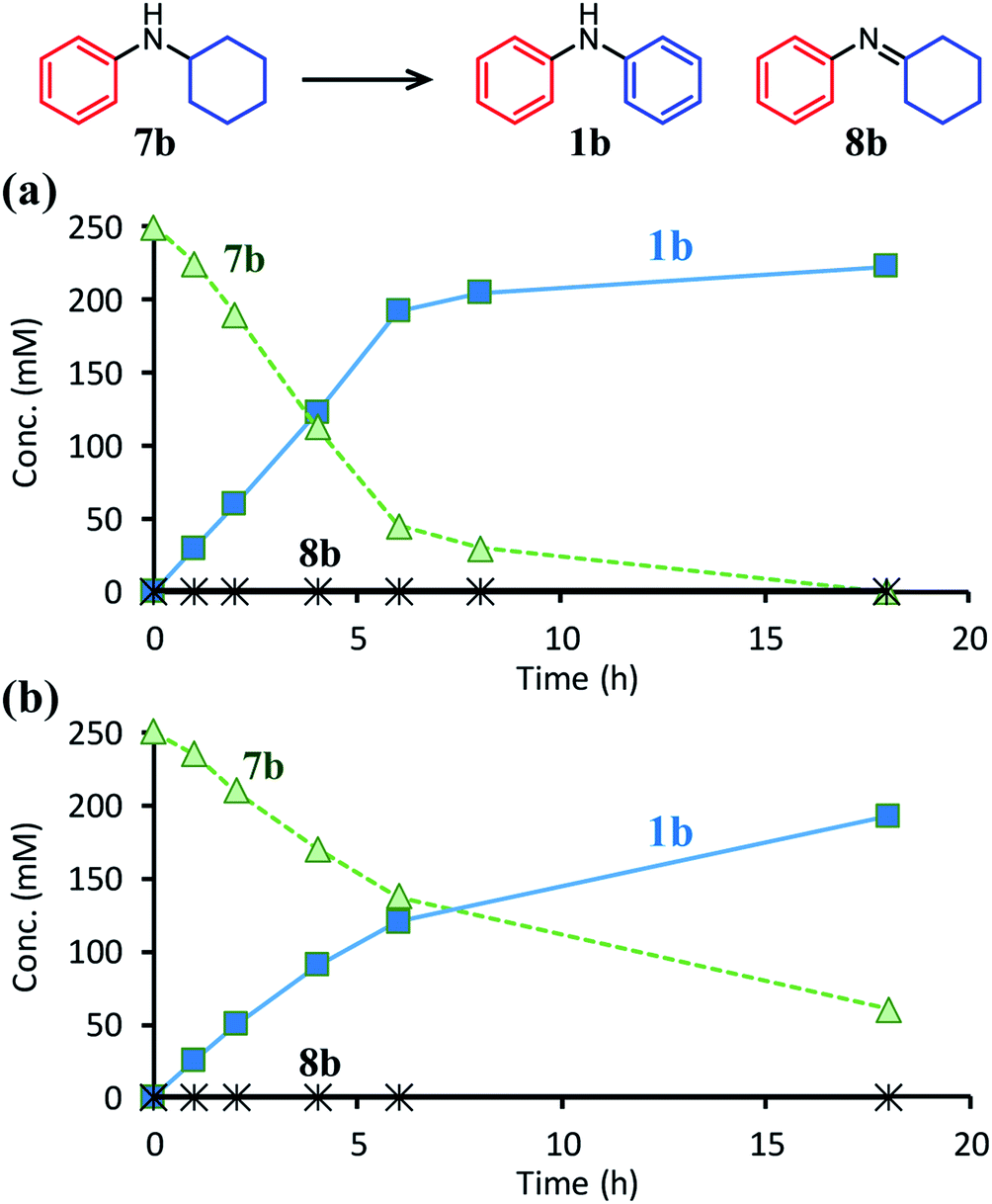 | ||
| Fig. 7 Time courses for the dehydrogenative aromatization of N-cyclohexylaniline (7b) (a) with TsOH or (b) without TsOH. Reaction conditions: Pd/C (Pd: 2 mol%), TsOH (0 or 10 mol%), 7b (0.5 mmol), mesitylene (2 mL), temperature 160 °C, Ar (1 atm). | ||
The positive effect of TsOH on the dehydrogenation of 7b to 1b was confirmed as follows. The reaction profiles for the Pd/C-catalyzed dehydrogenation of 7b clearly revealed that the dehydrogenation rate increased in the presence of TsOH (Fig. 7). Consequently, we conclude that TsOH promotes the rate-limiting amine dehydrogenation. In acceptorless alcohol dehydrogenation using several metal complexes, it has been reported that the activation energies of H2 liberation are relatively large and that the key to lowering them is to efficiently transfer H+ to metal hydride species.17 Therefore, we propose that one possible explanation for the role of TsOH on amine dehydrogenation is promoting H2 liberation via the reaction between TsOH (H+) and the hydride species generated on the Pd surface.
Next, the second step of the synthesis involving the reaction between diarylamines and cyclohexanones (steps 3 and 4 in Fig. 1c) was examined. Fig. 8a shows the time course for the reaction between diarylamine 1a and cyclohexanone 2a. The reductive amination product 4a and the corresponding enamine 9a were not detected during the reaction. In addition, no traces of partially dearomatized forms of 1a were also detected during the reaction.18,19 We separately synthesized N-cyclohexyldiphenylamine (4b) and N-(1-cyclohexenyl)diphenylamine (9b) and performed these reactions under the same conditions. However, the formation of triphenylamine was not observed when using 4b as the substrate. As for the reaction of 9b, although the hydrolysis of 9b initially progressed to form 1b and 2e, triphenylamine (3o) was finally obtained in good yield (Fig. 9). Therefore, enamines are probably the intermediates of triarylamines in this synthesis, but it is considered that triarylamines are formed by direct dehydrogenation rather than disproportionation, unlike the reaction in the first step (Fig. 8b). Since 9a was not observed at any time (Fig. 8a), it is presumed that the direct dehydrogenation from 9a to 3a is very fast. At present, although there is not enough experimental evidence, it seems possible that TsOH accelerates this dehydrogenation.
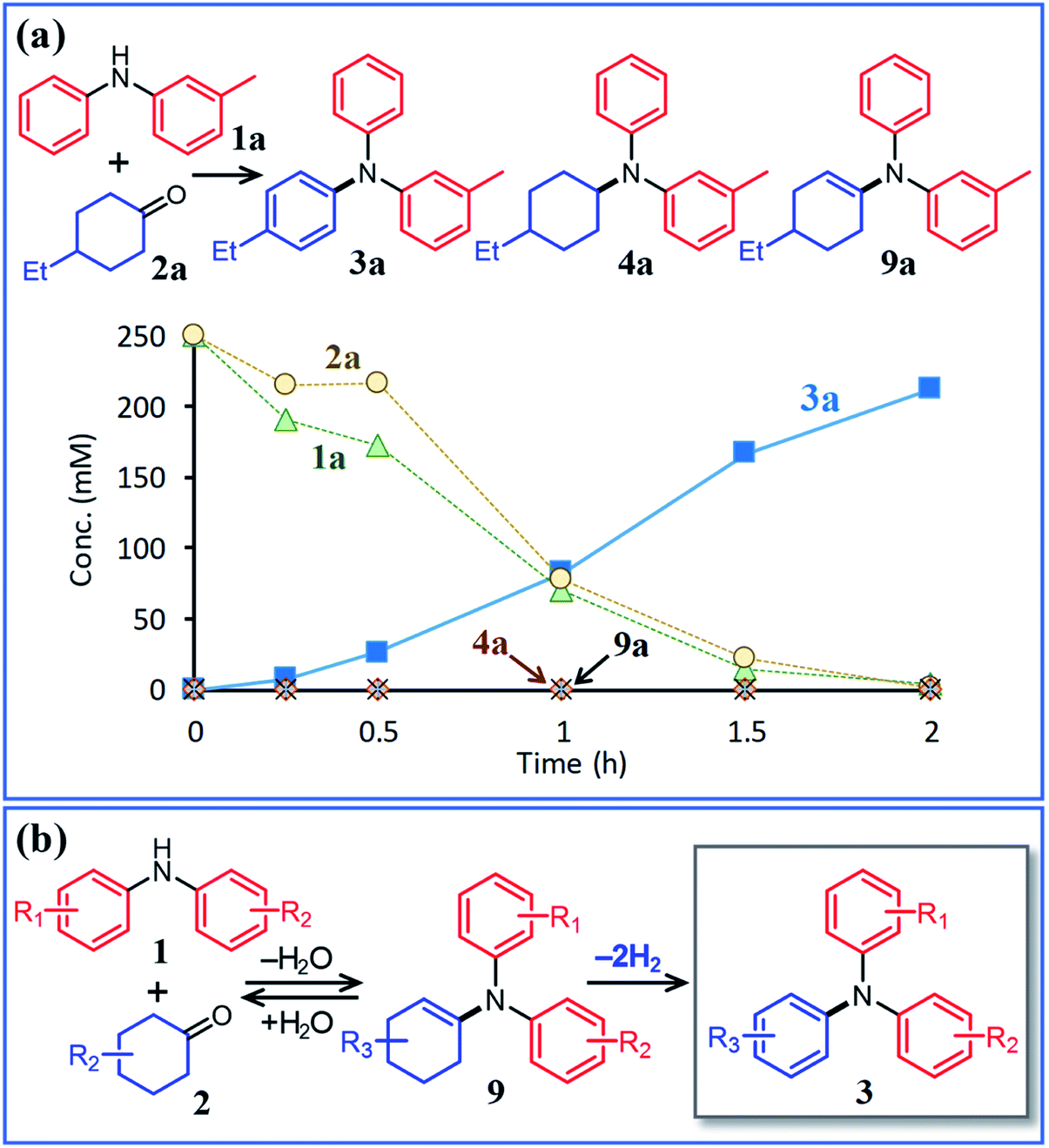 | ||
| Fig. 8 (a) Time course for the reaction between 3-methyldiphenylamine (1a) and 4-ethylcyclohexanone (2a) and (b) plausible reaction pathway. Reaction conditions: Pd/C (2 mol%), TsOH (10 mol%), 1a (0.5 mmol), 2a (0.5 mmol), mesitylene (2 mL), Ar (1 atm), and temperature 160 °C. | ||
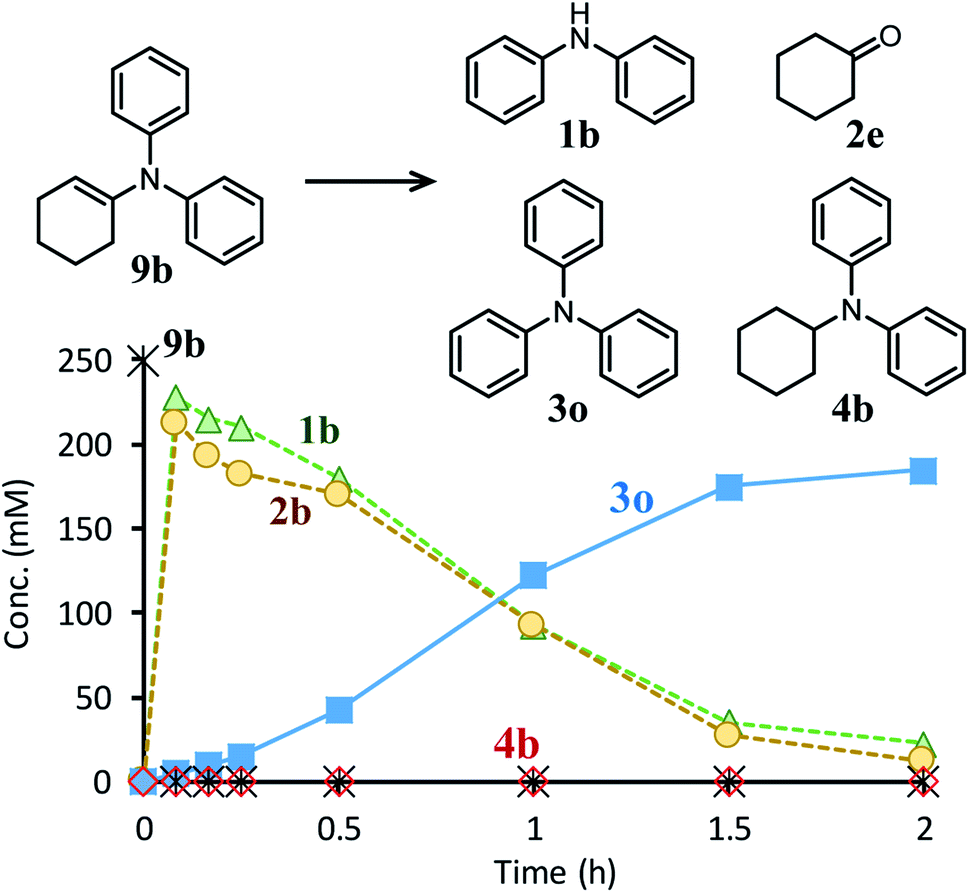 | ||
| Fig. 9 Time course for the reaction of N-(1-cyclohexenyl)diphenylamine (9b). Reaction conditions: Pd/C (2 mol%), TsOH (10 mol%), 9b (0.4 mmol), mesitylene (1.6 mL), temperature 160 °C, Ar (1 atm). | ||
Here arises a question why the tertiary amine N-cyclohexyldiphenylamine (4b) did not undergo dehydrogenation while the secondary amine N-cyclohexylaniline (7b) was dehydrogenated to 1b in Fig. 7. In the first step reaction, since imines (8) function as good hydrogen acceptors, it appears that diarylamines (1) and N-cyclohexylanilines (7) are formed by disproportionation (Fig. 6b). Thereafter, the dehydrogenations of 7 produce 8, and eventually, 1 become the major products (Fig. 6b). In contrast, in the second step reaction, enamines (9) do not act as hydrogen acceptors, so it appears that triarylamines (3) are formed by direct dehydrogenation (Fig. 8b). We consider that this difference arises because of a steric reason; the unsaturated bond in 8 can access the Pd NP surface but that in bulkier 9 is considered difficult. The lack of progress in the dehydrogenation of 4b is also considered to be caused by a similar steric reason. However, it may be also possible that 4b contained two aryl groups that the lone pair in nitrogen is very well delocalized to the aryl groups, causing the Pd cannot efficiently coordinate to the nitrogen to furnish the desired dehydrogenation process. To clarify this possibility, we separately synthesized N,N-dicyclohexylaniline and performed the reaction under the conditions described in Fig. 10 using Pd/C and TsOH. The reaction progressed marginally, affording 4b in 14% yield. In this case, triphenylamine was not detected. Therefore, it may be concluded that the reason for no reaction of 4b is due to not only the steric reason but also the effect of its lone pair delocalization. We also found during this work that when the Pd/C and TsOH hybrid relay catalyst was used, the dehydrogenative aromatization of several tertiary amines, such as N,N-dimethylcyclohexylamine (10a) and N,N-diethylcyclohexylamine (10b), which are less sterically crowded around the N positions or smaller delocalization effect than N-cyclohexyldiphenylamine and N,N-dicyclohexylaniline, proceeded effectively, giving the corresponding anilines (Fig. 10). To the best of our knowledge, this is the first report of dehydrogenative aromatization of tertiary amines.
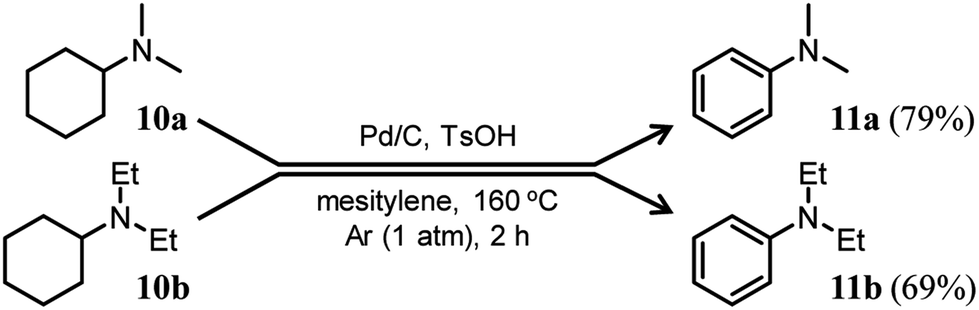 | ||
| Fig. 10 Dehydrogenative aromatization of N,N-dimethylcyclohexylamine (10a) and N,N-diethylcyclohexylamine (10b). Reaction conditions: Pd/C (2 mol%), TsOH (10 mol%), 10 (0.5 mmol), mesitylene (2 mL), Ar (1 atm), temperature 160 °C, and time 2 h. Yields are relative to 10 and determined by GC. | ||
Conclusion
We have successfully developed efficient catalytic procedures for synthesizing triarylamines via acceptorless dehydrogenative aromatization. In the presence of a Pd/C and TsOH hybrid relay catalyst, various types of structurally diverse unsymmetrically substituted triarylamines could be synthesized starting from diarylamines and cyclohexanones. Moreover, a one-pot sequential triarylamine synthesis from anilines and two different cyclohexanones was also successful. Owing to their practical and environmentally benign nature, we hope that the catalytic transformations developed in this study will find wide applications in the synthesis of arylamine derivatives and related compounds. The discovery that this type of dehydrogenation is promoted in the presence of a suitable Brønsted acid co-catalyst is also very interesting and can be expected to apply to a variety of related functional transformations, which may be exploited in future synthetic schemes.Experimental section
Instruments and reagents
Gas chromatography (GC) analyses were performed using a Shimadzu GC-2014 equipped with a flame ionization detector using an InertCap5 capillary column. GC-MS spectra were recorded using a Shimadzu GCMS-QP2010 equipped with an InertCap5 capillary column at an ionization voltage of 70 eV. Liquid-state nuclear magnetic resonance (NMR) spectra were recorded using a JEOL JNM-ECA-500. 1H and 13C NMR spectra were measured at 500.2 and 125.8 MHz, respectively, using tetramethylsilane as an internal standard (δ = 0 ppm). ICP-AES analyses were performed using a Shimadzu ICPS-8100. Transmission electron microscopy observations were performed using a JEOL JEM-2000EX. Column chromatography on silica gel was performed using a Biotage Isolera system. Elemental analyses for C, H, and N were performed using an Elementar vario MICRO cube. Pd/C (lot. no. 217-024030, 217-172450, N.E. CHEMCAT), Ru/C (lot. no. 417-020160, N.E. CHEMCAT), Rh/C (lot. no. MCM7367, FUJIFILM Wako Pure Chemical), and Pt/C (lot. no. 117-061210, N.E. CHEMCAT) were commercially available. Al2O3 (KHS-24, Sumitomo Chemical), TiO2 (ST-01, Ishihara Sangyo Kaisya), CeO2 (544841-25G, Aldrich), ZrO2 (37022, Nacalai Tesque), and Mg6Al2(OH)16CO3·4H2O (LDH, Tomita Pharmaceutical Co., Ltd.) were commercially available. Pd/support catalysts were prepared according to a literature procedure.20 Substrates (except for 4b and 9b) and solvents were obtained from TCI, Aldrich, Kanto Chemical, FUJIFILM Wako Pure Chemical (reagent grade), or Combi-blocks, and purified prior to use, if necessary.21 Compounds 4b and 9b were synthesized according to a literature procedure.22N,N-Dicyclohexylaniline was synthesized according to a literature procedure.23Catalytic reactions
Catalytic reactions were typically carried out according to the following procedure. Pd/C (2 mol%), TsOH (10 mol%), diarylamine (1, 0.5 mmol), cyclohexanone (2, 0.5 mmol), n-hexadecane (0.1 mmol, internal standard), mesitylene (2 mL), and a Teflon-coated magnetic stir bar were successively placed into a ∼20 mL Schlenk flask reactor, and then the mixture was stirred at 160 °C under Ar (1 atm). Substrate conversions and product yields were periodically monitored by GC analysis. For the isolation of the products, the internal standard was not used. After the reaction, the catalyst was removed by simple filtration and the filtrate was concentrated by evaporation of the mesitylene solvent. The crude product was subjected to column chromatography on silica gel (typically using hexane and toluene as an eluent), yielding the pure triarylamine product. The products were identified by GC-MS, NMR (1H and 13C), and/or elemental analyses. Detection of H2 in the gas phase was carried out by GC-MS analysis. Quantification of the H2 formed was achieved by measurement of the volume of evolved gas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Kento Taniguchi for his help with preliminary experiments. This work was supported in part by JSPS KAKENHI Grant No. 15H05797 and 15H05796 in “Precisely Designed Catalysts with Customized Scaffolding” and 19H02509.Notes and references
- (a) M. Negwer, Organic Chemical Drugs and their Synonyms: An International Survey, Akad. Verlag, Berlin, 7th edn, 1994 Search PubMed; (b) Pigment Handbook, ed. P. A. Lewis, John Wiley & Sons, New York, 1988 Search PubMed; (c) W. Shi, L. Wang, M. Umar, T. Awut, H. Mi, C. Tana and I. Nurullaa, Polym. Int., 2009, 58, 800 CrossRef CAS.
- (a) Y. Shirota, Y. Kuwabara and H. Inada, Appl. Phys. Lett., 1994, 65, 807 CrossRef CAS; (b) Y. Kuwabara, H. Ogawa, H. Inada, N. Noma and Y. Shirota, Adv. Mater., 1994, 6, 677 CrossRef CAS; (c) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS; (d) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS PubMed; (e) Z. Ning and H. Tian, Chem. Commun., 2009, 37, 5483 RSC; (f) J. Wang, K. Liu, L. Ma and X. Zhan, Chem. Rev., 2016, 116, 14675 CrossRef CAS; (g) H.-J. Yen and G.-S. Liou, Polym. J., 2016, 48, 117 CrossRef CAS.
- (a) Y. Shirota, T. Kobata and N. Noma, Chem. Lett., 1989, 18, 1145 CrossRef; (b) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef; (c) J. Safaei-Ghomi, Z. Akbarzadeh and A. Ziarati, RSC Adv., 2014, 4, 16385 RSC.
- (a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969 CrossRef; (b) A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901 CrossRef; (c) M. Nishiyama, T. Yamamoto and Y. Koie, Tetrahedron Lett., 1998, 39, 617 CrossRef; (d) T. Yamamoto, M. Nishiyama and Y. Koie, Tetrahedron Lett., 1998, 39, 2367 CrossRef CAS; (e) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (f) J. P. Wolfe, H. Tomori, J. P. Sadighi, J. J. Yin and S. L. Buchwald, J. Org. Chem., 2000, 65, 1158 CrossRef CAS; (g) M. Watanabe, T. Yamamoto and M. Nishiyama, Chem. Commun., 2000, 133 RSC; (h) M. C. Harris and S. L. Buchwald, J. Org. Chem., 2000, 65, 5327 CrossRef; (i) M. I. Ranasinghe, O. P. Varnavski, J. Pawlas, S. I. Hauck, J. Louie, J. F. Hartwig and T. Goodson III, J. Am. Chem. Soc., 2002, 124, 6520 CrossRef; (j) D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354 CrossRef PubMed.
- (a) S. A. Girard, H. Huang, F. Zhou, G.-J. Deng and C.-J. Li, Org. Chem. Front., 2015, 2, 279 RSC; (b) A. V. Iosub and S. S. Stahl, ACS Catal., 2016, 6, 8201 CrossRef; (c) X. Liu, J. Chen and T. Ma, Org. Biomol. Chem., 2018, 16, 8662 RSC.
- (a) Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef PubMed; (b) Y. Izawa, C. Zheng and S. S. Stahl, Angew. Chem., Int. Ed., 2013, 52, 3672 CrossRef CAS; (c) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS; (d) X. Jin, K. Taniguchi, K. Yamaguchi and N. Mizuno, Chem. Sci., 2016, 7, 5371 RSC; (e) J. Zhang, Q. Jiang, D. Yang, X. Zhao, Y. Dong and R. Liu, Chem. Sci., 2015, 6, 4674 RSC.
- (a) M.-O. Simon, S. A. Girard and C.-J. Li, Angew. Chem., Int. Ed., 2012, 51, 7537 CrossRef CAS; (b) M. Sutter, N. Sotto, Y. Raoul, E. Métay and M. Lemaire, Green Chem., 2013, 15, 347 RSC.
- (a) S. A. Girard, X. Hu, T. Knauber, F. Zhou, M.-O. Simon, G.-J. Deng and C.-J. Li, Org. Lett., 2012, 14, 5606 CrossRef CAS; (b) A. Hajra, Y. Wei and N. Yoshikai, Org. Lett., 2012, 14, 5488 CrossRef CAS; (c) M. T. Barros, S. S. Dey, C. D. Maycock and P. Rodrigues, Chem. Commun., 2012, 48, 10901 RSC; (d) Y. Xie, S. Liu, Y. Liu, Y. Wen and G.-J. Deng, Org. Lett., 2012, 14, 1692 CrossRef CAS; (e) M. Sutter, M.-C. Duclos, B. Guicheret, Y. Raoul, E. Métay and M. Lemaire, ACS Sustainable Chem. Eng., 2013, 1, 1463 CrossRef CAS; (f) K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 14969 RSC; (g) K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2016, 6, 3929 RSC.
- (a) F. Zhou, M.-O. Simon and C.-J. Li, Chem.–Eur. J., 2013, 19, 7151 CrossRef CAS; (b) S. Chen, Y. Liao, F. Zhao, H. Qi, S. Liu and G.-J. Deng, Org. Lett., 2014, 16, 1618 CrossRef CAS.
- (a) F. Xiao, Y. Liao, M. Wu and G.-J. Deng, Green Chem., 2012, 14, 3277 RSC; (b) D. Kim, M. Min and S. Hong, Chem. Commun., 2013, 49, 4021 RSC; (c) J. Zhao, H. Huang, W. Wu, H. Chen and H. Jiang, Org. Lett., 2013, 15, 2604 CrossRef CAS; (d) X. Cao, X. Cheng, Y. Bai, S. Liu and G.-J. Deng, Green Chem., 2014, 16, 4644 RSC.
- (a) Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C.-J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487 CrossRef CAS; (b) J.-S. Li, Z. Qiu and C.-J. Li, Adv. Synth. Catal., 2017, 359, 3648 CrossRef CAS; (c) Z. Qiu, J.-S. Li and C.-J. Li, Chem. Sci., 2017, 8, 6954 RSC; (d) H. Zeng, D. Cao, Z. Qiu and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 3752 CrossRef CAS; (e) Z. Qiu, L. Lv, J. Li, C.-C. Li and C.-J. Li, Chem. Sci., 2019, 10, 4775 RSC.
- (a) K. Taniguchi, X. Jin, K. Yamaguchi, K. Nozaki and N. Mizuno, Chem. Sci., 2017, 8, 2131 RSC; (b) X. Jin, K. Taniguchi, K. Yamaguchi, K. Nozaki and N. Mizuno, Chem. Commun., 2017, 53, 5267 RSC; (c) Y. Koizumi, K. Taniguchi, X. Jin, K. Yamaguchi, K. Nozaki and N. Mizuno, Chem. Commun., 2017, 53, 10827 RSC; (d) X. Jin, Y. Koizumi, K. Yamaguchi, K. Nozaki and N. Mizuno, J. Am. Chem. Soc., 2017, 139, 13821 CrossRef CAS PubMed; (e) T. Oyama, T. Yatabe, X. Jin, N. Mizuno and K. Yamaguchi, Chem. Lett., 2019, 48, 517 CrossRef CAS.
- Shi and co-workers recently reported an efficient triarylamine synthesis from nitrobenzenes and cyclohexanones (8 equivalents with respect to nitrobenzenes) using Pd NPs on N/O-doped carbon. However, in their system, triarylamines with three different aryl groups cannot be synthesized. See: S. Pang, Y. Zhang, Y. Huang, H. Yuan and F. Shi, Catal. Sci. Technol., 2017, 7, 2170 RSC.
- Although Li and co-workers recently reported the synthesis of triphenylamine from diphenylamine and 2-cyclohexen-1-one using O2 as the oxidant, the yield of triphenylamine was low (33%), and the substrate scope was not mentioned. See: A. Dominguez-Huerta, I. Perepichka and C.-J. Li, ChemSusChem, 2019, 12, 2999 CrossRef CAS.
- We have recently reported that base co-catalysts efficiently promote the Pd NP-catalyzed dehydrogenation of cyclohexanones to phenols. See ref. 6d and 12b.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- X. Yang, ACS Catal., 2013, 3, 2684 CrossRef CAS.
- We consider that the most important role of TsOH in this triarylamine synthesis is to promote dehydrogenation. In addition, TsOH is also considered to help on the condensation between diarylamines and cyclohexanones. One referee pointed out that it may be also possible that TsOH could partially protonate the diarylamines, and that protonated species would be possible to be reduced to partially dearomatized forms, e.g., some kinds of N-cyclohexylaniline type species, possessing stronger nucleophilicities than diarylamines.19 However, as mentioned above, such dearomatized forms were not detected at all during the reaction between 1a and 2a. If such dearomatized forms are the real nucleophiles, N-cyclohexyldiphenylamines should be formed. However, the corresponding N-cyclohexyldiphenylamine 4a was not detected during the reaction between 1a and 2a, as shown in Fig. 8a. In addition, starting from N-cyclohexyldiphenylamine 4b, the corresponding triarylamine was not produced at all in the present system. These results suggest that the role of TsOH pointed out by the referee is less likely to be present.
- Z. Wang, H. Zemg and C.-J. Li, Org. Lett., 2019, 21, 2302 CrossRef CAS PubMed.
- Y. Koizumi, X. Jin, T. Yatabe, R. Miyazaki, J. Hasegawa, K. Nozaki, N. Mizuno and K. Yamaguchi, Angew. Chem., Int. Ed., 2019, 58, 10893 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 5th edn, 2003 Search PubMed.
- M. Prashed, X. Y. Mak, Y. Liu and O. Repič, J. Org. Chem., 2003, 68, 1163 CrossRef PubMed.
- C. R. Johnson, M. I. Ansari and A. Coop, ACS Omega, 2018, 3, 10886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06442g |
| This journal is © The Royal Society of Chemistry 2020 |