
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of cyclic DNA polymerization with lanthanide coordination nanomaterials for liquid biopsy†
Wenting
Zhou
a,
Lei
Wang
b,
Can
Liu
a,
Qiuyi
Teng
a,
Zhaoyin
Wang
 *a and
Zhihui
Dai
ab
*a and
Zhihui
Dai
ab
aJiangsu Collaborative Innovation Center of Biomedical Functional Materials, Jiangsu Key Laboratory of Biofunctional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, 210023, P. R. China. E-mail: zywang@njnu.edu.cn; Fax: +86-25-85891051; Tel: +86-25-85891051
bNanjing Normal University Center for Analysis and Testing, Nanjing, 210023, P. R. China
First published on 13th March 2020
Abstract
Quantification of circulating tumor DNA (ctDNA) is of great importance in liquid biopsy but difficult due to its low amount in bodily fluids. To meet this high demand, a novel method for ctDNA detection is established by quantifying cyclic DNA polymerization using lanthanide coordination polymers (Ln-CPs). Relying on the coordination between the pyrophosphate ion (PPi) and trivalent cerium ion (Ce3+), organic ligand-free PPi–Ce coordination polymer networks (PPi–Ce CPNs) with enhanced fluorescence are prepared for the first time. By surveying the optical properties of PPi–Ce CPNs, it is found that PPi regulates electric-dipole transition of Ce3+ to the lowest excited state, thus facilitating the emission of fluorescence. Therefore, fluorescence enhancement of PPi–Ce CPNs originates from the ligand field effect rather than the normal antenna effect. Moreover, a new strategy to quantify DNA polymerization is developed based on PPi–Ce CPNs. By introducing multifold cyclic DNA polymerization, a small amount of ctDNA triggers the exponential generation of PPi to form plenty of PPi–Ce CPNs. Accordingly, a biosensor is constructed for sensitive ctDNA detection by measuring the intense fluorescence of PPi–Ce CPNs. The biosensor is capable of sensing ctDNA at the sub-femtomolar level, which is far better than the analytical performances of commercial dyes. Besides, the analytical method is able to detect single nucleotide polymorphism and determine ctDNA in real samples. Considering that DNA polymerization is widely used in bio-recognition, bio-assembly and biomineralization, the work provides a versatile quantitative strategy of making relevant processes precise and controllable.
Introduction
DNA polymerization is one of the most basic processes in living organisms.1,2 In the past few decades, various DNA polymerization-based approaches, including polymerase chain reaction (PCR) and nucleic acid isothermal amplification, have demonstrated their great value in molecular assembly,3–5 target recognition,6–8 and signal acquisition.9,10 However, quantitative methods for DNA polymerization are still lacking in diversity. It is well known that the most remarkable outcome of DNA polymerization is the generation of new DNA strands. Accordingly, current quantitative methods for DNA polymerization are mainly established by detecting the generated DNA strands (strand-based methods). In order to achieve quantification with these methods, the generated DNA strands have to be stained with fluorescent probes in most cases.10,11 Besides, fluorescent probes are normally toxic and thus unfriendly to experimental operations. Additionally, due to the insertion of fluorescent probes, the generated DNA strands are damaged and cannot be further used in subsequent applications. More importantly, fluorescent probes are sequence- or structure-dependent, and can only realize relative quantification. Therefore, strand-based methods are not able to compare and analyze the polymerization of different DNA sequences. Actually, besides DNA strands, the pyrophosphate ion (PPi) is also produced as a by-product in the procedure of DNA polymerization.12,13 Theoretically, the amount of produced PPi is much larger than that of generated DNA strands. If analytical methods are designed based on PPi, DNA polymerization is likely to be quantified more sensitively. Besides, PPi-based quantifications can be real-time, keeping the generated DNA strands undamaged. According to the basic principle of DNA polymerization, once DNA is polymerized, PPi will be produced. That is to say, PPi-based methods work in an absolute manner, and are tolerant of any sequence and structure.Rare earth elements possess excellent luminescence properties owing to their special valence electron structures.14,15 In particular, lanthanide (Ln) elements are capable of synthesizing luminescent materials with strong intensity, long lifetime and large Stokes shifts.16–18 However, the optical properties of pure Ln elements are greatly affected by solvents,19,20 and even aqueous solution can quench Ln elements due to the presence of OH groups.21 Recently, lanthanide coordination polymers (Ln-CPs) have been developed to address the above issue.22,23 In Ln-CPs, Ln elements are protected by coordination ligands against quenching by solvents. Typically, organic ligands are employed to synthesize Ln-CPs and induce the enhancement of fluorescence by the so-called antenna effect.24,25 For deficiencies in biocompatibility and biodegradability, these organic Ln-CPs are not suitable for bio-applications. To our knowledge, fluorescent Ln-CPs formed without organic ligands have not been reported to date.
In the process of tumor development, circulating tumor DNA (ctDNA) is released from cancer cells into the circulatory system.26 Since ctDNA contains the information of genome mutations, ctDNA has been considered as an ideal biomarker for liquid biopsy.27 Notably, ctDNA comes from mutations of the genome, which ensures the accuracy of ctDNA-based diagnosis.28 Moreover, ctDNA can be directly obtained from bodily fluids, making detection of ctDNA simple and painless.29 Although important, quantification of ctDNA in bodily fluids is difficult, mainly because the abundance of target ctDNA is low, while that of interferential DNA is high.30 Currently, apart from digital PCR and targeted deep sequencing, some other methodologies including surface enhanced Raman scattering, electrochemistry, and localized surface plasmon resonance have been employed in detecting ctDNA.31–35 However, the aforementioned methods still suffer from poor sensitivity. Therefore, more efforts should be devoted to exploring new strategies of ctDNA detection.
Herein, organic ligand-free Ln-CPs with enhanced fluorescence were synthesized for the first time and employed in response to ctDNA sensitively. By surveying the interactions between Ln elements and phosphate species, the trivalent cerium ion (Ce3+) and PPi were identified to prepare fluorescent PPi–Ce coordination polymer networks (PPi–Ce CPNs). The ligand field effect of PPi facilitated the energy transfer from the lowest excited state to the ground state of Ce3+, resulting in a dramatic enhancement of fluorescence. During the procedure of DNA polymerization, PPi was accumulated to form PPi–Ce CPNs. Accordingly, quantification of DNA polymerization was achieved by measuring the fluorescence of the formed PPi–Ce CPNs. The PPi–Ce CPN-based quantitative strategy was further used to design a ctDNA biosensor. Benefitting from the strong fluorescence of PPi–Ce CPNs and signal amplification of cyclic DNA polymerization, the biosensor was capable of detecting target ctDNA (KRAS G12DM, colon carcinoma-related ctDNA) in a wide linear range with a low detection limit. In addition, ctDNA in serum and wild DNA can also be determined accurately. Meanwhile, two commercial dyes were chosen to sense ctDNA as well. By comparing their analytical performances, it can be found that the detection limit of the proposed biosensor was at least eight orders of magnitude lower than that of commercial dyes.
Results and discussion
Principle of a fluorescent ctDNA biosensor based on cyclic DNA polymerization and PPi–Ce CPNs
Aiming to detect ctDNA at low concentration, a fluorescent biosensor was fabricated based on PPi–Ce CPNs and cyclic DNA polymerization. The biosensor consists of two DNA oligonucleotides, i.e., primer DNA (P-DNA) and hairpin DNA (H-DNA), a polymerase (Klenow Fragment, KF) and a restriction endonuclease (Nt.BbvCI) (Fig. 1). H-DNA contains two domains: one is the complementary sequence of target ctDNA, while the other is the cut site of Nt.BbvCI. In the initial phase, H-DNA is self-hybridized. In this case, P-DNA cannot hybridize with H-DNA, thus impeding the initiation of DNA polymerization. However, ctDNA may induce the conformational change of H-DNA. Therefore, in the presence of ctDNA, P-DNA hybridizes with H-DNA and triggers the DNA polymerization under the participation of KF. As a result, P-DNA is lengthened, releasing ctDNA from H-DNA. At the same time, PPi, as the by-product of DNA polymerization, is accumulated (Part I). Released ctDNA can change the conformation of H-DNA once again, resulting in cyclic DNA polymerization (Cycle I). Due to the elongation of P-DNA, the cleavage site in double-stranded H-DNA is recognized and cut by Nt.BbvCI, causing a nick in double-stranded H-DNA. In this case, P-DNA is further lengthened to achieve cyclic polymerization (Cycle II), releasing auxiliary DNA (A-DNA) and PPi (Part II). Released A-DNA works like ctDNA that can hybridize with H-DNA and initiate cyclic DNA polymerization (Cycles III and IV) with the aid of KF and Nt.BbvCI. During Cycles III and IV, PPi is also produced (Parts III and IV). Through the multifold cyclic DNA polymerization, a lot of PPi from Parts I, II, III and IV is accumulated, and is further employed to prepare Ln CPs. Owing to coordination of PPi and Ce3+, PPi–Ce CPNs are formed and emit strong fluorescence. Accordingly, by acquiring the signal from PPi–Ce CPNs, ctDNA is quantified sensitively.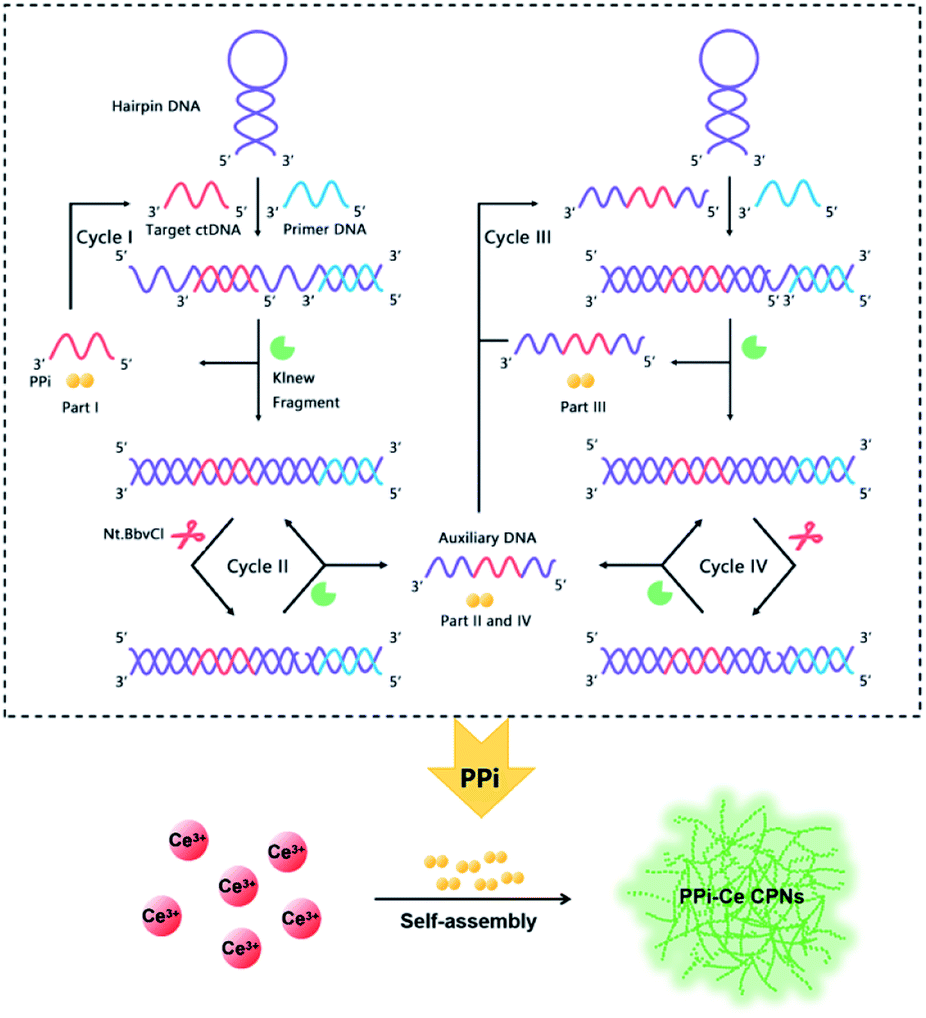 | ||
| Fig. 1 Schematic illustration of ctDNA detection via quantification of cyclic DNA polymerization using PPi–Ce CPNs. | ||
Characterization of PPi–Ce CPNs
PPi–Ce CPNs were prepared by the self-assembly of PPi and Ce3+, and the morphology of the obtained PPi–Ce CPNs was firstly characterized with scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 2A and B). PPi–Ce CPNs show a network structure, and abundant nodes with a diameter of about 15 nm can be found in the nanomaterials. In the energy dispersive X-ray spectroscopy (EDS) spectrum, elementary peaks of Ce, P and O can be observed (Fig. 2D). Besides, the scanning transmission electron microscope (STEM) image and EDS mapping indicate that Ce, P and O elements are uniformly distributed in PPi–Ce CPNs (Fig. 2C). In addition, Fourier transform infrared (FT-IR) spectra were recorded to investigate the formation of PPi–Ce CPNs (Fig. 2E). As reported in previous studies, the FT-IR peaks at 1655 cm−1, 1126 cm−1 and 903 cm−1 represent the P–OH stretching band (νP–OH), phosphate antisymmetric vibration bands (νasPO3), and phosphate symmetric vibration bands (νsPO3).36,37 Relevant peaks in the FT-IR spectrum of PPi–Ce CPNs emerge at 1666 cm−1, 1140 cm−1 and 897 cm−1, respectively. The shift of peak wavelength demonstrates that PPi interacts with Ce3+, which provides the basis for the formation of PPi–Ce CPNs.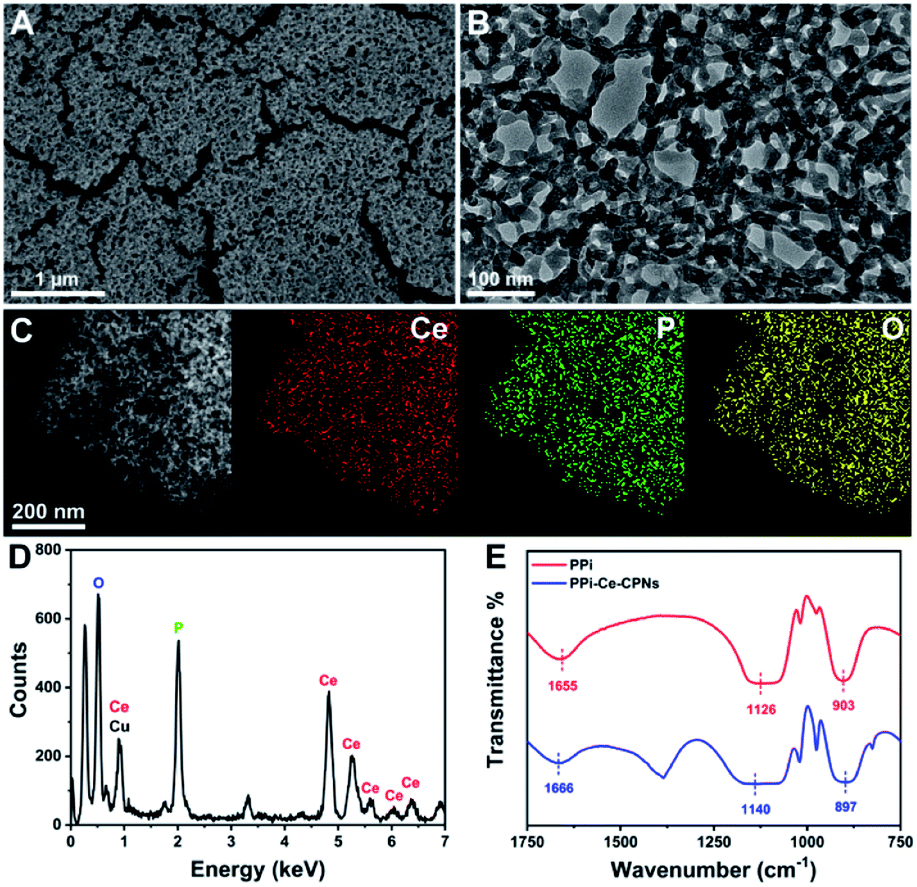 | ||
| Fig. 2 (A) SEM and (B) TEM images of PPi–Ce CPNs. (C) STEM image of PPi–Ce CPNs and EDS mapping images of Ce in red, P in green and O in yellow. (D) EDS spectrum of PPi–Ce CPNs. (E) FT-IR spectra of PPi and PPi–Ce CPNs. | ||
Optical properties of PPi–Ce CPNs
The optical properties of PPi–Ce CPNs were further studied with ultraviolet-visible (UV-vis) absorption and fluorescence spectrometry. Ce3+ reveals typical absorption peaks at 252 and 298 nm due to electric-dipole transitions of a ground state (2F5/2) to a doublet of excited states (2D5/2 and 2D3/2) (Fig. 3A).38 In contrast to Ce3+, there is no peak in the absorption spectrum of PPi. However, by mixing Ce3+ with PPi, the absorption at 298 nm is dramatically increased together with a slight red-shift, while the peak at 252 nm is depressed with a blue-shift. The results indicate that the ligand field effect of PPi generates a larger energy difference of 2D5/2 and 2D3/2 excited states of Ce3+. Therefore, the energy difference between 2F5/2 and 2D3/2 is decreased, resulting in the red-shift of the peak at 298 nm, while the energy difference between 2F5/2 and 2D5/2 is increased, leading to a blue-shift of the peak at 252 nm. Meanwhile, the electric-dipole transition to a low excited state (2D3/2) is greatly promoted, and the transition to a high excited state (2D5/2) is impeded. Furthermore, it is found that the fluorescence of Ce3+ is evidently enhanced (about 10 times) by the formation of PPi–Ce CPNs together with the reduction of average lifetime (Fig. 3B and S1†). As demonstrated above, the ligand field effect of PPi promotes the transition from 2F5/2 to 2D3/2. Accordingly, more Ce3+ reaches the lowest excited state (2D3/2). Therefore, the energy loss of nonradiative transition from 2D5/2 to 2D3/2 is reduced. Spontaneously, radiative transition from 2D3/2 to ground states (2F7/2 and 2F5/2) is facilitated, causing the fluorescence enhancement of Ce3+. The relevant energy level diagram and energy transfer process in absorption and fluorescence emission of Ce3+ and PPi–Ce CPNs are illustrated in Fig. 3C. Fig. S2† depicts the correlation between the fluorescence intensity of PPi–Ce CPNs and the ratio of PPi to Ce3+. With the increase of PPi, more PPi–Ce CPNs are assembled, leading to the enhancement of fluorescence. However, if excessive PPi is employed, PPi–Ce CPNs may be destroyed, causing the decrease of fluorescence. On the basis of our data, equimolar PPi and Ce3+ is the optimal ratio to obtain intense fluorescence emission. Under similar conditions, the effects of phosphate species on fluorescence of Ce3+ were studied. As shown in Fig. 3D, PPi and P3O105− can enhance the fluorescence of Ce3+, while the effects of H2PO4−, HPO42− and PO43− are negligible. In comparison with monophosphates, polyphosphates possess more terminal phosphates and negative charges, thus enhancing the binding affinity and electrostatic field intensity of ligands.39 Considering that the ligand field effect is highly relevant to the binding affinity and electrostatic field intensity of ligands, enhancement of fluorescence has a positive correlation with the number of phosphate groups in phosphate species, and polyphosphates are essential to the formation of Ce-based coordination polymers with strong fluorescence. To investigate the effect of pH on the fluorescence of Ce3+, PPi–Ce CPNs are prepared under different pH values (from 5 to 10). In these cases, fluorescence of the formed PPi–Ce CPNs is steady (Fig. S3†), indicating that fluorescence enhancement of Ce3+ caused by PPi is independent of the pH of the solution. It is reported that ATP can also be used to form polymers with Ce3+.22,40,41 Actually, in these studies, polymers are prepared in the presence of ATP as well as tris(hydroxymethyl)aminomethane (Tris), and Tris plays a key role in regulating the fluorescence of Ce3+ depending on the antenna effect. If Tris is absent, ATP can only induce a slight fluorescence enhancement (Fig. S4†). In contrast, by replacing ATP with PPi, fluorescence of Ce3+ can be significantly increased due to the ligand field effect. In addition to Ce3+, other Ln elements (except for non-fluorescent La3+ and Lu3+, and radioactive Pm3+) are employed to assemble coordination polymers with PPi. According to the fluorescence of different PPi–Ln mixtures (Fig. 3E), only Ce3+ can be coupled with PPi to form fluorescent Ln-CPs, which should be attributed to the special electron structure of Ce3+ and ligand field effect of PPi.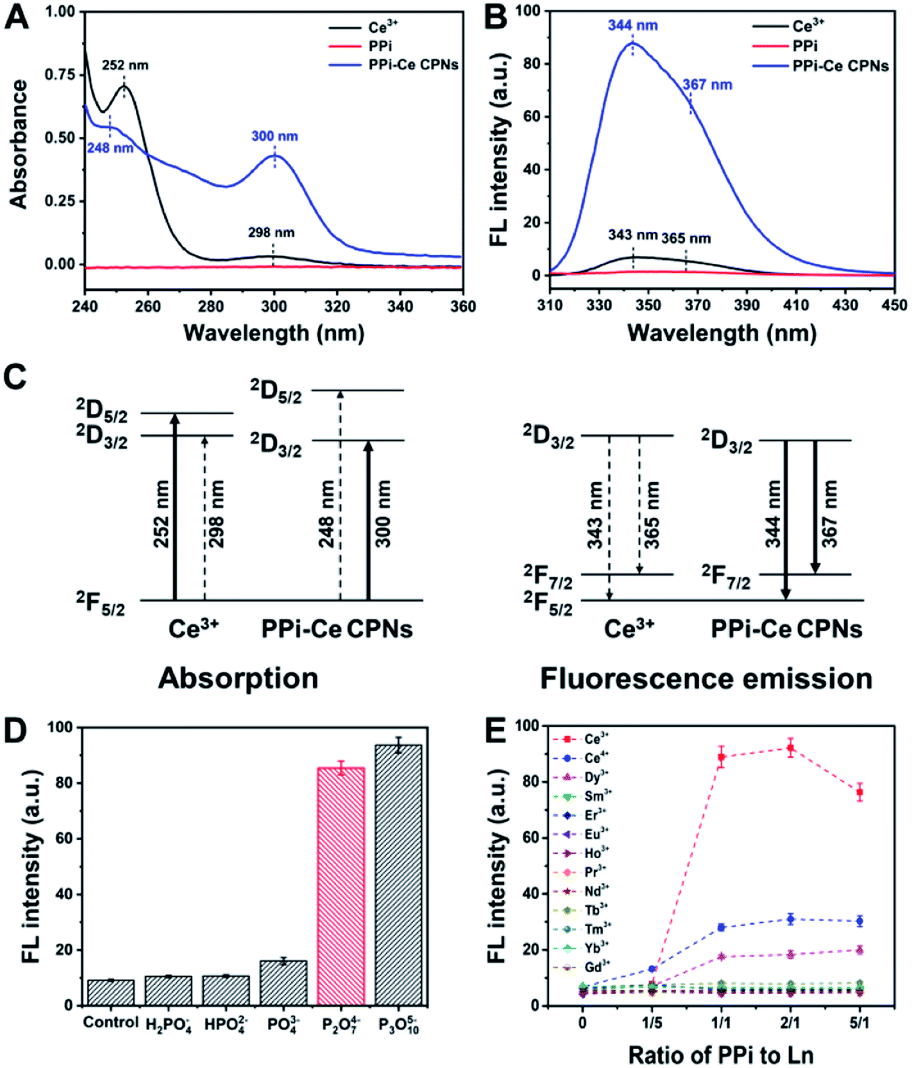 | ||
| Fig. 3 (A) UV-vis absorption and (B) fluorescence spectra of Ce3+, PPi and PPi–Ce CPNs. (C) Schematic energy level diagram and energy transfer process in absorption and fluorescence emission of Ce3+ and PPi–Ce CPNs. (D) Fluorescence intensity of Ce3+ mixed with different phosphate species. (E) Fluorescence intensity of PPi mixed with different Ln elements. | ||
Feasibility of the bioanalytical method
In order to verify the feasibility of the designed method, fluorescence spectra under different conditions were recorded (Fig. 4A). As expected, in the absence of ctDNA, P-DNA, H-DNA or KF, DNA amplification is impeded. In these cases, PPi is not produced, and thus PPi–Ce CPNs are not formed. Therefore, only weak background fluorescence of Ce3+ can be detected (curves a–d and f in Fig. 4A). In the presence of all reactants but Nt.BbvCI, one of multifold cyclic DNA polymerization (Cycle I) is triggered, while Cycles II, III and IV are still blocked. As a result, some PPi (Part I) is accumulated to form PPi–Ce CPNs that emit enhanced fluorescence (curve e in Fig. 4A). However, if all reactants are present, ctDNA initiates multifold cyclic DNA polymerization that produces a large amount of PPi from Parts I, II, III and IV. Theoretically, the approximate ratio of PPi/DNA strand is 28, since lengths of H-DNA and P-DNA are 43 and 15 nt, respectively. Accordingly, plenty of PPi–Ce CPNs are prepared and intense fluorescence is obtained (curve g in Fig. 4A). As shown in Fig. S5,† the effect of dNTPs on fluorescence of Ce3+ is slight. Accordingly, ctDNA can be accurately determined without purification to remove excess dNTPs. Electrophoresis was further carried out to study the detection process in view of DNA operation (Fig. 4B). In the polyacrylamide gel electrophoresis (PAGE) image, bands 1 and 2 in lane a represent H-DNA and P-DNA, respectively. In the presence of ctDNA, band 3 appears in lane b, manifesting the hybridization of ctDNA and H-DNA. The mixture of P-DNA and ctDNA results in band 4 in lane c. In lane d, conformation of H-DNA is changed by ctDNA. Accordingly, ctDNA and P-DNA can co-hybridize with H-DNA, leading to the appearance of band 5. In lane e, KF causes the elongation of P-DNA, forming the double-stranded H-DNA. Consequently, a new band (band 6) emerges in lane e. In the presence of Nt.BbvCI, A-DNA is obtained by cutting in the cleavage site of double-stranded H-DNA. Band 7 in lane g should be attributed to the hybridization of A-DNA and H-DNA. The above results demonstrate that ctDNA can trigger cyclic DNA polymerization, leading to the generation of PPi–Ce CPNs. Therefore, ctDNA can be sensitively quantified by the fluorescence of PPi–Ce CPNs.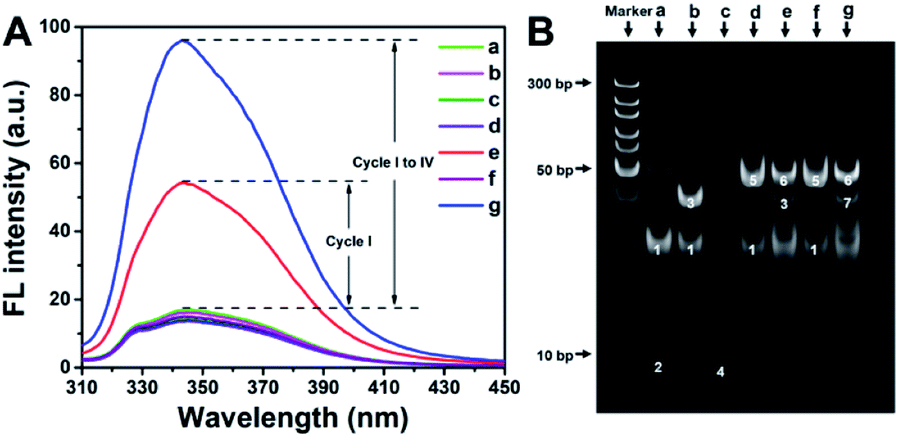 | ||
| Fig. 4 (A) Fluorescence spectra and (B) PAGE image obtained under different conditions: (a) H-DNA, P-DNA, KF and Nt.BbvCI, (b) H-DNA, ctDNA, KF and Nt.BbvCI, (c) P-DNA, ctDNA, KF and Nt.BbvCI, (d) H-DNA, P-DNA and ctDNA, (e) H-DNA, P-DNA, ctDNA, and KF, (f) H-DNA, P-DNA, ctDNA, and Nt.BbvCI, and (g) H-DNA, P-DNA, ctDNA, KF and Nt.BbvCI. | ||
Analytical performances of the proposed biosensor
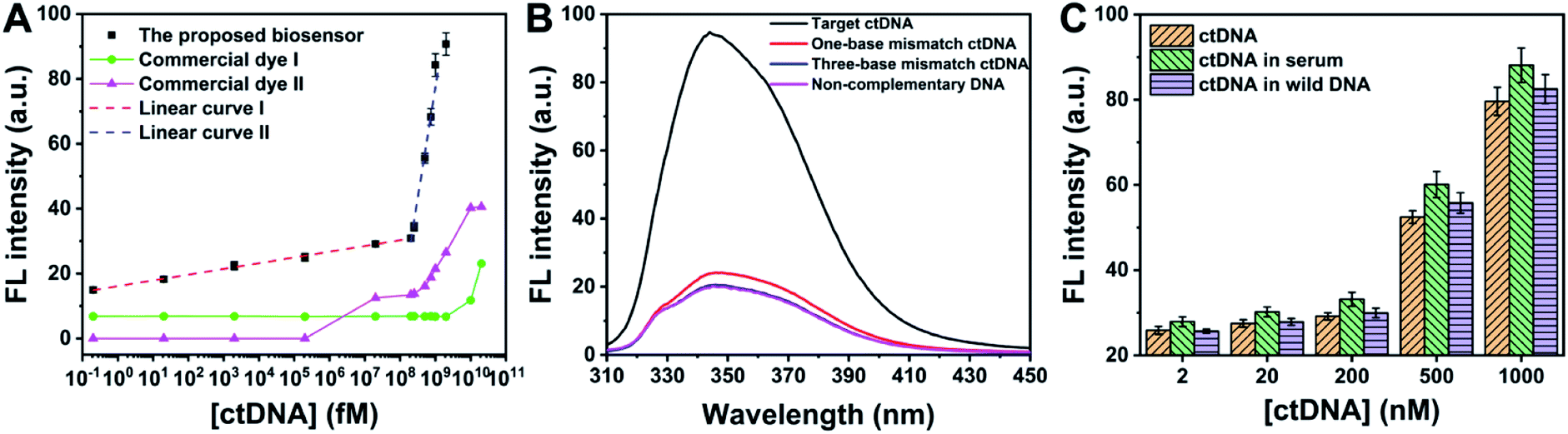
To pursue better analytical performances, dNTPs, KF and Nt.BbvCI used in the biosensor were optimized (Fig. S6A–C†). Meanwhile, 120 min was chosen as the best incubation time to detect ctDNA (Fig. S6D†). Under the optimal conditions, with the increase of ctDNA, more PPi is produced to form PPi–Ce CPNs, leading to the increase of fluorescence (Fig. S7†). As shown in Fig. 5A, the fluorescence intensity of PPi–Ce CPNs at 344 nm (F344 nm) is proportional to the concentration of ctDNA ([ctDNA]) over a wide range. In the range from 0.2 fM to 200 nM, the linear regression equation is F344 nm = 1.67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lg[ctDNA] (fM) + 15.2 with a correlation coefficient (R) of 0.998. In the range from 200 nM to 1 μM, the linear regression equation is F344 nm = 64.2lg[ctDNA] (fM) − 505 (R = 0.992). According to the parameters in the low concentration region (<200 nM), the detection limit of this biosensor is calculated to be 0.16 fM (S/N = 3). As mentioned above, quantification of DNA polymerization is normally achieved based on the detection of generated DNA strands using commercial dyes. In this work, two kinds of commercial dyes (4S Red Plus Nucleic Acid Stain and ssDNA dye) were employed to detect the same ctDNA. Although commercial dyes can also respond to ctDNA, fluorescence enhancement is observed only at high concentration. The detection limit of the biosensor is eight orders of magnitude lower than that of commercial dyes. It should be noted that besides commercial dyes, the detection limit and linear range of this biosensor are better than or at least comparable to those of different ctDNA biosensors (Table S1†).
lg[ctDNA] (fM) + 15.2 with a correlation coefficient (R) of 0.998. In the range from 200 nM to 1 μM, the linear regression equation is F344 nm = 64.2lg[ctDNA] (fM) − 505 (R = 0.992). According to the parameters in the low concentration region (<200 nM), the detection limit of this biosensor is calculated to be 0.16 fM (S/N = 3). As mentioned above, quantification of DNA polymerization is normally achieved based on the detection of generated DNA strands using commercial dyes. In this work, two kinds of commercial dyes (4S Red Plus Nucleic Acid Stain and ssDNA dye) were employed to detect the same ctDNA. Although commercial dyes can also respond to ctDNA, fluorescence enhancement is observed only at high concentration. The detection limit of the biosensor is eight orders of magnitude lower than that of commercial dyes. It should be noted that besides commercial dyes, the detection limit and linear range of this biosensor are better than or at least comparable to those of different ctDNA biosensors (Table S1†).
 | ||
| Fig. 5 (A) Fluorescence responses of the proposed biosensor, commercial dye I (4S Red Plus Nucleic Acid Stain) and commercial dye II (ssDNA dye) to various concentrations of ctDNA. (B) Fluorescence intensity of the biosensor in response to ctDNA, one-base mismatch ctDNA, three-base mismatch ctDNA and non-complementary DNA. The concentration of the different DNA sequences is 2 μM. (C) Detection of pure ctDNA, ctDNA in 1% serum and ctDNA in 2 μM wild DNA with the proposed biosensor. | ||
Besides ctDNA, three other DNA sequences including one-base mismatch ctDNA, three-base mismatch ctDNA and non-complementary DNA were chosen to challenge the proposed biosensor. According to the data shown in Fig. 5B, even one base mutation in ctDNA cannot induce significant fluorescence enhancement, demonstrating that the biosensor is capable of single nucleotide polymorphism detection. In combination with the data in Fig. 5A, it can be concluded that to obtain equal intensity, the concentration of mismatch DNA is 1 million times higher than that of ctDNA. The excellent selectivity should be ascribed to the strict Watson–Crick complementary matching principle and super-specific recognition function of the restriction endonuclease. In practical applications, detection of ctDNA is often confronted with serious interferential components such as proteins and oligonucleotides. Accordingly, performances of determining ctDNA in human serum and wild DNA were studied (Fig. 5C). Although the components of serum are complex, ctDNA in serum can still be detected accurately using our methods. Similar results are obtained in the case where concentration of interferential DNA is 100 times higher than that of ctDNA. Apart from the low detection limit, wide linear range and satisfactory selectivity, the fluorescent biosensor possesses other strengths, such as simple operation, low cost and being free of modification, suggesting the great potential of this bioanalytical platform in liquid biopsy.
Conclusions
In summary, a new type of Ln-CP is prepared according to the self-assembly of PPi and Ce3+. Besides the novel morphology, the PPi–Ce CPNs are able to emit strong fluorescence. Different from former Ln-CPs, the optical properties of PPi–Ce CPNs are improved without the participation of organic ligands, and the fluorescence enhancement is the result of the ligand field effect rather than the antenna effect. Based on these findings, a PPi–Ce CPN-based method is established to quantify DNA polymerization. Since the amount of produced PPi is much larger than that of generated strands in the procedure of DNA polymerization, the PPi–Ce CPN-based approach reveals overwhelming superiority over strand-based methods in sensitivity and versatility. To make full use of the excellent properties of PPi–Ce CPNs, a biosensor is further fabricated in a combination of multifold cyclic DNA polymerization. Benefitting from the intense fluorescence of PPi–Ce CPNs and signal amplification of cyclic DNA polymerization, ctDNA in the range from 0.2 fM to 1 μM can be detected, which is valuable in ctDNA-based clinical diagnosis. Meanwhile, single nucleotide polymorphism of ctDNA and ctDNA in real samples can be accurately determined with the designed biosensor. Relying on the quantitative strategy proposed in this work, various PPi-related processes can be controlled and utilized precisely, which is of great significance in biomedicine and biotechnology.Experimental
Chemicals and materials
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O), holmium nitrate pentahydrate (Ho(NO3)3·5H2O), samarium nitrate hexahydrate (Sm(NO3)3·6H2O), erbium nitrate hexahydrate (Er(NO3)3·6H2O), dysprosium nitrate hexahydrate (Dy(NO3)3·6H2O), europium nitrate hexahydrate (Eu(NO3)3·6H2O), ceric sulphate (Ce(SO4)2), terbium nitrate hexahydrate (Tb(NO3)3·6H2O), thulium nitrate pentahydrate (Tm(NO3)3·5H2O), ytterbium nitrate pentahydrate (Yb(NO3)3·5H2O), potassium pyrophosphate (K4P2O7), mono-potassium phosphate (KH2PO4), di-potassium phosphate (K2HPO4), potassium phosphate (K3PO4), sodium tripolyphosphate (Na5P3O10), and deoxy-ribonucleoside triphosphates (dNTPs) were obtained from Sigma-Aldrich Co., Ltd. (China). Praseodymium nitrate hexahydrate (Pr(NO3)3·6H2O), neodymium nitrate hexahydrate (Nd(NO3)3·6H2O), and gadolinium nitrate hexahydrate (Gd(NO3)3·6H2O) were obtained from Aladdin Bio-Chem Technology Co., Ltd. (Shanghai, China). The Klenow Fragment (KF) and Nt.BbvCI were provided by New England Biolabs (NEB) Ltd. (Beijing, China). 4S Red Plus Nucleic Acid Stain (1000×) was obtained from Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (China). Single-stranded DNA (ssDNA) dye was obtained from Promega Biotech Co., Ltd. (Beijing, China). All other reagents in this experiment were of analytical grade, and were used directly without extra purification.The oligonucleotides used in this work were synthesized and purified via HPLC by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (China). Detailed sequences of these oligonucleotides are shown in Table 1.
| Name | Sequence from 5′ to 3′ |
|---|---|
| a The sequence used to form a hairpin structure is underlined. The italic and bold sequences were used to hybridize with P-DNA and ctDNA, respectively. The mismatch bases are marked with a wavy line. | |
| Hairpin DNA (H-DNA) |
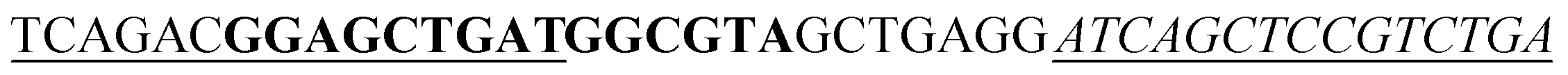
|
| Primer DNA (P-DNA) | TCAGACGGAGCTGAT |
| Target ctDNA | TACGCCATCAGCTCC |
| One-base mismatch ctDNA |
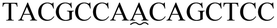
|
| Three-base mismatch ctDNA |
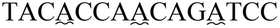
|
| Non-complementary DNA | TACGACTCACTATAG |
Apparatus
The fluorescence spectra were recorded using a Fluoromax-4 spectrometer (Horiba, France). The fluorescence emission spectra of the trivalent cerium ion (Ce3+) and PPi–Ce CPNs were measured from 310 to 450 nm with an excitation wavelength of 295 nm. The scanning electron microscopy (SEM) image was obtained on a JEOL JSM-7600F instrument (Japan). The transmission electron microscopy (TEM) image, scanning transmission electron microscope (STEM) image and energy dispersive X-ray spectroscopy (EDS) mapping images were obtained on a JEM-200CX (200 kV) instrument (Japan). The polyacrylamide gel electrophoresis (PAGE) image was obtained using a Model DYY-8C electrophoretic device. Fourier transform infrared (FT-IR) spectra were obtained using a gas chromatography Fourier transform infrared spectrometer (NEXUS670). Ultraviolet-visible (UV-vis) absorption spectra were measured with a Cary 60 spectrophotometer (Agilent, USA).Interactions between Ln elements and phosphate species
On the one hand, the interactions between Ce3+ and phosphate species were studied by mixing 20 μL of 0.5 mM Ce(NO3)3·6H2O with 10 μL of 1 mM KH2PO4 (pH 5.68), K2HPO4 (pH 8.37), K3PO4 (pH 10.94), K4P2O7 (pH 9.74) and Na5P3O10 (pH 9.81), respectively. On the other hand, to investigate the interactions between the pyrophosphate ion and Ln elements, 10 μL of 1 mM K4P2O7 was mixed with 20 μL of 0.5 mM Dy(NO3)3·6H2O, Sm(NO3)3·6H2O, Er(NO3)3·6H2O, Eu(NO3)3·6H2O, Ho(NO3)3·5H2O, Ce(SO4)2, Pr(NO3)3·6H2O, Nd(NO3)3·6H2O, Tb(NO3)3·6H2O, Tm(NO3)3·5H2O, Yb(NO3)3·5H2O and Gd(NO3)3·6H2O, respectively. After being diluted to 300 μL with ultrapure water, these compounds were kept at room temperature for 15 minutes. Finally, fluorescence spectra of the above samples were recorded.Construction of a fluorescent ctDNA biosensor
The biosensor was constructed based on cyclic DNA polymerization and PPi–Ce CPNs. First, the H-DNA was dissolved in 10 mM Tris–HCl (pH 7.6, 500 mM NaCl, 100 mM MgCl2). Then the H-DNA solution was heated at 95 °C for 3 min, and cooled down gently to room temperature to form a hairpin structure. After that, DNA cyclic polymerization was carried out in a series of 100 μL solutions containing 3 μM H-DNA, 2 μM P-DNA, 50 μM dNTPs, 2 U KF, 1.5 U Nt.BbvCI, and different concentrations of ctDNA. After being incubated at 37 °C for 2 hours, each of these solutions was mixed with 180 μL of 25 μM Ce(NO3)3·6H2O. After that, PPi–Ce CPNs were prepared by keeping these solutions at room temperature for 15 min. Finally, to achieve quantification of ctDNA, the fluorescence spectrum of each sample was obtained under an excitation light of 295 nm.To verify the selectivity of the biosensor, three other DNA sequences (one-base mismatch ctDNA, three-base mismatch ctDNA and non-complementary DNA) were employed to replace ctDNA, and detected with the PPi–Ce CPN-based quantitative method. To verify the analytical performances of the biosensor, two commercial dyes, i.e., 4S Red Plus Nucleic Acid Stain and ssDNA dye, were chosen to detect the same ctDNA. Various concentrations of ctDNA were mixed with 100 × 4S Red Plus Nucleic Acid Stain and 1% ssDNA dye, respectively. These solutions were incubated at room temperature for 15 min, and then fluorescence spectra of the samples were recorded with an excitation wavelength of 300 or 492 nm. The applicability of the analytical method was clarified by detecting ctDNA in real samples. To be specific, different concentrations of ctDNA were diluted with 1% human serum and 2 μM wild DNA, respectively. Subsequently, ctDNA in real samples was quantified using the proposed biosensor.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China for the projects under grant No. 21625502 and 21974070, and the Natural Science Foundation of Jiangsu Province of China (No. BK20192008 and BK20191367). All experiments involving human serum were performed in accordance with the guidelines of Safety and Welfare of Human Research of Nanjing Normal University and approved by the Ethics Committee of Nanjing Normal University. Informed consent was obtained from human participants of this study.Notes and references
- C. Marchal, J. Sima and D. M. Gilbert, Nat. Rev. Mol. Cell Biol., 2019, 20, 721–737 CrossRef CAS PubMed.
- J. Kottur and D. T. Nair, Nucleic Acids Res., 2018, 46, 5875–5885 CrossRef CAS PubMed.
- S. Li, L. Xu, W. Ma, X. Wu, M. Sun, H. Kuang, L. Wang, N. A. Kotov and C. Xu, J. Am. Chem. Soc., 2016, 138, 306–312 CrossRef CAS PubMed.
- S. Tanwar, K. K. Haldar and T. Sen, J. Am. Chem. Soc., 2017, 139, 17639–17648 CrossRef CAS PubMed.
- Y. Liu, X. Hu, T. Fu, R. Wang and W. Tan, Sci. China: Chem., 2019, 62, 407–408 CrossRef CAS.
- R. Yamashige, M. Kimoto, R. Okumura and I. Hirao, J. Am. Chem. Soc., 2018, 140, 14038–14041 CrossRef CAS PubMed.
- N. Melnychuk and A. S. Klymchenko, J. Am. Chem. Soc., 2018, 140, 10856–10865 CrossRef CAS PubMed.
- X. Wang, F. Chen, D. Zhang, Y. Zhao, J. Wei, L. Wang, S. Song, C. Fan and Y. Zhao, Chem. Sci., 2017, 8, 4764–4770 RSC.
- M. G. Mohsen, D. Ji and E. T. Kool, Chem. Sci., 2019, 10, 3264–3270 RSC.
- L. J. Wang, X. Han, C. C. Li and C. Y. Zhang, Chem. Sci., 2018, 9, 6053–6061 RSC.
- C. C. Li, W. X. Liu, J. Hu and C. Y. Zhang, Chem. Sci., 2019, 10, 8675–8684 RSC.
- T. Nakamura, Y. Zhao, Y. Yamagata, Y. J. Hua and W. Yang, Nature, 2012, 487, 196–201 CrossRef CAS PubMed.
- L. T. Da, D. Wang and X. Huang, J. Am. Chem. Soc., 2012, 134, 2399–2406 CrossRef CAS PubMed.
- L. Dai, W. S. Lo, Y. Gu, Q. Xiong, K. L. Wong, W. M. Kwok, W. T. Wong and G. L. Law, Chem. Sci., 2019, 10, 4550–4559 RSC.
- M. Hatanaka, Y. Hirai, Y. Kitagawa, T. Nakanishi, Y. Hasegawa and K. Morokuma, Chem. Sci., 2017, 8, 423–429 RSC.
- F. Chen, Y. M. Wang, W. Guo and X. B. Yin, Chem. Sci., 2019, 10, 1644–1650 RSC.
- J. Y. Hu, Y. Ning, Y. S. Meng, J. Zhang, Z. Y. Wu, S. Gao and J. L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC.
- X. L. Zheng, Y. Liu, M. Pan, X. Q. Lu, J. Y. Zhang, C. Y. Zhao, Y. X. Tong and C. Y. Su, Angew. Chem., Int. Ed., 2007, 46, 7399–7403 CrossRef CAS PubMed.
- C. L. Liu, R. L. Zhang, C. S. Lin, L. P. Zhou, L. X. Cai, J. T. Kong, S. Q. Yang, K. L. Han and Q. F. Sun, J. Am. Chem. Soc., 2017, 139, 12474–12479 CrossRef CAS PubMed.
- O. Guzman-Mendez, F. Gonzalez, S. Bernes, M. Flores-Alamo, J. Ordonez-Hernandez, H. Garcia-Ortega, J. Guerrero, W. Qian, N. Aliaga-Alcalde and L. Gasque, Inorg. Chem., 2018, 57, 908–911 CrossRef CAS PubMed.
- H. H. Zeng, W. B. Qiu, L. Zhang, R. P. Liang and J. D. Qiu, Anal. Chem., 2016, 88, 6342–6348 CrossRef CAS PubMed.
- X. Wu, Q. DeGottardi, I. C. Wu, J. Yu, L. Wu, F. Ye, C. T. Kuo, W. W. Kwok and D. T. Chiu, Angew. Chem., Int. Ed., 2017, 56, 14908–14912 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Angew. Chem., Int. Ed., 2017, 56, 7171–7175 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Angew. Chem., Int. Ed., 2016, 55, 12059–12062 CrossRef CAS PubMed.
- C. Abbosh, N. J. Birkbak and C. Swanton, Nat. Rev. Clin. Oncol., 2018, 15, 577–586 CrossRef CAS PubMed.
- J. Wang, K. M. Koo, Y. Wang and M. Trau, Adv. Sci., 2019, 6, 1900730 CrossRef PubMed.
- L. A. Diaz Jr and A. Bardelli, J. Clin. Oncol., 2014, 32, 579–586 CrossRef PubMed.
- E. Heitzer, I. S. Haque, C. E. S. Roberts and M. R. Speicher, Nat. Rev. Genet., 2019, 20, 71–88 CrossRef CAS PubMed.
- J. Das, I. Ivanov, T. S. Safaei, E. H. Sargent and S. O. Kelley, Angew. Chem., Int. Ed., 2018, 57, 3711–3716 CrossRef CAS PubMed.
- A. M. Newman, S. V. Bratman, J. To, J. F. Wynne, N. C. Eclov, L. A. Modlin, C. L. Liu, J. W. Neal, H. A. Wakelee, R. E. Merritt, J. B. Shrager, B. W. Loo Jr, A. A. Alizadeh and M. Diehn, Nat. Med., 2014, 20, 548–554 CrossRef CAS PubMed.
- M. Murtaza, S.-J. Dawson, D. W. Y. Tsui, D. Gale, T. Forshew, A. M. Piskorz, C. Parkinson, S.-F. Chin, Z. Kingsbury, A. S. C. Wong, F. Marass, S. Humphray, J. Hadfield, D. Bentley, T. M. Chin, J. D. Brenton, C. Caldas and N. Rosenfeld, Nature, 2013, 497, 108–112 CrossRef CAS PubMed.
- Q. Zhou, J. Zheng, Z. Qing, M. Zheng, J. Yang, S. Yang, L. Ying and R. Yang, Anal. Chem., 2016, 88, 4759–4765 CrossRef CAS PubMed.
- J. Das, I. Ivanov, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2016, 138, 11009–11016 CrossRef CAS PubMed.
- A. H. Nguyen and S. J. Sim, Biosens. Bioelectron., 2015, 67, 443–449 CrossRef CAS PubMed.
- P. Huang, F. Wu and L. Mao, Anal. Chem., 2015, 87, 6834–6841 CrossRef CAS PubMed.
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS PubMed.
- C. Tang, Y. Bando, D. Golberg and R. Ma, Angew. Chem., Int. Ed., 2005, 117, 582–585 CrossRef.
- X. Z. Wang, A. Lopez and J. W. Liu, Langmuir, 2018, 34, 7899–7905 CrossRef CAS PubMed.
- C. X. Chen, Q. Yuan, P. J. Ni, Y. Y. Jiang, Z. L. Zhao and Y. Z. Lu, Analyst, 2018, 143, 3821–3828 RSC.
- H. H. Zeng, L. Zhang, L. Q. Rong, R. P. Liang and J. D. Qiu, Biosens. Bioelectron., 2017, 89, 721–727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of PPi on the lifetime of Ce3+, effect of ratio of PPi to Ce3+ and pH on fluorescence intensity of PPi–Ce CPNs, effect of ATP and dNTPs on fluorescence intensity of Ce3+, optimization of detection conditions, fluorescence spectra obtained at different concentrations of ctDNA, and a comparison of different methods for the determination of ctDNA and related references. See DOI: 10.1039/c9sc06408g |
| This journal is © The Royal Society of Chemistry 2020 |