
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoft interactions with hard Lewis acids: generation of mono- and dicationic alkaline-earth metal arene-complexes by direct oxidation†
Marcel
Schorpp
 and
Ingo
Krossing
*
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
First published on 13th January 2020
Abstract
The synthesis of the first unsupported dicationic arene complexes of calcium and strontium [(η6-HMB)AE(oDFB)4]2+ is reported (HMB = hexamethylbenzene; AE = alkaline earth metal; oDFB = ortho-difluorobenzene). They were prepared by direct oxidation of the elemental metals employing the ligand-forming radical cation salt [HMB][WCA] as an oxidant (WCA = [Al(ORF)4] or [μF-{Al(ORF)3}2]; RF = C(CF3)3). In addition, monocationic η6-HMB complexes of calcium, strontium and barium supported by coordination of the monodentate anion [F–Al(ORF)3]− are reported. In all examples, almost undistorted η6-HMB coordination is observed with rather short M-arenecentroid distances approaching those observed with the isoelectronic but negatively charged pentamethylcyclopentadienyl ligand. The structure and bonding, thermodynamic stability and Lewis acidity (fluoride/hydride ion affinities, FIA/HIA) of the generated complexes were assessed by DFT methods. It followed that the gaseous dications [(η6-HMB)AE(oDFB)4]2+ are extremely hard Lewis acids that retain FIAs close to superacidity in solution.
While coordination of anionic 6π-aromatics such as group 2 metallocenes has been known for more than 60 years, the coordination of neutral arenes to heavier Alkaline Earth (AE) metals is scarce and neutral arene complexes of AE metal dications without a supporting ligand framework are still unprecedented.1 Hitherto, mainly the enforced coordination of flanking aryl rings of anionic polydentate ligand scaffolds in neutral or monocationic complexes was observed. The first structurally characterized example of an isolated arene coordinating to a heavier AE metal dates back to 2001. Here Ba2+ formed neutral dinuclear complexes with a very strong σ-chelating porphyrinogen. Its central metal is perfectly η6 coordinated by arenes (benzene, toluene, naphthalene, and durene).2 Formally cationic complexes were later discovered within the tightly bound (BaCl2Ga)∞ coordination-network of [Ba(η6-C6H6)2(GaCl4)2]∞. Related dimeric motifs [Ba(C7H8)2(MEt4)2]2 with tightly bound formal [MEt4]− (M = Al, Ga) counterions were reported.3 The directed binding of isolated arenes to the hard and highly Lewis acidic metal centers was only observed within rigid [(ligand)AE(ηx-arene)]0,+ pockets. Thus, Hill and Harder paired monoanionic chelating beta-diketiminate ligands (NacNac) with Mg and Ca to form the complex salts [M(NacNac)][WCA] (where WCA = [B(C6F5)4] or [Al(ORF)4] with ORF = –OC(CF3)3). They include highly Lewis acidic cations that are capable of coordinating benzene, toluene, m-xylene, and mesitylene, as well as the generally poorly coordinating silyl ethers.4,5 In addition, cationic Lewis base-free [M(NacNac)][B(C6F5)4] complexes compete with B(C6F5)3 in their Lewis acidity. By addition of difluoro-benzene, a [Mg(NacNac)(1,4-F2C6H4)3]+ complex cation with κ1-F–M interactions is generated. It was also possible to isolate further complex cations with rather unconventional ligands for AE metal chemistry, for example triphenylphosphine and 3-hexyne adducts. Due to their high Lewis acidity, the π-electron density of the hexyne-adduct is highly polarized allowing for activation and subsequent attack of the C–C multiple bond.6,7 Related is the reaction between [Ca(NacNac)][WCA] and AlI(NacNac) in the presence of benzene. Originally the Lewis donor–acceptor complex was aimed for, but instead reduction of benzene to an alumina-norbornadiene derivative was observed.8 These results generated renewed interest in group 2 chemistry and their possible application in catalysis to exploit their pronounced Lewis acidity. Useful examples include monocationic and neutral AE complexes that were used in the transfer hydrogenation or hydrosilylation of alkenes and imines. Concomitantly, the underlying reactivity of intermediary and selectively generated M-hydride species has been elucidated.7,9
Inspired by the recent synthesis of isolated cationic mono-arene group 13 complexes [MI(HMB)]+ (M = Ga, In) with the ligand-forming oxidant [HMB]+˙[Al(ORF)4]− (eqn (1), HMB = hexamethylbenzene),10 we were interested in transferring this methodology to group 2, i.e. the formation of unsupported mono-/bis(arene)-complexes by direct oxidation of the elemental metals as shown in eqn (2).
 | (1) |
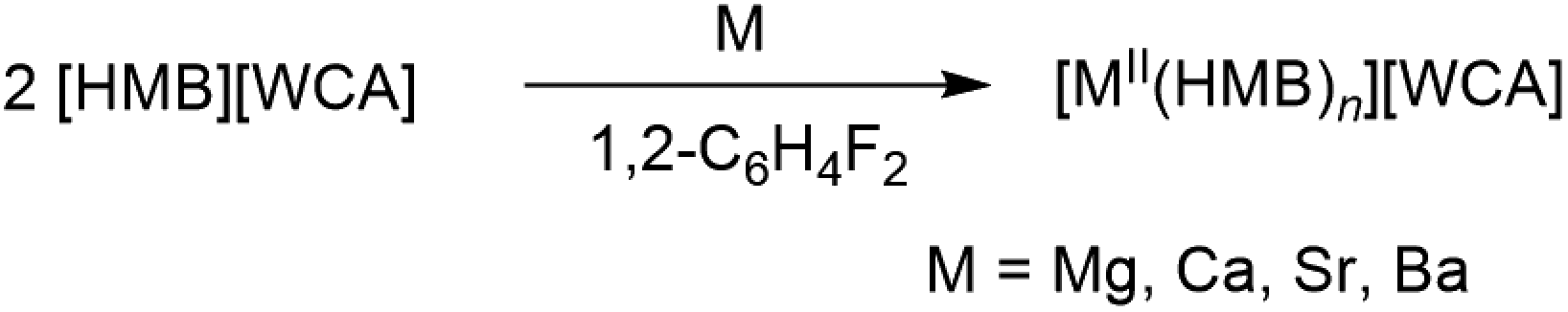 | (2) |
Results and discussion
Orienting investigations towards Mg and Ca salts using [HMB][Al(ORF)4]
The first reactions between magnesium and the ligand-forming oxidant [HMB][Al(ORF)4] in ortho-difluorobenzene (oDFB) were futile; instead the deep red color attributed to the HMB radical slowly faded over weeks, while the only isolable products were the already reported10 long-term degradation products of [HMB]+˙: [C6Me7]+ and the cycloaddition product of [HMB]+˙ and oDFB (see Scheme 1a). However, upon spiking the reaction mixture with a few drops of MeCN, immediate loss of the deep red color was observed. Single crystals suitable for X-ray diffraction analysis revealed a hexacoordinate acetonitrile magnesium salt, [Mg(MeCN)6][Al(ORF)4]21 (see Scheme 1b & Fig. S1†). The formal potential of the oxidant [HMB]+˙ is therefore most likely not high enough to oxidize magnesium in non-polar media and only upon addition of the donor solvent MeCN does the formation of the stable hexacoordinate MgII salt give the thermodynamic driving force for the reaction to proceed.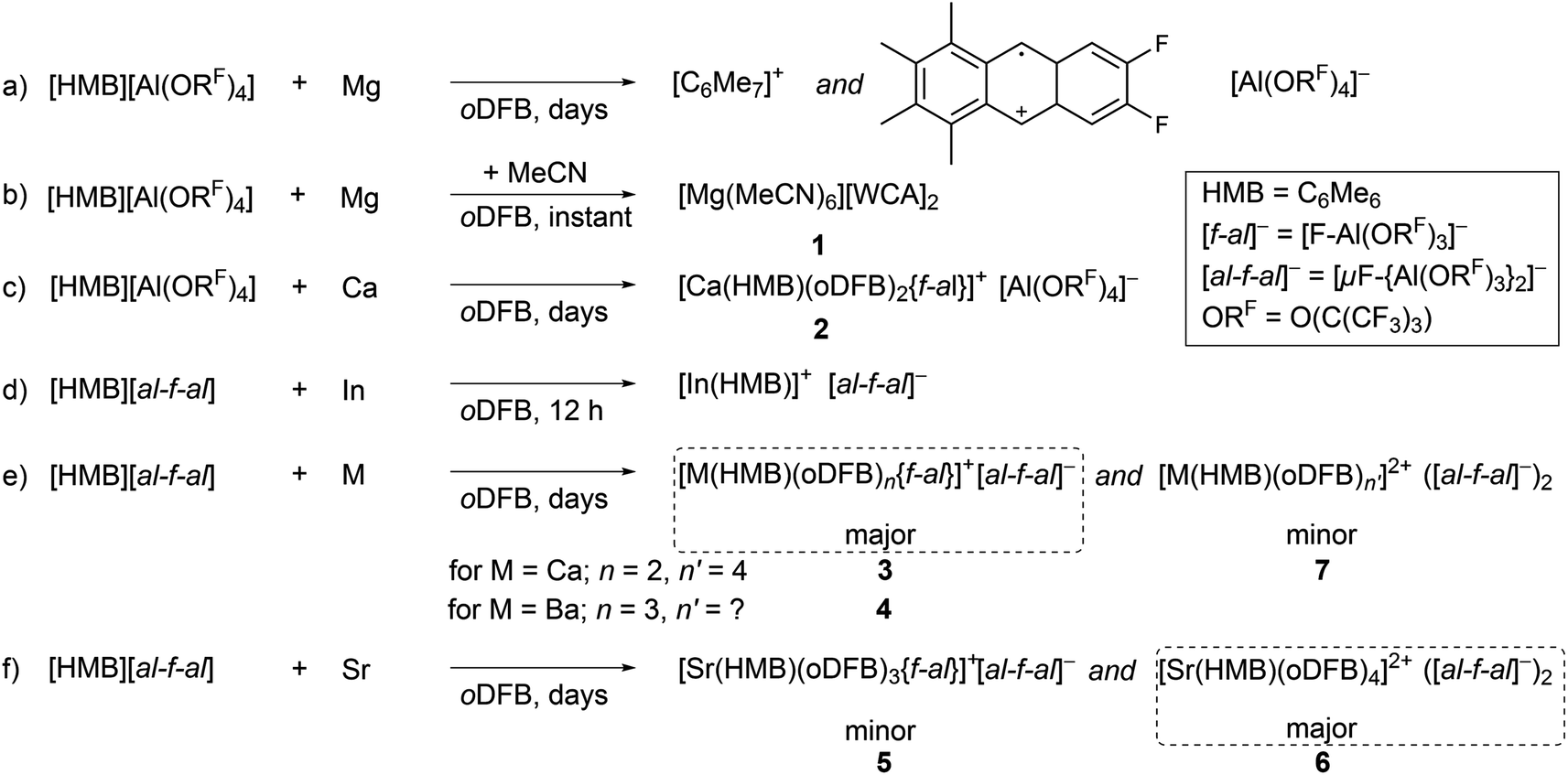 | ||
| Scheme 1 | ||
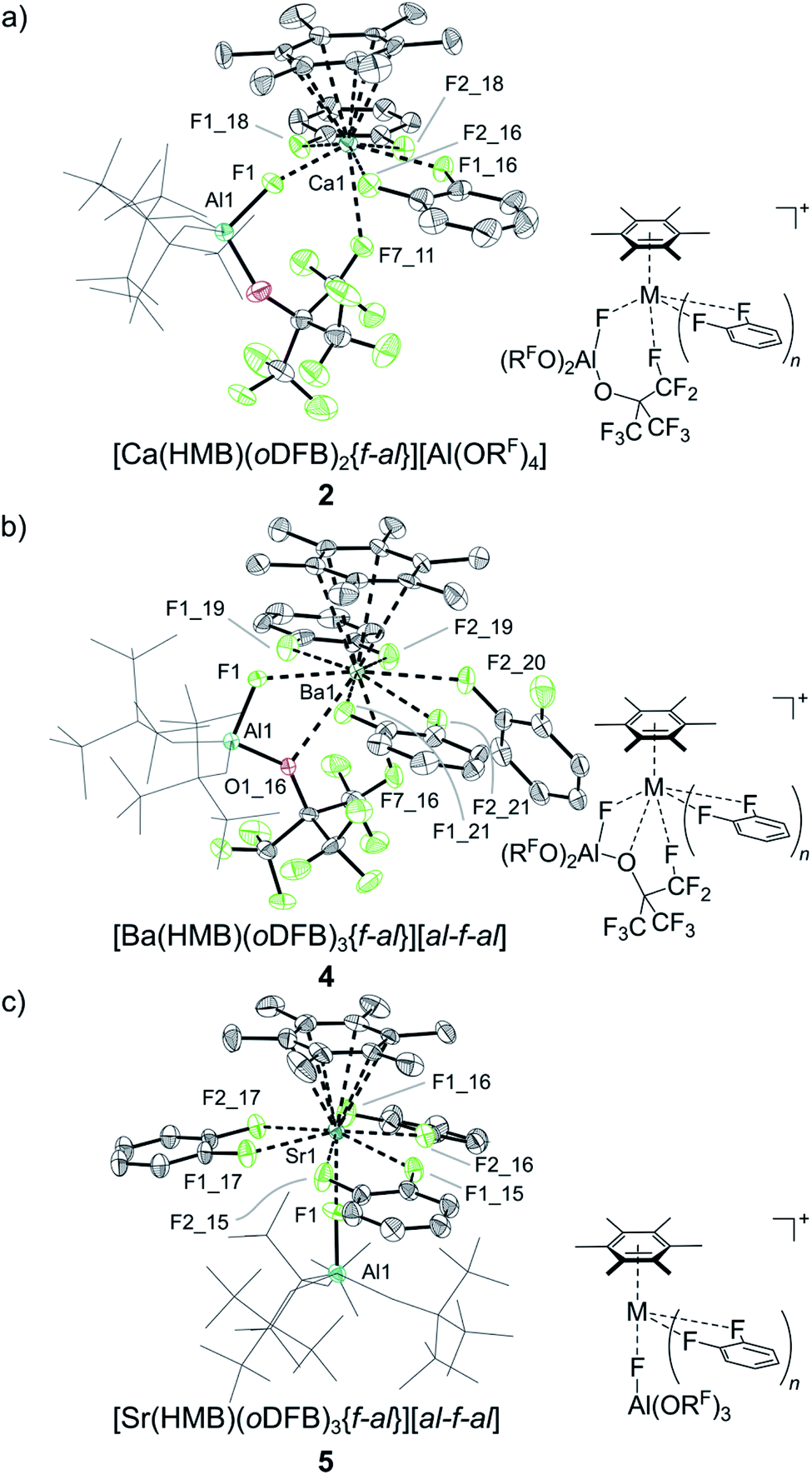
The reaction between less noble calcium and [HMB][Al(ORF)4] in oDFB proceeds via color loss over five days. By layering the filtrate of the reaction mixture with n-pentane, we obtained a crystalline material of the form [Ca(HMB)(oDFB)2{f-al}][Al(ORF)4] 2 ([f-al]− = [F–Al(ORF)3]−, see Scheme 1c, Fig. 1a & Table 1).
 | ||
| Fig. 1 Molecular structures of the cationic parts of (a) 2, (b) 3, and (c) 5 (due to disorder only the majority component of bound [f-al]− is shown for clarity). Counterions and protons are omitted and parts of [f-al]− are drawn as wireframes for clarity. Thermal displacement ellipsoids are shown at 50% probability. | ||
| M= | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| Ca | Ca | Ba | Sr | Sr | Ca | ||
| HMB | M–Crange | 2.82(1)–2.88(1) | 2.787(9)–2.906(9) | 3.134(7)–3.193(7) | 2.997(6)–3.016(6) | 2.997(3)–3.025(3) | 2.813(3)–2.916(3) |
| M–Cavg. | 2.84(1) | 2.843(9) | 3.170(7) | 3.006(5) | 3.015(3) | 2.872(3) | |
| oDFB | M–Frange | 2.415(7)–2.483(7) | 2.445(6)–2.503(6) | 2.791(4)–2.939(5) | 2.625(3)–2.703(3) | 2.595(2)–2.799(2) | 2.360(2)–2.565(2) |
| {0.17–0.20; Σ = 0.72} | {0.11–0.20; Σ = 0.83} | {0.15–0.20; Σ = 1.03} | {0.15–0.21; Σ = 1.35} | {0.14–0.25; Σ = 1.13} | |||
| M–Favg. | 2.441(7) | 2.471(6) | 2.865(4) | 2.661(3) | 2.683(2) | 2.466(2) | |
| C–Frange | 1.36(1)–1.39(1) | 1.376(3)–1.379(3) | 1.364(9)–1.389(9) | 1.362(5)–1.393(5) | 1.376(2)–1.386(2) | 1.371(3)–1.392(3) | |
| C–Favg. | 1.38(1) | 1.378(3) | 1.378(9) | 1.375(5) | 1.381(2) | 1.381(3) | |
| [f-al]− | M–F | 2.185(7) | 2.189(6) | 2.589(4) | 2.338(3) | — | — |
| {0.39} | {0.34} | {0.42} | |||||
| Al–F | 1.714(8) | 1.712(6) | 1.705(4) | 1.699(3) | — | — | |
| {0.64} | {0.65} | {0.66} | |||||
| M–O | — | 3.05(2) | 2.941(5) | — | — | — | |
| {0.05} | {0.17} | ||||||
| Al–Oavg. | 1.704(9) | 1.69(2) | 1.709(5) | 1.70(1) | — | — | |
| Al–OM bound | — | 1.77(2) | 1.769(5) | — | — | — |
The cationic part constitutes Ca in the +II oxidation state ion-paired with an anionic [f-al]− moiety, giving an overall monocationic complex. The [f-al]− fragment herein stems from a decomposition reaction of the [Al(ORF)4]− counterion, presumably proceeding via fluoride abstraction by the generated Ca2+ species, loss of OC4F8 epoxide and re-coordination to the liberated Al(ORF)3 Lewis acid – a well-documented degradation path of this anion with extreme electrophiles like small silylium ions.11,12 Half of the by electron transfer (ET) generated neutral HMB is η6-bound to the central Ca with a Ca arene distance of dCa-centroid = 2.459(6) Å (structures and metric data in Fig. 1 and Table 1). This is the shortest coordination of an isolated neutral arene reported in the literature (cf. [Ca(NacNac) (C6H6)][Al(ORF)4] dCa-centroid = 2.596 Å (ref. 13)). Moreover, it is in the range of Ca-pentamethylcyclopentadienide (Cp*) distances with the anionic [Cp*]− ligand in CaCp*(ODipp)(THF)3 (Dipp = 2,6-iPr-C6H3), (avg. dCa-centroid = 2.453 Å (ref. 14)) and only by 0.1 Å longer as in 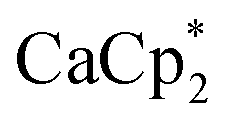 (avg. dCa-centroid = 2.351 Å (ref. 15)). The remaining space in the coordination sphere around Ca in 2 is filled by coordination of two difluorobenzene molecules with κ2-F–M coordination mode with rather short M–F contact distances ranging from dCa–F = 2.415(7) to 2.483(7) Å. Generally, the coordination of halobenzenes via their halogen atoms is scarce and κ2-F coordination of difluorobenzene was only structurally characterized in
(avg. dCa-centroid = 2.351 Å (ref. 15)). The remaining space in the coordination sphere around Ca in 2 is filled by coordination of two difluorobenzene molecules with κ2-F–M coordination mode with rather short M–F contact distances ranging from dCa–F = 2.415(7) to 2.483(7) Å. Generally, the coordination of halobenzenes via their halogen atoms is scarce and κ2-F coordination of difluorobenzene was only structurally characterized in  (M = Sc, Ti). This underlines the high Lewis acidity of the central calcium atom.16 The shortest M–F distance with dCa–F = 2.185(7) Å is formed with the coordinated [f-al]− moiety, with further M–F interactions with peripheral CF3 fluorine atoms of this anionic ligand (dCa–F = 2.620(8) Å). The Al–F and Al–O bonds (dAl–F = 1.714(8) & avg. dAl–O = 1.704(9) Å) are well in the range of the reported bond lengths for [f-al]−.12
(M = Sc, Ti). This underlines the high Lewis acidity of the central calcium atom.16 The shortest M–F distance with dCa–F = 2.185(7) Å is formed with the coordinated [f-al]− moiety, with further M–F interactions with peripheral CF3 fluorine atoms of this anionic ligand (dCa–F = 2.620(8) Å). The Al–F and Al–O bonds (dAl–F = 1.714(8) & avg. dAl–O = 1.704(9) Å) are well in the range of the reported bond lengths for [f-al]−.12
Switching to the fluoride-bridged anion [μF-{Al(ORF)3}2]−; reactions with Ca–Ba
Similar results have been obtained when reacting the heavier congener Ba with [HMB][al-f-al], yielding [Ba(HMB)(oDFB)3{f-al}][a-f-al] 4 (see Scheme 1e and Fig. 1b). Again HMB is η6-coordinated with a Ba-centroid distance of dBa-centroid = 2.850(3) Å (cf. 15 BaCp*X dBa-centroid = 2.752 Å;20 X = C(N(iPr)2)N(H)iPr). The cationic structure of 4 is closely related to that of 3, except for an additional, κ1-F bound oDFB (with Ba–F distances ranging from dBa–F = 2.791(4) Å to 2.939(5) Å) accounting for the larger metal atom. The [f-al] moiety is bound similarly to 3 with a short Ba–F contact distance of dBa–F = 2.589(4) Å, as well as coordination to oxygen on one of the ORF sidearms (dBa–O = 2.941(5) Å). It is noteworthy that the bound [f-al]− moiety in 3 and 4 does not stem from fluoride abstraction but rather dissociation of the anion. This may be understood as the result of a competition between the two Lewis acids AE2+ and Al(ORF)3 for the Lewis basic anionic [f-al]− fragment. We note that the dissociation of [al-f-al]− to give the coordinated [f-al]− anion and the free Lewis acid Al(ORF)3 may be supported by complexation of the latter with oDFB giving the known adduct (oDFB)Al(ORF)3.21 Therefore, this route provides a controlled synthesis of complexes 3 and 4.
15 BaCp*X dBa-centroid = 2.752 Å;20 X = C(N(iPr)2)N(H)iPr). The cationic structure of 4 is closely related to that of 3, except for an additional, κ1-F bound oDFB (with Ba–F distances ranging from dBa–F = 2.791(4) Å to 2.939(5) Å) accounting for the larger metal atom. The [f-al] moiety is bound similarly to 3 with a short Ba–F contact distance of dBa–F = 2.589(4) Å, as well as coordination to oxygen on one of the ORF sidearms (dBa–O = 2.941(5) Å). It is noteworthy that the bound [f-al]− moiety in 3 and 4 does not stem from fluoride abstraction but rather dissociation of the anion. This may be understood as the result of a competition between the two Lewis acids AE2+ and Al(ORF)3 for the Lewis basic anionic [f-al]− fragment. We note that the dissociation of [al-f-al]− to give the coordinated [f-al]− anion and the free Lewis acid Al(ORF)3 may be supported by complexation of the latter with oDFB giving the known adduct (oDFB)Al(ORF)3.21 Therefore, this route provides a controlled synthesis of complexes 3 and 4.
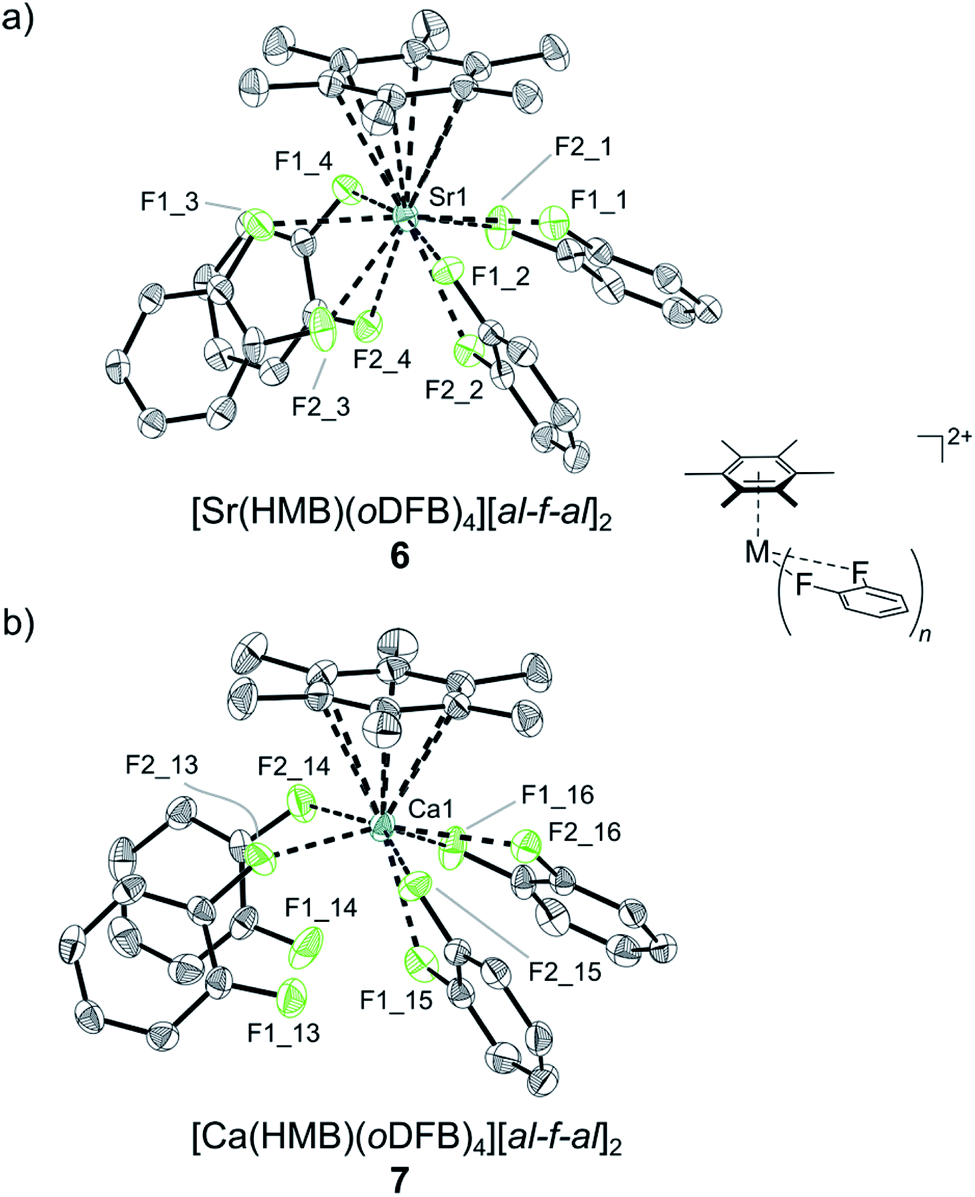 | ||
| Fig. 2 Molecular structures of the cationic parts of (a) 6 and (b) 7. Counterions and protons are omitted for clarity. Thermal displacement ellipsoids are shown at 50% probability. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 3 and 4. However, in all collected samples a distinct excess of the fluoride-bridged anion was detected ([f-al]−:[al-f-al]−; 1:2.1 for 3 and 1:1.7 for 4), which is not accounted for by cationic trace impurities from HMB radical degradation reactions. NMR spectra of isolated crystalline materials from reactions with both Ca and Ba showed similar outcomes. Furthermore, the sum of both anionic moieties and HMB, as determined by referencing to oDFB as an internal standard, shows a 2:1 ratio. Since free HMB is soluble in both oDFB and pentane, and the respective 13C resonances of the quaternary ring carbon in HMB show a significant downfield shift for all samples by Δδ = 15 ppm compared to those of free HMB, it is likely that the excess fluoride bridged anion is part of a second dicationic HMB complex containing two intact fluoride bridged anions. Yet, all attempts to selectively synthesize or crystallize the minor products have failed and only by a serendipitous finding could a dicationic Ca complex, [Ca(HMB)(oDFB)4][al-f-al]27 (see Scheme 1e and Fig. 2b), be isolated from the layering ampoule showing a similar coordination to the already-described 6 (for bond metric data see Table 1).
:1 for 3 and 4. However, in all collected samples a distinct excess of the fluoride-bridged anion was detected ([f-al]−:[al-f-al]−; 1:2.1 for 3 and 1:1.7 for 4), which is not accounted for by cationic trace impurities from HMB radical degradation reactions. NMR spectra of isolated crystalline materials from reactions with both Ca and Ba showed similar outcomes. Furthermore, the sum of both anionic moieties and HMB, as determined by referencing to oDFB as an internal standard, shows a 2:1 ratio. Since free HMB is soluble in both oDFB and pentane, and the respective 13C resonances of the quaternary ring carbon in HMB show a significant downfield shift for all samples by Δδ = 15 ppm compared to those of free HMB, it is likely that the excess fluoride bridged anion is part of a second dicationic HMB complex containing two intact fluoride bridged anions. Yet, all attempts to selectively synthesize or crystallize the minor products have failed and only by a serendipitous finding could a dicationic Ca complex, [Ca(HMB)(oDFB)4][al-f-al]27 (see Scheme 1e and Fig. 2b), be isolated from the layering ampoule showing a similar coordination to the already-described 6 (for bond metric data see Table 1).
Since with identical reaction procedures only Sr2+ complex 6 was easily isolable as the major product, we were intrigued to understand the underlying thermodynamics, as this suggests an equilibrium state between dicationic and monocationic [f-al]− bound complexes. However, the generation of [f-al]− from [al-f-al]− inevitably generates one equivalent of Al(ORF)3, the fate of which is unclear. Its fluoro- and difluorobenzene adducts are known and rather soluble in oDFB, but are only marginally stable at room temperature and were not clearly assignable by NMR spectroscopy.21 Attempts to generate the dications by abstraction of [f-al]− from 4 with the stable Al(ORF)3 equivalent and strong Lewis acid Me3Si–F–Al(ORF)3 (fluoride ion affinity FIA = 458 kJ mol−1) did not increase the [al-f-al]− content, underlining the tight ion-pairing of the anionic [f-al]− moiety. We conclude that the concentration of unbound [f-al]− must be very minimal, making a true equilibrium and thus the reaction of free [f-al]− with Me3Si–F–Al(ORF)3 to give [al-f-al]− and Me3SiF unlikely. Therefore, the formation of the monocationic complexes is irreversible and apparently only slightly favored over the formation of the dication in 7 and maybe 6. Furthermore, the resonances in 19F-NMR spectra attributable to M-bound [f-al]− moieties at about −130 to −152 ppm are distinctly low field-shifted compared to those of “free” [f-al]− (e.g. in [CPh3][f-al], δ = −186.1 ppm; in Ag[f-al], δ = −189.9 ppm) and are more comparable to that in Me3Si–F–Al(ORF)3 (δ = −156.2 ppm), suggesting tight bonding of this moiety. Additionally, the binding of the anionic [f-al]− differs between the isolated complexes 3, 4 and 5 as the 19F-NMR chemical shift of aluminum bound fluorine in [f-al]− is distinctly low-field shifted by Δδ = 20 ppm in the barium complex (δ = −130.3 ppm) 4, compared to 3 (δ = −151.5 ppm) and 5 (δ = −149.3 ppm). This suggests a very tight bonding interaction of this moiety in 4.
DFT calculations
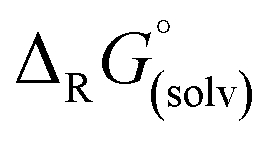 is small and lies within the error of the method but is apparently not linear when descending down the group. For strontium, the formation of the monocationic complex is least favorable, while barium again shows an increased tendency for this reaction. As the fate of the Al(ORF)3 generated during the formation of the [f-al]− bound complexes is unknown, the respective known21oDFB adduct was included to provide a balanced equation.‡ Thus, it appears that all compounds are thermodynamic products and only Sr dication 6 is the kinetic product that is metastable at RT in oDFB solution against further reaction to the [f-al]− coordinated product.
is small and lies within the error of the method but is apparently not linear when descending down the group. For strontium, the formation of the monocationic complex is least favorable, while barium again shows an increased tendency for this reaction. As the fate of the Al(ORF)3 generated during the formation of the [f-al]− bound complexes is unknown, the respective known21oDFB adduct was included to provide a balanced equation.‡ Thus, it appears that all compounds are thermodynamic products and only Sr dication 6 is the kinetic product that is metastable at RT in oDFB solution against further reaction to the [f-al]− coordinated product. | (3) |
To evaluate the reaction energies for the exchange of the X-type ligand [f-al]− between the different mono- and dicationic AE complexes, they were investigated according to eqn (4a–c).
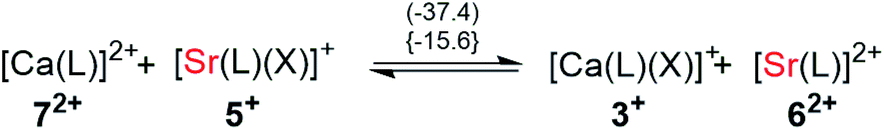 | (4a) |
 | (4b) |
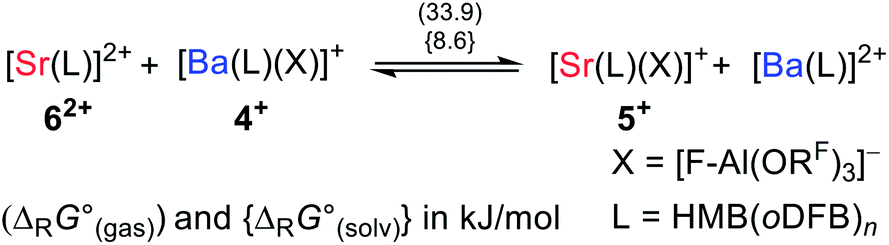 | (4c) |
Abstraction of [f-al]− by [M(HMB)(oDFB)4]2+ from [M′(HMB)(oDFB)n{f-al}]+ is exergonic for M = Ca and M′ = Sr (−15.6 kJ mol−1; eqn (4a)) and Ba (−7.7 kJ mol−1; eqn (4b)), while the abstraction for M = Sr and M′ = Ba is counterintuitively endergonic (+8.6 kJ mol; eqn (4c)). Although all of the obtained reaction energies are rather close, a trend supporting the experimental findings is visible. The increased stability of the monocationic Ba complex 3+ in comparison to the monocationic Sr complex 5+ causes the abstraction of [f-al]− by 72+ to be more exergonic for the latter, and the abstraction by 62+ to be endergonic for the former. These findings are also backed by NMR spectroscopic investigations as described above, showing stronger coordination by [f-al]− in 4. To understand the stability of isolated dicationic complexes 7 and especially 6, a dismutation of the strontium complex 6 to hypothetical [Sr(HMB)2]2+ and [Sr(oDFB)8]2+ was modeled (see eqn (5)). This transformation was found to be unfavorable by 176 kJ mol−1 in ΔRG° 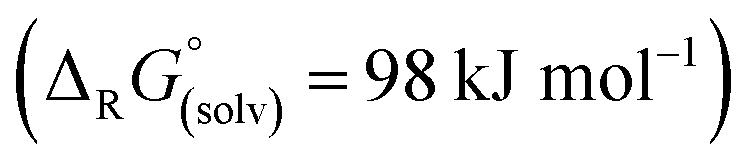 , underlining the stability of this unique coordination environment.
, underlining the stability of this unique coordination environment.
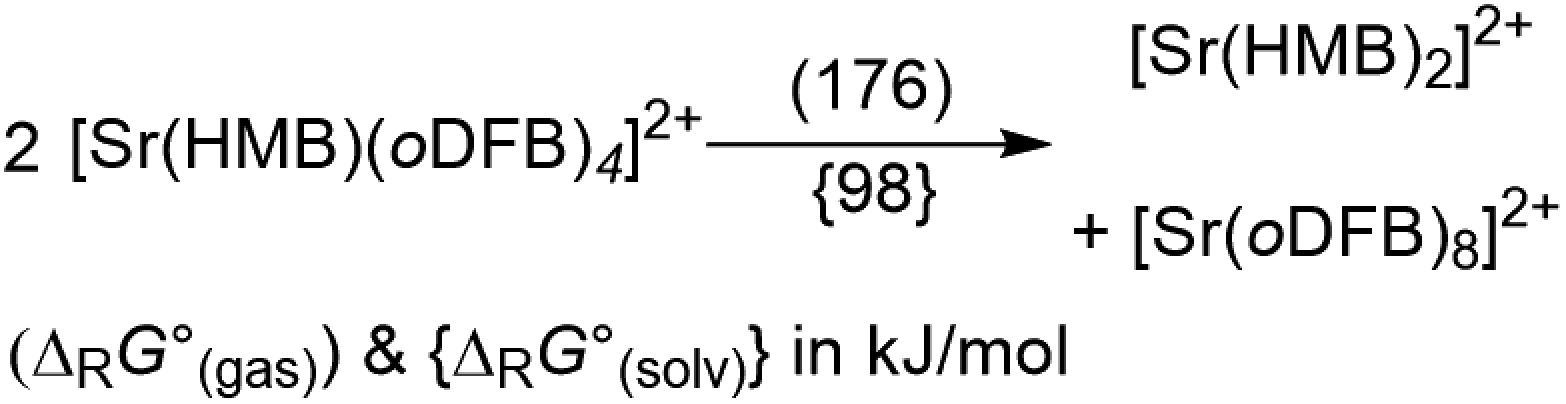 | (5) |
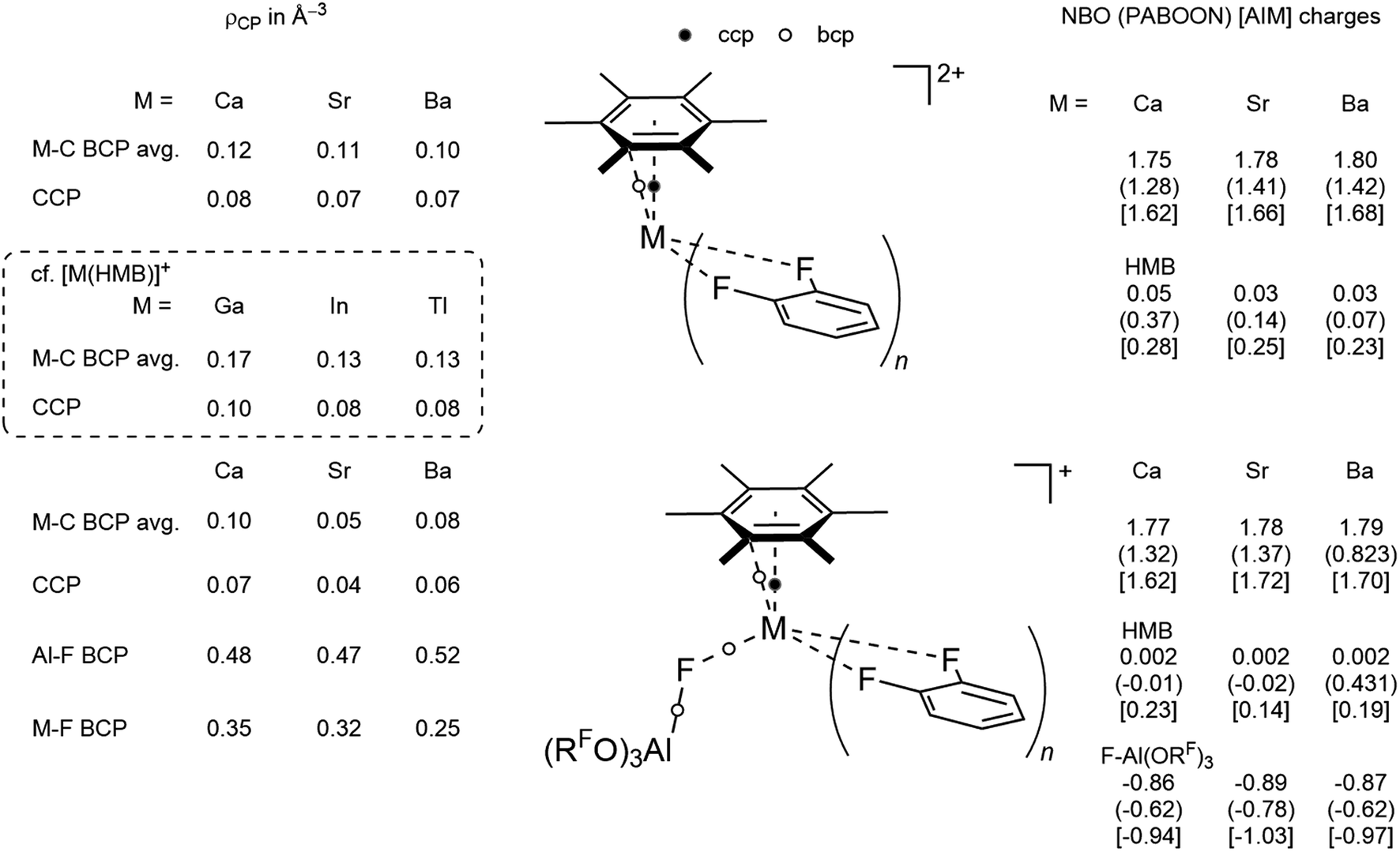 | ||
| Scheme 2 Population analysis by NBO,24 PABOON25 and AIM26 and electron density at bond and cluster critical points. | ||
For the monocationic complexes, the same behavior is observed for Ca and Sr in 3+ and 5+. However, the positive charge on the central Ba in 4+ decreases noticeably for both PABOON and AIM compared to the Sr complex 5+, while the charge on bound HMB increases, suggesting more π-donation by HMB in this complex, in agreement with the best 5d acceptor capacity of Ba2+. AIM analysis of the electron density residing at critical points reveals an increase for the M–C BCP and CCP located in the center of the Ba(HMB) subunit with decreasing electron density on the BCP of the M–F bond. The presence of the CCP in the M(HMB) subunit allows for its description as a nido-cluster analogous to the reported cationic group 13 HMB complexes [M(HMB)]+ (ρCP(CCP) = 0.10 (Ga), 0.08 (In) and 0.08 e Å−3 (Tl)).
Investigation of the Lewis acidity
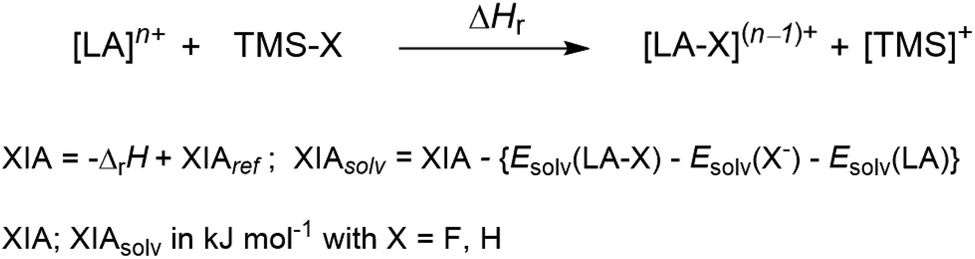 | (6) |
All of the dicationic complexes 72+, 62+ and [Ba(HMB)(oDFB)4]2+ show expectedly very high FIAs in both the gas phase (923–879 kJ mol−1) and in solution (316–283 kJ mol−1), comparable only to those of a recently reported also dicationic phosphorandiylium [R3P]2+ species (FIA = 904 kJ mol−1; FIAsolv = 318 kJ mol−1).31 Although the gas phase values exceed those of B(C6F5)3 (FIA = 456 kJ mol−1; FIAsolv = 240 kJ mol−1) and SbF5 (FIA = 494 kJ mol−1; FIAsolv = 330 kJ mol−1) – allowing for their declaration as Lewis superacids – their solvated FIAs in DCM are considerably lower. However, they are still much higher than that of B(C6F5)3 and comparable in size to that of SbF5 or Al(ORF)3 (FIA = 539 kJ mol−1; FIAsolv = 323 kJ mol−1). These solvated values underline the above-mentioned competition between AE2+ and Al(ORF)3 around the [f-al]− moiety.
The calculated XIAs decrease when descending down group 2, although non-linearly. The difference between Sr and Ba containing complexes 5 & 4 is in both cases smaller than the gap between Ca and Sr containing complexes 3 & 5. By contrast, the calculated HIAs are not as pronounced as the FIA values and especially the solvated HIAs are rather small and in some cases even exothermic. This is in line with the Pearson hard character of the cations.
The experimental findings together with data gathered from DFT analysis allow for interpretation of the stability of the complexes found. The monocationic Ca complex is predominantly formed over 7 because of the very high Lewis acidity of the smallest central atom in this series. When going down the group this effect should lessen, in line with the isolation of the almost exclusively formed dicationic strontium complex 6. A similar reactivity would be expected for the even less polarizing larger barium. However, here again the monocationic complex 4 is predominantly formed. NMR and DFT analysis revealed tighter bonding of the [f-al]− moiety and higher stability of 4, respectively. The decreased charge on the central barium suggests higher covalent character in Ba–F–Al and especially Ba–HMB bonds. However, only the latter is backed by electron density analysis by AIM theory.
Conclusion
By use of the ligand-forming oxidant [HMB][WCA] we were able to generate a series of mono- and dicationic AE metal arene complexes [(η6-HMB)M(oDFB)n{f-al}]+ (M = Ca, Sr, Ba) and [(η6-HMB)M(oDFB)4]2+ (M = Ca, Sr) by direct oxidation of the elemental metals. Thus far, no evidence for generation of the single oxidation products, the respective AE(+I) complexes, was observed – even at lower temperature. It is unclear, if the generation of the respective AE(+II) is due to overoxidation of AE(+I) species in solution or its disproportionation. For oxidative synthesis of such low valent AE complexes further studies concerning the type and sterics of the employed ligand and chosen oxidant have to be performed. The highly Lewis acidic AE metal centers show a polarized soft interaction with the η6-coordinated HMB, while the rest of the coordination sphere is filled with hard interactions with coordinated oDFB and in the case of the monocationic complexes [f-al]−. The modeled dismutation of dicationic 6 to hypothetical [Sr(HMB)2]2+ and [Sr(oDFB)8]2+ was shown to be unfavorable in ΔRG°, underlining the stability of this unique mixed hard/soft coordination environment. The so generated dicationic AE arene complexes are unprecedented and show high and hard Lewis acidities in line with other Lewis superacidic, neutral,32 mono-33 and dicationic31,34 main-group Lewis acids published in recent years.The coordination by weak neutral σ/π-donors to generate other dicationic complexes is currently explored. However, orienting reactions to explore the coordination chemistry of isolable [(η6-HMB)Sr(oDFB)4]2+6 has furthermore shown the sensitivity of this complex; disturbance by strong σ-donors (e.g. PPh3) has shown partial rearrangement of the counterion to form 5 [(η6-HMB)Sr(oDFB)3{f-al}]+ among other products. Yet, the thereby or directly generated monocationic complexes [(η6-HMB)M(oDFB)n{f-al}]+ (M = Ca, Sr, Ba) may open interesting reactivities due to stabilization – in the presence of stronger donors – by the potentially hemi-labile [f-al]− moiety bearing an ideal platform for application of this class of compounds for Lewis acid catalyzed transformations, which will be addressed in the future.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg, the Deutsche Forschungsgemeinschaft (KR2046/37-1) and the Carl-Zeiss-Stiftung. We thank Dr H. Scherer and F. Bitgül for performing NMR measurements, Dr T. Ludwig for measuring pXRD, Dr D. Kratzert for help with scXRD measurements, and the Magres Core Facility for instrument support.References
- (a) K. Ziegler, H. Froitzheim-Kühlhorn and K. Hafner, Chem. Ber., 1956, 89, 434 CrossRef CAS; (b) R. Zerger and G. Stucky, J. Organomet. Chem., 1974, 80, 7 CrossRef CAS.
- L. Bonomo, E. Solari, R. Scopelliti and C. Floriani, Chem.–Eur. J., 2001, 7, 1322 CrossRef CAS.
- M. Lindsjö, A. Fischer and L. Kloo, Dalton Trans., 2010, 39, 1467 RSC.
- (a) A. Friedrich, J. Pahl, H. Elsen and S. Harder, Dalton Trans., 2019, 48, 5560 RSC; (b) J. Pahl, H. Elsen, A. Friedrich and S. Harder, Chem. Commun., 2018, 54, 7846 RSC; (c) J. Pahl, A. Friedrich, H. Elsen and S. Harder, Organometallics, 2018, 37, 2901 CrossRef CAS.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685 RSC.
- (a) M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC; (b) J. Pahl, T. E. Stennett, M. Volland, D. M. Guldi and S. Harder, Chem.–Eur. J., 2019, 25, 2025 CrossRef CAS PubMed.
- J. Penafiel, L. Maron and S. Harder, Angew. Chem., Int. Ed., 2015, 54, 201 CrossRef CAS PubMed.
- S. Brand, H. Elsen, J. Langer, W. A. Donaubauer, F. Hampel and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 14169 CrossRef CAS PubMed.
- (a) M. D. Anker and M. S. Hill, in Encyclopedia of inorganic and bioinorganic chemistry, ed. R. A. Scott, Wiley, Chichester, 2012, vol. 475, pp. 1–23 Search PubMed; (b) H. Bauer, M. Alonso, C. Fischer, B. Rösch, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 15177 CrossRef CAS PubMed; (c) H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. de Proft and S. Harder, Nat. Catal., 2018, 1, 40 CrossRef CAS; (d) H. Bauer, K. Thum, M. Alonso, C. Fischer and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 4248 CrossRef CAS PubMed; (e) B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396 CrossRef PubMed; (f) A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351 CrossRef CAS; (g) M. S. Hill, M. F. Mahon, A. S. S. Wilson, C. Dinoi, L. Maron and E. Richards, Chem. Commun., 2019, 55, 5732 RSC; (h) D. Mukherjee, T. Höllerhage, V. Leich, T. P. Spaniol, U. Englert, L. Maron and J. Okuda, J. Am. Chem. Soc., 2018, 140, 3403 CrossRef CAS PubMed; (i) D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367 CrossRef CAS PubMed; (j) D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 310 CrossRef CAS PubMed.
- M. Schorpp, S. Rein, S. Weber, H. Scherer and I. Krossing, Chem. Commun., 2018, 54, 10036 RSC.
- (a) I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721 CrossRef CAS; (b) M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem.–Eur. J., 2014, 20, 1218 CrossRef CAS PubMed.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058 RSC.
- L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684 RSC.
- M. J. Harvey, D. J. Burkey, S. C. Chmely and T. P. Hanusa, J. Alloys Compd., 2009, 488, 528 CrossRef CAS.
- R. A. Williams, T. P. Hanusa and J. C. Huffman, Organometallics, 1990, 9, 1128 CrossRef CAS.
- M. W. Bouwkamp, P. H. M. Budzelaar, J. Gercama, I. Del Hierro Morales, J. de Wolf, A. Meetsma, S. I. Troyanov, J. H. Teuben and B. Hessen, J. Am. Chem. Soc., 2005, 127, 14310 CrossRef CAS PubMed.
- (a) A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem.–Eur. J., 2004, 10, 5041 CrossRef CAS PubMed; (b) A. Martens, M. Kreuzer, A. Ripp, M. Schneider, D. Himmel, H. Scherer and I. Krossing, Chem. Sci., 2019, 10, 2821 RSC; (c) I. Krossing and I. Raabe, Chem.–Eur. J., 2004, 10, 5017 CrossRef CAS PubMed.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC.
- I. D. Brown, J. Appl. Crystallogr., 1996, 29, 479 CrossRef CAS.
- T. M. Cameron, C. Xu, A. G. Dipasquale and A. L. Rheingold, Organometallics, 2008, 27, 1596 CrossRef CAS.
- A. Kraft, N. Trapp, D. Himmel, H. Böhrer, P. Schlüter, H. Scherer and I. Krossing, Chem.–Eur. J., 2012, 18, 9371 CrossRef CAS PubMed.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659 CrossRef PubMed.
- X. Wu, L. Zhao, J. Jin, S. Pan, W. Li, X. Jin, G. Wang, M. Zhou and G. Frenking, Science, 2018, 361, 912 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- C. Ehrhardt and R. Ahlrichs, Theor. Chim. Acta, 1985, 68, 231 CrossRef CAS.
- (a) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 2, 799 RSC.
- L. Greb, Chem.–Eur. J., 2018, 24, 17881 CrossRef CAS PubMed.
- (a) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765 RSC; (b) E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712 CrossRef CAS; (c) V. Fasano, J. H. W. LaFortune, J. M. Bayne, M. J. Ingleson and D. W. Stephan, Chem. Commun., 2018, 54, 662 RSC; (d) J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 5407 CrossRef CAS PubMed; (e) E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem.–Eur. J., 2013, 19, 2462 CrossRef CAS PubMed; (f) J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC; (g) B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564 CrossRef CAS PubMed; (h) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS; (i) C. B. Caputo, D. Winkelhaus, R. Dobrovetsky, L. J. Hounjet and D. W. Stephan, Dalton Trans., 2015, 44, 12256 RSC.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139 CrossRef CAS PubMed.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717 CrossRef CAS PubMed.
- (a) H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984 CrossRef CAS PubMed; (b) P. Weis, D. C. Röhner, R. Prediger, B. Butschke, H. Scherer, S. Weber and I. Krossing, Chem. Sci., 2019, 10, 10779 RSC.
- J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Chem, 2018, 4, 2699 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, procedures, weights, 1D- and 2D-NMR spectra, IR spectra and powder diffraction patterns of the reactions, and details of the quantum chemical calculations together with crystallographic details. CCDC 1969777–1969784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06254h |
| ‡ Note that oDFB·Al(ORF)3 is unstable in solution at RT over prolonged periods of time as necessary for the syntheses. |
| This journal is © The Royal Society of Chemistry 2020 |