
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction of chloroauric acid with histidine in microdroplets yields a catalytic Au–(His)2 complex†
Kai
Luo‡
a,
Jia
Li‡
a,
Yufei
Cao
b,
Chengyuan
Liu
a,
Jun
Ge
 b,
Hao
Chen
c and
Richard N.
Zare
*a
b,
Hao
Chen
c and
Richard N.
Zare
*a
aDepartment of Chemistry, Fudan University, Shanghai 200438, China. E-mail: rnz@fudan.edu.cn
bDepartment of Chemical Engineering, Key Lab for Industrial Biocatalysis, Ministry of Education, Tsinghua University, Beijing 100084, China
cDepartment of Chemistry and Environmental Science, New Jersey Institute of Technology, Newark, NJ 07102, USA
First published on 31st January 2020
Abstract
An aqueous solution containing histidine (His, 100 μM) and chloroauric acid (HAuCl4, 10 μM) is electrosprayed (−4.5 kV) from a capillary (50 μm in diameter) with N2 nebulizing gas (120 psi). The resulting microdroplets entered a mass spectrometer with a 2 cm flight path. The mass spectrum recorded in negative ion mode showed several peaks including the Au5 nanocluster with the major one being [Au + 2His–2H]−, which is a catalytically active species. The reaction time was less 1 ms, and the yield of [Au + 2His–2H]− was 76%. In contrast, the bulk reaction for the same concentration run at room temperature for 2 h did not produce this species but instead formed Au10 nanocluster. When a solution of water and acetonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing indoline (100 mM) and the phenylacetylene (200 mM) as well as histidine and chloroauric acid at the same concentrations as above was electrosprayed, the mass spectrum showed the formation of the intermediate [Au + 2His + phenylacetylene + H]+. Upon collecting the microdroplets, the 4-methyl-4,6-diphenyl-1,2-dihydro-4H-pyrrolo[3,2,1-ij] quinolone product was observed by 1H nuclear magnetic resonance and liquid chromatography with a yield of 44%. The microdroplet synthesis using the Au–(His)2 complex as a catalyst was scaled up using room-temperature ultrasonic nebulization to produce the product at the rate of 35 mg min−1, which is semi-preparative and demonstrates the promise of using microdroplet reactions for chemical synthesis.
:1) containing indoline (100 mM) and the phenylacetylene (200 mM) as well as histidine and chloroauric acid at the same concentrations as above was electrosprayed, the mass spectrum showed the formation of the intermediate [Au + 2His + phenylacetylene + H]+. Upon collecting the microdroplets, the 4-methyl-4,6-diphenyl-1,2-dihydro-4H-pyrrolo[3,2,1-ij] quinolone product was observed by 1H nuclear magnetic resonance and liquid chromatography with a yield of 44%. The microdroplet synthesis using the Au–(His)2 complex as a catalyst was scaled up using room-temperature ultrasonic nebulization to produce the product at the rate of 35 mg min−1, which is semi-preparative and demonstrates the promise of using microdroplet reactions for chemical synthesis.
Introduction
Metal-based N-heterocyclic carbenes (NHCs) have found remarkable use as pre-catalysts and active intermediates in many catalytic reactions.1–4 The most important challenge is to capture effectively pre-catalysts and active intermediates and achieve maximum catalytic efficiency.5–7 This problem has been reviewed by Hahn,8 Kuhn,9 Braunstein,10 and Tamm11 from 2017 to 2019. In order to stabilize the metal-NHCs, it is usually necessary to find stable electron-donating ligands. Although there are some special ligands that have proved useful in NHC organic catalysis,12,13 the classical NHC, mainly derived from substituted imidazole rings,14 fails to be effective. Water is considered to be a green solvent and it is often employed as a reaction medium, but it is frequently considered the “natural enemy” of organometallic species. Consequently, most NHC reactions have been carried out in dry organic solvents.15 We present here an alternative approach. We use water microdroplets containing imidazole rings coordinated to gold cations to explore a new catalytic metal-based NHC synthesis procedure. We find that this approach has many advantages.Microdroplet chemistry has emerged as a unique platform for studying and performing chemical reactions.16–22 The ESI/ESSI as the main setup to generate charged microdroplets is usually accompanied by special redox reactions, which have been used for some synthesis reaction and nanomaterials.23,24 Microdroplets as microreactors combined with on-line MS have been used to capture and identify transient intermediates of organic reactions, and microdroplet chemistry has been broadly applied in the field of bioanalytical detection.25–27 Microdroplets show a strikingly different environment for reactions than that of the corresponding bulk phase.28,29 Recently, some studies showed that reasons for the acceleration of microdroplet reactions are evaporation,30–34 the distribution of pH,35 size change, surface polarization, reagent confinement,36 high surface-to-volume ratio, and redox reactions at or near the surface of the micro-droplet.30,37 There have some reports about the some special catalytic reaction in the microdroplet reaction, like Suzuki reaction and Suzuki–Miyaura coupling reaction.38,39 However, for these results to have practical value, the microdroplet reactions need to be scaled up in practice to improve the reaction yield. Some factors, like size, with or without high voltage, solvent, and lasting reaction time of the microdroplet will have a direct effect on the high yields.40 Recently, there have some technology explored for increasing the yield of the product and scaling-up the microdroplet reaction.21,38,40–42 Heated ultrasonic nebulization (HUN) has been used successfully for this purpose in our group.43 We report here the use of ultrasonic nebulization to make a scaled-up preparation the catalytic Au–(His)2 complex.
Methods
Chemicals
Gold(III) chloride trihydrate (HAuCl4·3H2O), gold trichloride (AuCl3), histidine (His), glutathione (GSH), triphenylphosphine gold chloride (TPPGC), indoline, and benzyl acetylene were purchased from Sigma-Aldrich (St. Louis, MO). Water was purified using a Milli-Q system (Millipore, Bedford, MA, USA) with a resistivity of 18.2 MΩ cm and used throughout.Electrosonic spray ionization (ESSI)
The sample solution was sprayed from the tip of a fused silica capillary (148 μm o.d., 50 μm i.d., Polymicro Technologies, China) of a homemade sprayer, which is assisted by a nebulizing gas of dry N2 with a pressure of 120 psi (Fig. 1a). The sample solution containing histidine (100 μM) and chloroauric acid (10 μM) was pushed by a micropump (KD Scientific) through a 1 mL syringe (Hamilton), which was connected by a fused silica capillary between the needle and the ESSI capillary emitter (Fig. 1b). Different high voltages were applied on the needle to form microdroplets at the tip of the homemade ESSI. The flow rate was precisely set to 5 μL min−1 by the micropump. By adjusting different distances between the tip of sprayer and the inlet of a high-resolution Q-TOF mass spectrometer (Synapt G2, Waters, USA), the MS spectrum was recorded in the mass range of 100–2000 Da. The MS inlet capillary was always maintained at 150 °C and the capillary voltage at 0 V. No other nebulizing gases were used when performing microdroplet reactions.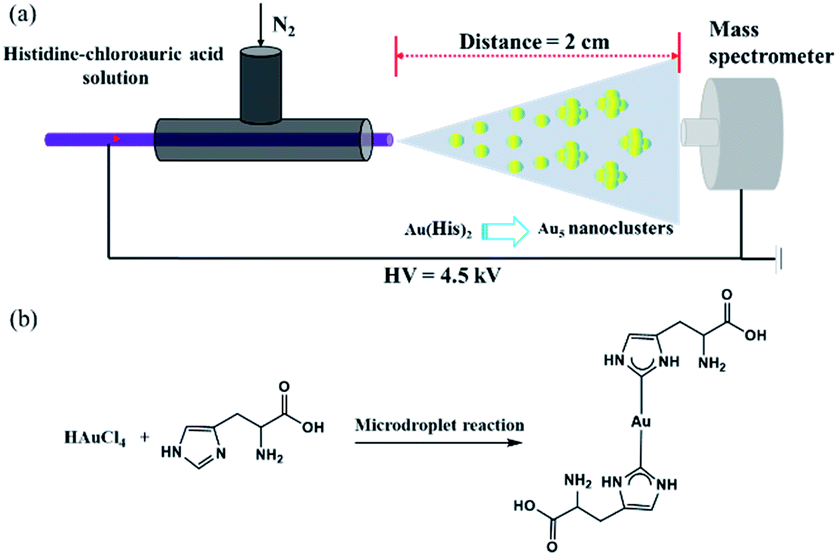 | ||
| Fig. 1 (a) Schematic of the experimental apparatus for the online microdroplet reaction coupled with mass spectrometry (microdroplet-MS) with N2 gas pressure of 120 psi and high voltage of −4.5 kV. The travel distance of the microdroplets from the spray source to the heated inlet of the mass spectrometer is 2 cm. (b) Schematic of the microdroplet reaction with histidine (100 μM) and chloroauric acid (10 μM). | ||
Ultrasonic nebulization microdroplets
The scaled-up preparation of Au–(His)2 nanocluster from ultrasonic nebulization microdroplet reaction was carried out using a homemade ultrasonic nebulizer (Fig. 2a). The aqueous solution was injected into the homemade glass nebulization cell, which consisted of a cylindrical upper part (I.D. 35 mm, height 90 mm) and lower part (I.D. 22 mm, height 15 mm) and a thin polyethylene membrane covering the bottom of the nebulization cell to transport ultrasonic waves. When the ultrasonic nebulizer (Model 402AI, Yuwell Medical Equipment & Supply Corp., Suzhou, China) is switched on, the reactant solution in the nebulization cell can be atomized into a large amount of microdroplets. The working frequency of the transducer of the ultrasonic nebulizer is 1.7 MHz ± 10%. The distance between the cover layer of nebulization cell and the water surface was set to 2 cm to ensure maximum efficiency of reagent microdroplet formation in the nebulization cell. The solution of HAuCl4 (10 mM, 100 μL) and histidine (100 mM, 100 μL) was first injected into the UN cell containing a 900 μL water solution for a reaction time of 12 min to generate Au–(His)2 complex. Then, a solution of phenylacetylene (200 mM, 100 μL) and indoline (100 mM, 100 μL) in acetonitrile solution was added to the same glass nebulization cell, which contained the in situ generation of Au–(His)2, for further ultrasonic nebulization to cause the in situ catalysis of the enamine reaction (Fig. 2b).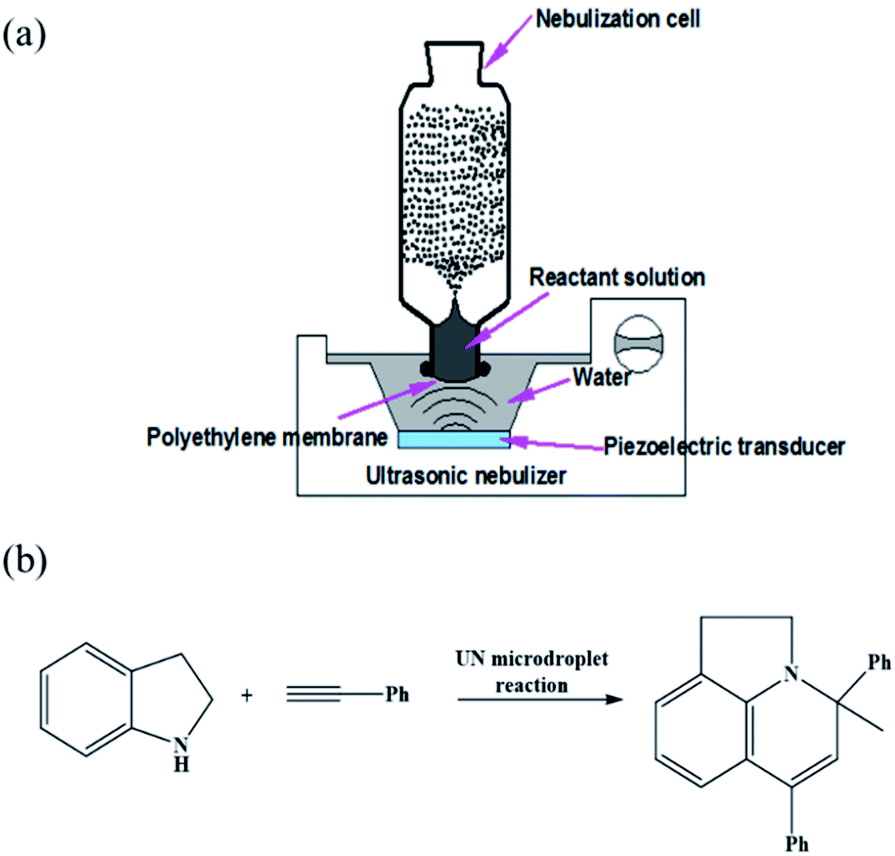 | ||
| Fig. 2 (a) Schematic of the experimental apparatus for the ultrasonic nebulization microdroplet reaction. (b) Schematic of the reaction for studying the catalytic activity of Au–(His)2 for the solution of phenylacetylene (200 mM, 100 μL) and indoline (100 mM, 100 μL) in acetonitrile by ultrasonic nebulization. | ||
Results and discussion
Microdroplet reaction for Au–(His)2
We find that the aqueous microdroplet reaction of 10 μM chloroauric acid (HAuCl4) and 100 μM histidine (C6H9N3O2) generates a transient intermediate in which Au bridges two histidine ligands, which we call Au–(His)2. Some key ions were identified in negative mode of MS (Fig. 3a), including [2His–H]− (m/z = 309.2650), [Au + His + Cl–H]− (m/z = 385.1173), [Au + 2His–2H]− (m/z = 505.0853), and [Au + 2His + Cl–H]− (m/z = 541.3378). As mentioned in the Methods section, the voltage on the spray tip and the distance between the spray tip and the mass spectrometer were optimized to −4.5 kV and 2 cm, respectively, to produce the highest yield of Au–(His)2 (Fig. 3b and c). Interestingly, some new ion peaks appear in the range of 600–1600 Da. When increasing the distance to 7 cm, the intensity of [Au + 2His + Cl–H]− gradually decreased and the intensity of some ions in the range of 600–1600 Da increased (Fig. S1†).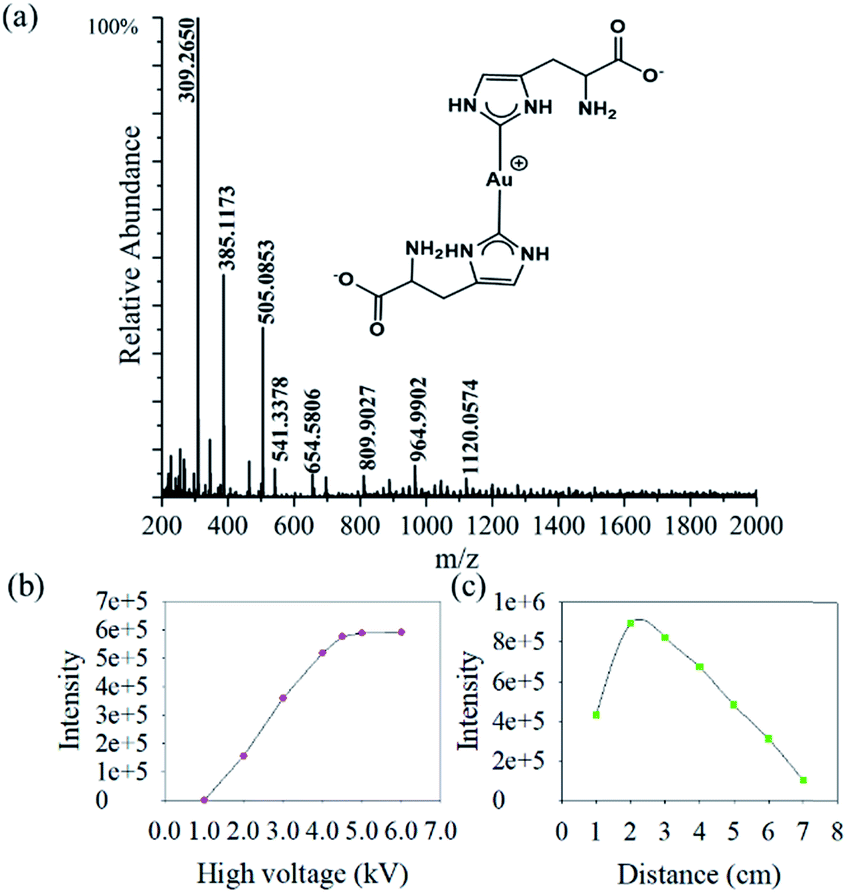 | ||
| Fig. 3 Microdroplet reaction of HAuCl4 (10 μM) and histidine (100 μM): (a) mass spectrum; (b) Au–(His)2 intensity as a function of different high voltages; and (c) Au–(His)2 intensity as a function of travel distance from the tip of sprayer to the inlet of the mass spectrometer. | ||
Compared with the bulk reaction under the same conditions, there was little intensity of [Au + 2His–H]− and mainly ions appeared in the m/z range of 600–1600 Da. A series of peaks which matched to the Au5 nanocluster were analyzed with a spacing of 77.5 Da (Fig. S2 and Table S1†). These results indicate histidine as the ligand could be attached one by one to the Au core to form the Au5 nanocluster with different numbers of histidine consistent with bulk reaction for forming the Au10 nanocluster.44 In order to verify further that the ion peaks belong to Au5 nanocluster, we examined the isotope distribution. We found that the measured isotope peaks coincide almost exactly with the theoretical simulated isotopes peaks (Fig. S3 and S4†), which indicates that 5 gold atoms could be accumulated into gold nanocluster by the microdroplet reaction. According to the reported velocity of microdroplets (84 m s−1),45 the reaction time is estimated to be 0.24 × 10−3 s, which is 3.0 × 107 times shorter than the bulk reaction of 2 h to make the Au10 nanocluster. We suspect that the different reaction times are mainly responsible for the different effects of Au atom accumulation. Here, the [Au + 2His + Cl–H]− may act as a nucleation seed in the formation of larger Au nanoclusters or nanoparticles with the passage of time, as proposed by Xie and co-workers.46 According to this process for nucleation, we speculate that Au–(His)2 is the transient intermediate for forming different Au atom nanocluster depending on the reaction time.
Characterization of Au–(His)2
In order to confirm further the structure information of [Au + 2His–2H]−, collision induce dissociation (CID) was employed. Under an applied collisional energy of 25 eV, there are hardly any fragmented ions observed and only with the parent ion of Au–(His)2 appears. When the collisional energy increases to 75 eV, the overall view of fragment ion was observed, as shown in Fig. 4a. The main fragmentation channels of [Au + 2His–2H]− correspond to the consecutive losses of 44 (CO2), 29 (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH), 14 (CH2), 67 (imidazole), 44 (CO2) to form the quasi-2,4-cyclopentadiene-1-ethanamine–Au (Au–C7H11N) complex. Similarly, we notice when the His fragment loses 44 (CO2) it gives rise to 2,4-cyclopentadiene-1-ethanamine (C7H11N). Fig. 4b presents what we believe is the possible fragment path. Thus, we propose that the structure of [Au + 2His–2H]− is that of Au–(His)2, in which the single Au+ is connected to the carbene carbon of the imidazole rings in the structure of His. Here, His acts as the ligand of NHC. We notice that the collisional energy of 75 eV for Au–(His)2 is higher than the normal collisional energy of 25–35 eV, which indicates that the bond energy of carbene carbon and Au+ is large, which is consistent with the bond of Au–S as proposed by Glorius,1 Crudden,47 Ravoo,48 and Wu.49
NH), 14 (CH2), 67 (imidazole), 44 (CO2) to form the quasi-2,4-cyclopentadiene-1-ethanamine–Au (Au–C7H11N) complex. Similarly, we notice when the His fragment loses 44 (CO2) it gives rise to 2,4-cyclopentadiene-1-ethanamine (C7H11N). Fig. 4b presents what we believe is the possible fragment path. Thus, we propose that the structure of [Au + 2His–2H]− is that of Au–(His)2, in which the single Au+ is connected to the carbene carbon of the imidazole rings in the structure of His. Here, His acts as the ligand of NHC. We notice that the collisional energy of 75 eV for Au–(His)2 is higher than the normal collisional energy of 25–35 eV, which indicates that the bond energy of carbene carbon and Au+ is large, which is consistent with the bond of Au–S as proposed by Glorius,1 Crudden,47 Ravoo,48 and Wu.49
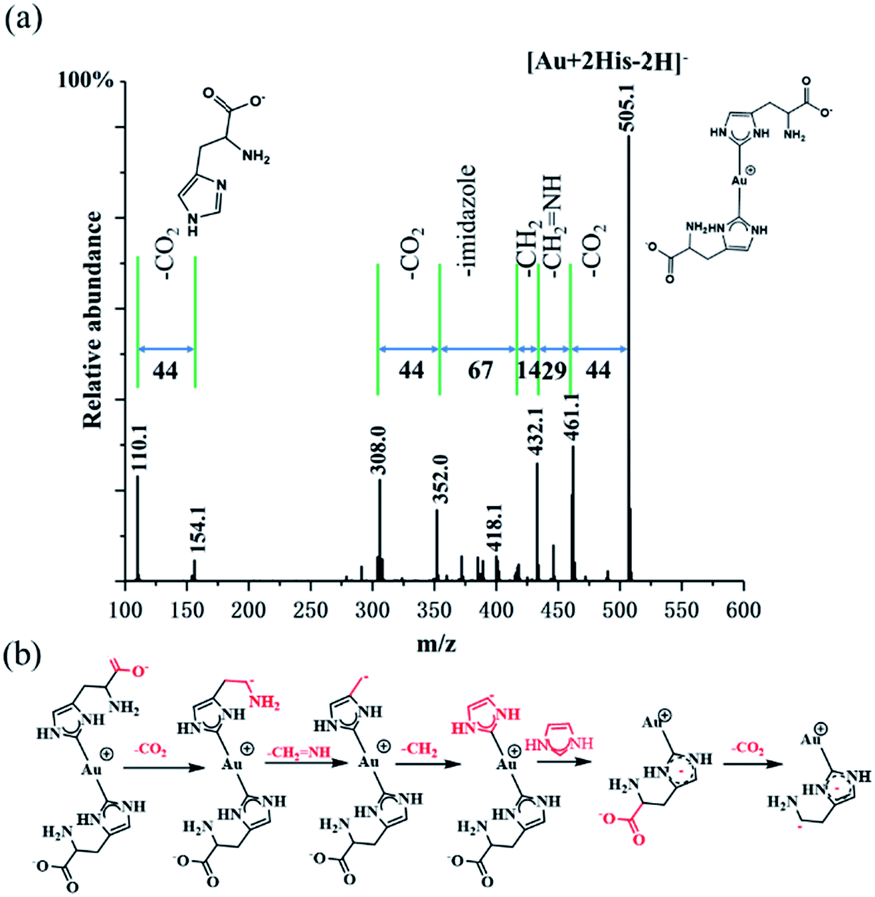 | ||
| Fig. 4 Microdroplet reaction of histidine (100 μM) and HAuCl4 (10 μM) dissolved in water: (a) CID mass spectrum of Au–(His)2 at an energy of 75 eV and (b) proposed fragmentation pathway for Au–(His)2 based on (a). | ||
DFT calculation of Au–(His)2
The coordination between the Au atom and His was further investigated via density functional theory (DFT) calculations. All calculations were performed with Gaussian 09.50 Geometries were optimized with B3LYP and a standard 6-31++G (d, p) basis set (SDD basis set for gold). At first, we speculated that Au–(His)2 was a sandwich compound in which the Au atom is centered between two aromatic rings, such as ferrocene (Fig. S5†). However, calculations indicated otherwise. Previous studies have shown that in the structure of an metal-NHC complex, the adjacent σ-electron-withdrawing and p-electron-donating nitrogen atoms stabilize this structure both inductively by lowering the energy of the occupied s-orbital and monomerically by donating electron density into the empty p-orbital. This ground-state structure is reflected in the C2–N bond lengths. We focused on obtaining the optimized structural of Au–(His)2. As shown in Fig. 5a, the Au atom is directly connected to the carbene carbon of imidazole ring with a bond length of 2.3 Å. Furthermore, it forms a large delocalized π bond between the carbene carbon and the two-nitrogen atom to reduce the electronic cloud density. This result is reflected in Fig. 5b, and obtained the low energy for the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is 3.66 eV, which is consistent with the NHC ligand coordinated with other metal ion.51–53 Combined with the optimized structural and the electronic cloud density, the Au–(His)2 molecule exhibits a singlet ground-state electronic configuration with HOMO and the LOMO best described as a formally sp2-hydridized lone pair and an unoccupied p-orbital at the carbene carbon. The adjacent σ-electron withdrawing and π-electron-donating nitrogen atoms stabilize this structure both inductively by donating electron density into the empty p-orbital. This bonding picture for Au–(His)2 is consistent with the characterization of a metal-NHC complex.1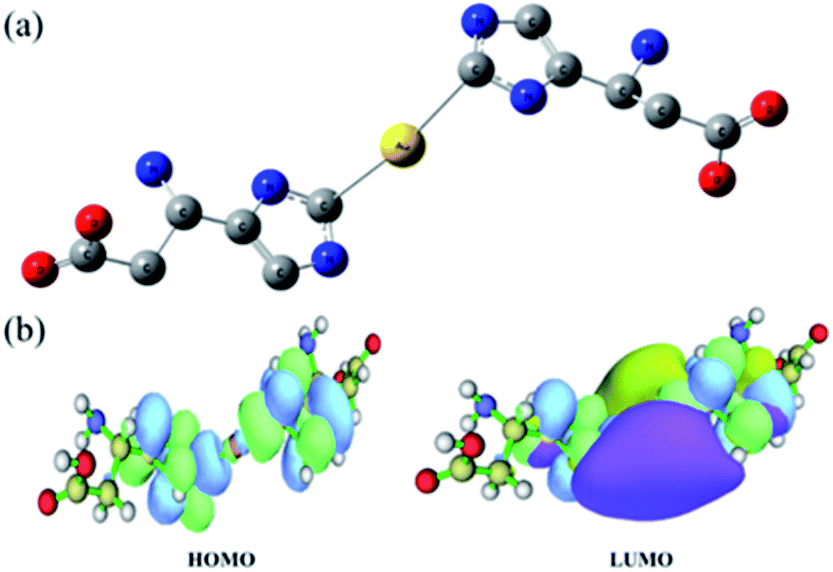 | ||
| Fig. 5 DFT calculations for Au–(His)2 showing (a) the optimized structure and (b) the energy gap distribution of HOMO and LUMO. Illustrations were made using the software of Multiwfn.54 | ||
Possible mechanism for Au–(His)2 formation
As mentioned before, the microdroplet as the microreactor could markedly accelerate some chemical reactions compared to the same reactions in bulk solution due to the microenvironment between the water–air interface.30 So, it has the natural advantage for capturing the transient intermediate of the reaction. Recently, it has been proposed that as the size of a water microdroplet decreases, water molecules at and near the air–water interface autoionize more readily into H+ and OH− than in bulk water.55 This strong electric field formed at the air–water interface is expected to promote the reduction of AuCl4− to Au in which gold goes from the +3 valence state to zero.56 The source of the electrons causing this reduction is believed to be from the OH− anion.18 Fig. S6† presents the mass spectrum obtained from aqueous microdroplets containing HAuCl4. Gold ions with an oxidation number of +1 and +2 are clearly evident.According to the previous report33 about the gradient distribution of pH in a microdroplet, we investigated the pH effect for the reaction of HAuCl4 and histidine. As a control experiment, we use gold trichloride (AuCl3) instead of chloroauric acid (HAuCl4) in the microdroplet reaction with histidine. Unfortunately, we did not detect the peak of Au–(His)2 and some peaks of Au5 nanocluster in the microdroplet reaction of AuCl3 with histidine (Fig. S7†). The AuCl3 solution (100 μM) falls within the neutral range (pH ≈ 6.5), but for HAuCl4 (100 μM), the pH value of the solution falls within the range of 2.8–3.5. Histidine has an imidazole ring with two nitrogen atoms, one nitrogen that is ‘pyrrole-like’ and one that is ‘pyridine-like’. In the bulk water solution, histidine has the pKa value of ≈6.5, which falls within the physiological range.57 As such, it can undergo protonation/deprotonation of one of the nitrogen atoms.58 The ‘pyrrole-like’ nitrogen atom has a much high pKa of >14. When losing a proton, the imidazole ring will take the negatively charge form (Fig. S8†). In order to further verify the pH effect, we compared the cyclic voltammogram (CV) curve of reaction of HAuCl4 and histidine under the pH of 2.0, 6.5, and 12 (Fig. S9†). The results showed that under the pH of 12, it has the higher electrochemical response with the potential difference (ΔE) of 1.299 V, which is smaller than 1.706 V and 1.764 V for pH of 6.5 and 2.0, respectively. We could conclude that the alkaline environment at the interface promotes the reaction of HAuCl4 and histidine.
Recently, there have some reports about the air–water interface of microdroplets having a more acidic and more basic microenvironment at the same time.50,59–61 This behaviour will help to promote more hydrogen ion and hydroxyl ion accumulation at the inner and outer interface to form a double layer. In this double layer, the chloroauric acid was reduced from Au3+ to Au+ and histidine lost one hydrogen from the carbene carbon of imidazole to ultimately form the NHC ligand with one negative charge (Fig. S10†). Under the double layer interaction of the air–water interface, it promotes the formation of a coordination bond between Au+ and negative NHC ligand with the large π bond between the two nitrogen atoms of ‘pyrrole-like’ and ‘pyridine-like’ character, which is consistent with what Thiel62 reported and recently reviewed by Glorious.1 Most commonly, this strategy involves a redox process for chloroauric acid18 and α-elimination or oxidative addition at the carbene carbon of histidine,7 to form the metal-based NHC complex with the base-promoted transformation of imidazole complex to carbene derivatives (Fig. S11†).8
We could verify indirectly this behaviour from the chemical shift of 1H-NMR (Fig. S12†) and the Au valence of XPS (Fig. S13†). With the exception of the bulk reaction, histidine usually acts as a chelating ligand that preferentially coordinates to transition metals via the N atoms from the amino group and from the imidazole ring as well as O atom from the carboxylate group in the bulk reaction.63–65 Here, we speculate that under the bulk reaction, the Au3+ was absorbed first on histidine via the ‘pyridine-like’ N atom on the imidazole ring and N atom from the amino group to form the chelating complex and then underwent reduction to the Au10 nanocluster by the amino group of histidine for the bulk reaction,44 which is twice the size compared with the Au5 nanocluster observed in the microdroplet reaction (Fig. S14†).
Comparing the possible mechanisms for the bulk reaction and microdroplet reaction, it is expected that hydroxyl anions (OH−) and hydronium cations (H+) will both be found in high concentration at the air–water interface of the microdroplet forming an electrical double layer. Although this might appear at first to be paradoxical, many experiments support the creation of a double layer and the production of a strong electric field at the periphery of the microdroplet. We suggest that these features help to explain the behavior we have observed in the formation of the Au–(His)2 complex. Clearly, more work is needed to establish in detail the factors that account for these markedly different mechanisms for Au–(His)2 in the aqueous microdroplet reaction. But according to this distinct steric and electronic influence of the NHC on the metal center, catalytic activity of Au–(His)2 may also be improved under the microdroplet environment.
Catalytic activity of Au–(His)2 in microdroplets
In order to ensure the potential catalytic activity of Au–(His)2 complex, we employed the enamine reaction between phenylacetylene (1) and indoline (2) as the model reaction to test it using the homemade reactive DESI-MS setup (Fig. S15†).66 The microdroplet reaction was conducted with an aqueous solution of HAuCl4 (10 μM) and His (100 μM) combined with an acetonitrile solution of phenylacetylene (100 μM), and indoline (100 μM). Under the proposed MS condition, intermediates and some product were detected. We just found the intermediate product at m/z of 222.1242 for the first step of amine reaction, which resulted from the addition reaction of phenylacetylene (1) and indoline (2) (Fig. S16†). We did not get any other information about the final product in the mass spectrum, evidently due to weak ionization. The intermediate of [2His + Au + 1]+, [2His + Au + 1 + ACN]+, and [3His + Au + 1 + ACN]+ were captured with the m/z of 609.1545 (theoretical mass = 609.1525, mass error = 3.61 ppm), 650.0653 (theoretical mass = 650.0623, mass error = 3.48 ppm), and 805.1383 (theoretical mass = 805.1355, mass error = 3.65 ppm), respectively (Fig. 6a). The intermediate of [2His + Au + 1 + ACN]+ had the maximum intensity, [3His + Au + 1 + ACN]+ was next, followed by [2His + Au + 1]+. Unfortunately, we did not detect other species, owing perhaps to their weak ionization ability.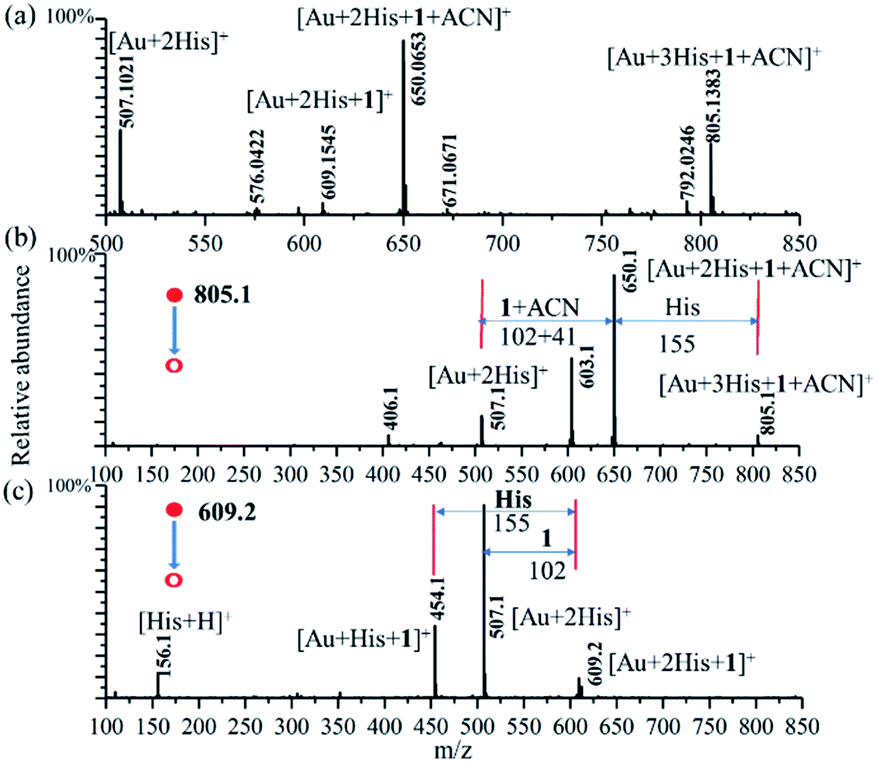 | ||
| Fig. 6 (a) Online reactive DESI-MS spectrum upon spraying the solution of HAuCl4 (10 μM) and His (100 μM) in water mixed with phenylacetylene (100 μM), and indoline (100 μM) in acetonitrile solution with the high voltage of +4.5 kV and the MS2 spectrum for the m/z values of (b) 805.1, (c) 609.2 with the energy of collision-induced dissociation of 25 eV. | ||
In order to confirm further the possible structure of the above intermediates, CID was employed to obtain more detailed information about [2His + Au + 1]+ (Fig. 6b), and [3His + Au + 1 + ACN]+ (Fig. 6c) with a CID energy of 25 eV. An analysis of the CID mass spectrum shows for [2His + Au + 1]+ the loss of m/z = 102 (1) to form the [2His + Au]+ (m/z = 507.1), and for [3His + Au + 1 + ACN]+ the loss of the m/z = 155 (His), and 146 (1 + ACN) to form also [2His + Au]+ (m/z = 507.1). The maximum fragment peak of the parent ion of [3His + Au + 1 + ACN]+ corresponds to the loss of m/z = 146 (1 + ACN) to form also [2His + Au + H]+ (m/z = 507.1). The m/z peaks at 603.1 and 454.1 are the second major fragmentation of MS2 for the m/z peaks at 805.1 and 609.1, respectively. For the m/z peak at 454.1, corresponding to [Au + His + 1]+, we assigned it to arising from the m/z peak at 609.1 corresponding to [Au + 2His + 1]+ through the loss of one of His with a m/z value of 155. Unfortunately, we did not get further information for the m/z peak at 603.1.
Catalytic activity of Au–(His)2via ultrasonic nebulization mode
Based on the result of the enamine reaction with Au–(His)2 by reactive DESI, the scale up of the microdroplet reaction was conducted using the ultrasonic nebulizer with commercial Au(I) catalyst (TPPGC) as a reference. Here, HPLC was employed to evaluate catalytic activity of Au–(His)2 in the enamine reaction. We noticed that in the HPLC chromatogram there are different retention times of 1.2 min, 2.4 min, 6.5 min, and 9.1 min for Au–(His)2, indoline, product, and phenylacetylene, respectively (Fig. S17†). During the UN microdroplet reaction time of 40 min at the room temperature, we noticed that the intensity of the product increased sharply at the initial stage of 25 min, and then gradually reached a plateau. We found that when the reaction time was extended to 120 min, the product yield of the microdroplet reaction increased from 40% to 68%. When we use the heating ultrasonic atomization method and set the temperature to 50 °C and the reaction time to 40 min, the reaction yield was 78% but reached 94% for 80 min (Fig. S18†). The intensity trend of phenylacetylene had the opposite behaviour (Fig. S19†). The product was confirmed to have the structure of 4-methyl-4,6-diphenyl-1,2-dihydro-4H-pyrrolo[3,2,1-ij]quinolone via the 1H-NMR and GC-MS (Fig. S20†), which is consistent with the work of Guzei and coworkers67 and Purkait and Blechert.68 As a control experiment, we checked whether phenylacetylene and indoline in the acetonitrile–water solution react via UN microdroplet setup. We observed no product. This result further confirms that the Au–(His)2 prepared by UN microdroplet has an extremely high catalytic activity and could promote the enamine reaction with a good yield of product.In the enamine bulk reaction, the Au–(His)2 was collected by the offline homemade-ESSI and then reacted with phenylacetylene (1) and indoline (2) under stirring for 780 min (Fig. S21†). We further evaluated the catalytic activity of Au–(His)2 for the enamine reaction under the proposed HPLC condition. Meanwhile, in order to obtain the optimized catalytic activity of Au–(His)2 in UN microdroplet reaction, the fluorescence (FL) intensity at Ex (396 nm) and Em (495 nm) was utilized as the evaluation index (Fig. S22†). We found that the optimized FL intensity of Au–(His)2 complex occurs in 12 min (Fig. S23†). Based on the above result, the catalytic activity of Au–(His)2 was further evaluated, compared with that a of commercial Au(I) catalyst (TPPGC). The result showed that the slope for the bulk reaction is 5.7 in 780 min (Fig. 7a) and 759.8 for UN microdroplet reaction in 40 min (Fig. 7b). For the UN microdroplet reaction, the reaction rate was 133 times higher than that of the bulk reaction. The yield of 92% for the bulk reaction is for 780 min (Fig. 7c), but 43% for the UN reaction is only for 40 min (Fig. 7d). Previous studies on the enamine reaction usually required more than 21 h to achieve over 90% yield, even with the assistance of a bi-catalyst under the strict acid–base system to promote the Au activity.
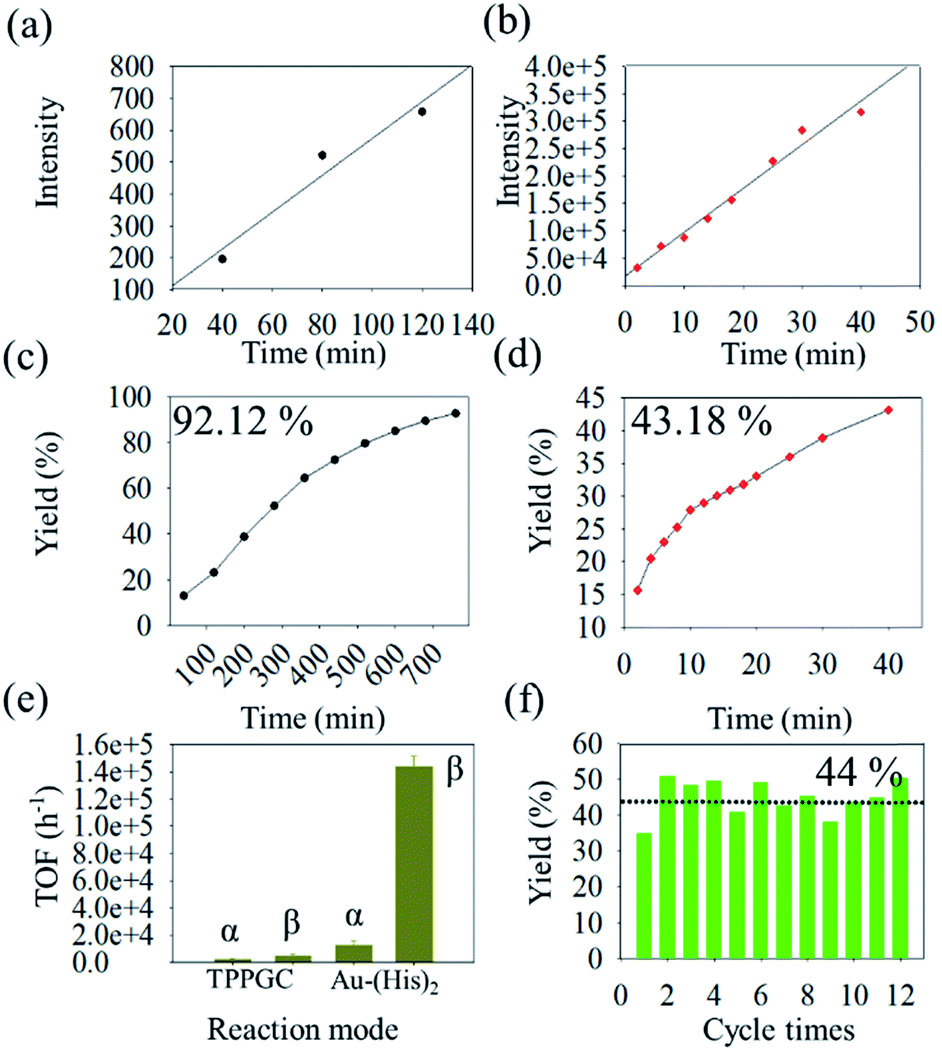 | ||
| Fig. 7 The dynamic study for enamine reaction via Au–(His)2 and the catalytic activity evaluation for Au–(His)2 and TPPGC. (a and b) the rates curve of Au–(His)2 in the mode of bulk reaction and the UN microdroplet reaction; (c and d) the yields curve for bulk reaction and the UN microdroplet reaction; (e) the TOF for enamines reaction via TPPGC and Au–(His)2 under the different reaction system (“α” and “β” denoted for bulk reaction and microdroplet reaction, respectively); and (f) the cycle times for Au–(His)2 in the UN microdroplet reaction. | ||
In order to further evaluate the catalytic activity of Au–(His)2, we have calculated the TOF (turnover frequency) of Au–(His)2 in bulk reaction and UN microdroplet reaction, compared with the TPPGC (commercial Au(I) catalyst) as the control. The results showed Au–(His)2 has the maximum catalytic activity with the TOF of 14235 h−1 in the UN microdroplet reaction, compared with the TOF of 480 h−1 for TPPGC in UN microdroplet reaction and 1260 h−1 for Au–(His)2 in bulk reaction (Fig. 7e). It was showed that the Au–(His)2 has good catalytic activity compared with the commercial Au(I) catalyst, TPPGC. Combined with the function of scale-up of UN microdroplet reaction, we finally achieved a product production rate of 35 mg min−1 with the catalytic activity of Au–(His)2via UN microdroplets, which is semi-preparative. Additionally, we found that the Au–(His)2 could be simply extracted from the EA–H2O solution and nebulized for next cycles. This process makes sure that the cumbersome separation and purification process can be omitted when we use precursors with maximum catalytic activity. The resulting Au–(His)2 exhibited about 44% yield for 12 batches without detachment or aggregation (Fig. 7f). Therefore, the obtained Au–(His)2 has the potential for industrial production of enamine reactions.
Conclusions
The transient intermediates, Au–(His)2, for the formation of gold nanocluster and gold nanoparticle were captured by an on-line microdroplet reaction combined with mass spectrometry with a reaction time that is less than 1 ms. Using a microdroplet as the microreactor with an ultra-short reaction time provides reaction acceleration and could effectively inhibit aggregation between Au–(His)2 during the metal crystal growth, therefore producing a rather uniform size of 0.6 nm Au–(His)2 based Au5 nanocluster. Due to the coordination bond between carbene carbon of imidazole ring from histidine and Au atom, the Au–(His)2 has excellent catalytic activity compared with the traditional commercial TPPGC. We suggest that this metal-NHC based on microdroplet reaction provides a promising strategy for further development of potential catalytic complex.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific Research Startup Foundation of Fudan University (Grant IDH1615113) and Natural Science Foundation of China (No. 2197040302)References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- J. Wang, X. Cao, S. Lv, C. Zhang, S. Xu, M. Shi and J. Zhang, Nat. Commun., 2017, 8, 14625–14635 CrossRef PubMed.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC.
- M. Matena, T. Riehm, M. Stohr, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 2414–2417 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- X. Li, Y. Cao, K. Luo, Y. Sun, J. Xiong, L. Wang, Z. Liu, J. Li, J. Ma, J. Ge, H. Xiao and R. N. Zare, Nat. Catal., 2019, 2, 718–725 CrossRef CAS.
- S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS PubMed.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kuhn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607–4692 CrossRef CAS PubMed.
- S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed.
- I. Nam, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 36–40 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Nat. Commun., 2018, 9, 1562 CrossRef PubMed.
- Y. H. Lai, S. Sathyamoorthi, R. M. Bain and R. N. Zare, J. Am. Soc. Mass Spectrom., 2018, 29, 1036–1043 CrossRef CAS PubMed.
- S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Analyst, 2017, 142, 1399–1402 RSC.
- X. Yan, H. Cheng and R. N. Zare, Angew. Chem., Int. Ed., 2017, 56, 3562–3565 CrossRef CAS PubMed.
- D. Gao, F. Jin, X. Yan and R. N. Zare, Chem.–Eur. J., 2019, 25, 1466–1471 CrossRef CAS PubMed.
- D. Sarkar, R. Singh, A. Som, C. K. Manju, M. A. Ganayee, R. Adhikari and T. Pradeep, J. Phys. Chem. C, 2018, 122, 17777–17783 CrossRef CAS.
- A. Li, Z. Baird, S. Bag, D. Sarkar, A. Prabhath, T. Pradeep and R. G. Cooks, Angew. Chem., Int. Ed., 2014, 53, 12528–12531 CAS.
- A. J. Ingram, D. Solis-Ibarra, R. N. Zare and R. M. Waymouth, Angew. Chem., Int. Ed., 2014, 53, 5648–5652 CrossRef CAS PubMed.
- H. Cheng, X. Yan and R. N. Zare, Anal. Chem., 2017, 89, 3191–3198 CrossRef CAS PubMed.
- T. A. Brown, H. Chen and R. N. Zare, J. Am. Chem. Soc., 2015, 137, 7274–7277 CrossRef CAS PubMed.
- Y. Q. Xue, X. C. Yang, Z. X. Cui and W. P. Lai, J. Phys. Chem. B, 2011, 115, 109–112 CrossRef CAS PubMed.
- S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, J. Chem. Phys., 2018, 148, 244704 CrossRef PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- J. N. Smith, R. C. Flagan and J. L. Beauchamp, J. Phys. Chem. A, 2002, 106, 9957–9967 CrossRef CAS.
- R. M. Bain, C. J. Pulliam, S. T. Ayrton, K. Bain and R. G. Cooks, Rapid Commun. Mass Spectrom., 2016, 30, 1875–1878 CrossRef CAS PubMed.
- D. Duft, T. Achtzehn, R. Muller, B. A. Huber and T. Leisner, Nature, 2003, 421, 128 CrossRef CAS PubMed.
- A. Gallo Jr, A. S. F. Farinha, M. Dinis, A. H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard III and H. Mishra, Chem. Sci., 2019, 10, 2566–2577 RSC.
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- Z. Zhou, X. Yan, Y. H. Lai and R. N. Zare, J. Phys. Chem. Lett., 2018, 9, 2928–2932 CrossRef CAS PubMed.
- J. K. Lee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2017, 50, e2 CrossRef PubMed.
- T. Muller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed., 2012, 51, 11832–11835 CrossRef PubMed.
- M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson and R. G. Cooks, Chem. Sci., 2018, 9, 1647–1653 RSC.
- X. Yan, Y. H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207–5211 RSC.
- Z. Wei, M. Wleklinski, C. Ferreira and R. G. Cooks, Angew. Chem., Int. Ed., 2017, 56, 9386–9390 CrossRef CAS PubMed.
- X. Zhu, W. Zhang, Q. Lin, M. Ye, L. Xue, J. Liu, Y. Wang and H. Cheng, ACS Sustainable Chem. Eng., 2019, 7, 6486–6491 CrossRef CAS.
- C. Liu, J. Li, H. Chen and R. N. Zare, Chem. Sci., 2019, 10, 9367–9373 RSC.
- X. Yang, M. Shi, R. Zhou, X. Chen and H. Chen, Nanoscale, 2011, 3, 2596–2601 RSC.
- J. K. Lee, S. Kim, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3898–3903 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D. E. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C. T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Hakkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- S. Engel, E. C. Fritz and B. J. Ravoo, Chem. Soc. Rev., 2017, 46, 2057–2075 RSC.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H. B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. Mishra, S. Enami, R. J. Nielsen, L. A. Stewart, M. R. Hoffmann, W. A. Goddard III and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18679–18683 CrossRef CAS PubMed.
- G. Prabusankar, N. Muthukumaran, M. Vaddamanu, G. Raju, K. Velappan, A. Sathyanarayana, Y. Masaya, S. Sugiyama, K. Hisano and O. Tsutsumi, RSC Adv., 2019, 9, 7543–7550 RSC.
- S. Shi, M. C. Jung, C. Coburn, A. Tadle, M. R. D. Sylvinson, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- Y. Ma, S. Bian, Y. Shi, X. Fan and X. Kong, ACS Omega, 2019, 4, 650–654 CrossRef CAS.
- S. Zechel, M. D. Hager, T. Priemel and M. J. Harrington, Biomimetics, 2019, 4, 20–41 CrossRef CAS PubMed.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- I. Nam, J. K. Lee, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12396–12400 CrossRef CAS PubMed.
- S. Enami and A. J. Colussi, J. Phys. Chem. A, 2017, 121, 5175–5182 CrossRef CAS PubMed.
- M. Kumar, J. Zhong, X. C. Zeng and J. S. Francisco, J. Am. Chem. Soc., 2018, 140, 4913–4921 CrossRef CAS PubMed.
- K. S. M. Salih, S. Bergner, H. Kelm, Y. Sun, A. Grün, Y. Schmitt, R. Schoch, M. Busch, N. Deibel, S. Bräse, B. Sarkar, M. Bauer, M. Gerhards and W. R. Thiel, Eur. J. Inorg. Chem., 2013, 1, 6049–6059 CrossRef.
- X. Huang, Y. Lin, J. Chen, Y. Chen, Y. Li and W. Gao, New J. Chem., 2015, 39, 8569–8575 RSC.
- Y. Liu, D. Ding, Y. Zhen and R. Guo, Biosens. Bioelectron., 2017, 92, 140–146 CrossRef CAS PubMed.
- B. Warzajtis, B. D. Glisic, N. D. Savic, A. Pavic, S. Vojnovic, A. Veselinovic, J. Nikodinovic-Runic, U. Rychlewska and M. I. Djuran, Dalton Trans., 2017, 46, 2594–2608 RSC.
- M. Lu, Y. Su, P. Zhao, X. Ye, Y. Cai, X. Shi, E. Masson, F. Li, J. L. Campbell and H. Chen, Chem.–Eur. J., 2018, 24, 2144–2150 CrossRef CAS PubMed.
- C. S. Yi, S. Y. Yun and I. A. Guzei, J. Am. Chem. Soc., 2005, 127, 5782–5783 CrossRef CAS PubMed.
- N. Purkait and S. Blechert, Adv. Synth. Catal., 2012, 354, 2079–2083 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06221a |
| ‡ K. Luo and J. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |