
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe enantioselective total synthesis of laurendecumallene B†
Cooper A.
Taylor‡
 ,
Yu-An
Zhang‡
and
Scott A.
Snyder
*
,
Yu-An
Zhang‡
and
Scott A.
Snyder
*
Department of Chemistry, University of Chicago, 5735 S. Ellis Avenue, Chicago, IL 60637, USA. E-mail: sasnyder@uchicago.edu
First published on 6th February 2020
Abstract
For decades, the Laurencia family of halogenated C15-acetogenins has served as a valuable testing ground for the prowess of chemical synthesis, particularly as it relates to generating functionalized 8-membered bromoethers. Herein, we show that a readily modified and predictable approach that generates such rings and an array of attendant stereocenters via a bromenium-induced cyclization/ring-expansion process can be used to synthesize laurendecumallene B and determine the configuration of two of its previously unassigned stereocenters. In particular, this work highlights how the use of the bromenium source BDSB (Et2SBr·SbCl5Br) in non-conventional solvents is essential in generating much of the target's complexity in optimal yields and stereoselectivity. Moreover, the final structural assignment of laurendecumallene B reveals that it has one element of bromine-based chirality that, to the best of our knowledge, is not shared with any other member of the class.
Introduction
Over the past half-century, chemists have isolated and structurally characterized more than 150 halogenated ethers from red algae of the Laurencia genus.1 Over a third of these C15-containing natural products possess an 8-membered ring, typically with a bromine atom on the carbon β-to the ethereal linkage either within that ring (as in 1, Scheme 1)2 or just outside it (as in 2–5);3 most possess additional fused ring systems and additional halogen atoms, particularly as part of exocyclic allenes.4 Unsurprisingly, such molecular complexity has long captivated the attention of the synthetic community with dozens of syntheses of varied members having been reported to date featuring an array of creative tactics to forge their medium-sized rings.5 However, only a few of these strategies (particularly from the Kim and Paton groups) have proven capable of delivering several members of the class.5g,i Our approach to addressing that challenge (bottom of Scheme 1) has been to use a tetrahydrofuran substrate (8 and 9) containing an exocyclic alkene.6 When activated by a powerful bromenium source such as BDSB (10, Et2SBr·SbCl5Br),7 an oxonium intermediate (11 and 12) is produced which subsequently undergoes ring-opening either by elimination of the neighboring silane or nucleophilic attack by the Boc-protected alcohol to afford an 8-membered ring containing either an alkene (13) or protected diol (14), respectively. To date, we have synthesized natural products 1–6, among others, concisely through this approach due to the ease of varying the chiral information encoded into the starting substrates. Excitingly, the Paton and Burton groups have recently shown that polycyclic oxonium species are likely part of Nature's constructions as well with even more complex precursors,8a with other uses of such species also leading to other natural products.8b–k Herein, we show how our general approach can lead to an efficient synthesis of laurendecumallene B (7), establishing its structure and absolute configuration as being one of the more distinct within the class. | ||
| Scheme 1 Structures of varied members of the Laurencia class of natural products which have been synthesized (1–6) as well as a new member (7) whose configuration at two sites (marked with a red star) are unassigned, and the general approach our group has developed for 8-membered ring formation to access such natural products. | ||
Laurendecumallene B (7) was isolated off the coast of Weizhou Island in China from Laurencia decumbens Kützing and characterized by Wang and co-workers, with 500 g of dried sample affording just 13.0 mg of the natural product.9 Although 7 contains the standard 15 carbon atoms of the class, its proposed structure includes a rare cis-disposition of hydrogen atoms at the blue starred positions (found in 1–3, but few others) and unknown stereochemical configurations about the bromine-bearing exocyclic stereocenter off the 8-membered ring, as well as the bromoallene moiety, both denoted by red stars in Scheme 1. To date, only one synthetic effort to laurendecumallene B has been reported by Fujii, Ohno, and co-workers;10 they obtained the full carbon and ring skeleton of 7 through a Pd-catalyzed ring closure over 26 steps, but were unable to complete the target definitively and elucidate its full structural assignment, particularly the exocyclic stereocenter adjacent to the 8-membered ring, following the final 2 operations of their sequence.
As part of a recent effort that led to the total synthesis of 7 different Laurencia natural products, including the non-natural iso-desepilaurallene (15, Scheme 2), we questioned whether or not simple dihydroxylation of its lone alkene could complete the ring-based functionality of one potential 8-membered stereoisomeric precursor of 7.6c However, such an endeavor afforded a diol (i.e.16) with the opposite stereochemistry desired (as determined by nOe). This outcome highlights the general challenge of appropriately installing functional groups post 8-membered ring formation due to conformational restraints. As such, given a desire to form the same exocyclic stereocenter as in 15 (since all other natural products with a similar 8-/5-fused ring system such as 4 and 5 have a 1,2-trans arrangement of drawn hydrogen and exocyclic bromine, cf.Scheme 1), we wondered whether the key 8-membered ring of 7 and its attendant diol functionality could arise directly from a cyclization using 22 or 23 as the key tetrahydrofuran-containing starting material. In this approach, bromenium-activation followed by attack of an oxygen from the Boc protecting group was projected to afford the desired array of functionality and stereochemical information as expressed within either 18 or 19. Although previous studies showed that having fused lactones on the core tetrahydrofuran ring systems did not diminish the efficacy and/or selectivity of similar processes involving silyl elimination, low-level DFT calculations of simplified models of the likely oxonium intermediates revealed that the lactone-opened 25 (a simplified form of 21 without a full exocyclic sidechain) had a significant relaxation of the C1–O–C2–C3 angle (Δϕ = 16.39°) versus its bicyclic counterpart (24); unclear was whether such a difference would be relevant to cyclization success. Following 8-membered ring formation, subsequent generation of the enyne (17) and a final bromenium-induced closure (usually only partially stereoselective) was then anticipated to deliver both bromoallene isomers in hopes that the desired target (7) would be present. If not, the sequence could be repeated using the cis-alkene isomers of both 22 and 23 to generate the opposite exocyclic stereoisomer.
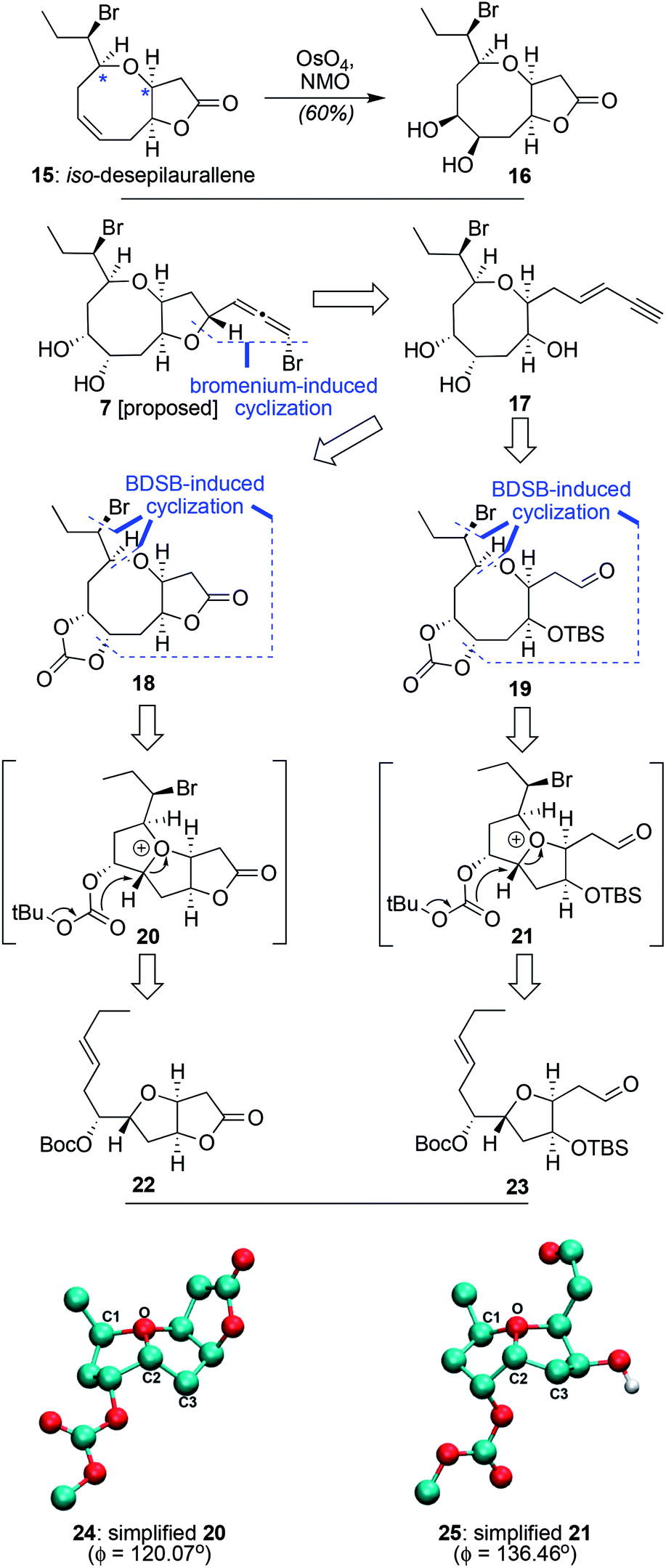 | ||
| Scheme 2 Challenges in forming laurendecumallene B (7) from previously prepared compounds, a general retrosynthetic analysis for 7 based on a distinct approach, and structures obtained from low level DFT analysis (24 and 25) to aid in the design of the key ring expansion step with geometries optimized using B3LYP/cc-pVDZ. | ||
Results and discussion
Our efforts began with the preparation of ring-expansion precursors 22 and 23; the sequence for the latter is shown in Scheme 3. First, cross metathesis of known chiral 26 (synthesized over 5 routine steps from (S)-(−)-glycidol as established by literature precedent; see ESI for full details†)11 with allyl acetate (used as solvent) smoothly furnished 27 in 54% yield (76% brsm). Subsequent NIS-induced cyclization then yielded tetrahydrofuran 28 in 69% yield after 8 h of stirring in CH2Cl2.12 From here, iodine removal and some protecting group and oxidation state changes as effected over 4 steps smoothly delivered aldehyde 29. Efforts to perform the following Sakurai allylation13 using bicoordinate TiCl4 to promote the shown chelation model 30 proved high yielding (89%), with only a trace of material generated of opposite chirality, likely via the monocoordinate Felkin–Ahn variant 31.14 Intriguingly, the use of monodentate Lewis acids such as BF3·OEt2 also gave 30 as the major product, but were low yielding, suggesting that further elements of substrate bias might favor the desired addition product. Next, a second cross metathesis and further minor structural manipulations then afforded cyclization precursor 23, noting that the yield for this sequence was significantly reduced if the Boc-protection step was performed prior to metathesis. | ||
Scheme 3 Total synthesis of laurendecumallene B (7): (a) allyl acetate (10.0 equiv.), Grubbs 2nd generation initiator (3 mol%), 25 °C, 4 h, 54%, 76% brsm; (b) NaHCO3 (6.0 equiv.), NIS (2.5 equiv.), CH2Cl2, 25 °C, 8 h, 69%; (c) TBSOTf (1.6 equiv.), 2,6-lutidine (4.0 equiv.), CH2Cl2, 0 °C, 1 h, 89%; (d) Raney® nickel (excess), EtOH, 25 °C, 10 min, 72%; (e) Pd/C (0.13 equiv.), H2 (1 atm), EtOAc, 25 °C, 2 h; (f) NaHCO3 (4.0 equiv.), Dess–Martin periodinane (1.1 equiv.), CH2Cl2, 25 °C, 30 min, 92% over 2 steps; (g) TiCl4 (1.0 equiv.), allyltrimethylsilane (1.5 equiv.), CH2Cl2, -78 °C, 30 min, 89%; (h) trans-3-hexene, Grubbs 2nd generation initiator (5 mol%), CH2Cl2, 25 °C, 8 h, 69%, 79% brsm; (i) Boc2O (2.0 equiv.), LiHMDS (1.3 equiv.), THF, −78 to 25 °C, 8 h, 96%; (j) K2CO3 (6.0 equiv.), MeOH, 25 °C, 2 h, 96%; (k) NaHCO3 (4.0 equiv.), Dess–Martin periodinane (1.1 equiv.), CH2Cl2, 25 °C, 30 min, 93%; (l) BDSB (1.2 equiv.), CH2Cl2, 0 °C, 10 min, 38%, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr; (m) 33 (1.8 equiv.), n-BuLi (1.6 equiv.), THF, 0 °C, 30 min, then 19, THF, 0 °C, <5 min, 58%, 80% brsm, 2:1 E/Z; (n) TBAF (2.0 equiv.), THF, 0 °C, 5 min, 81%; (o) BDSB (1.2 equiv.), MeCN, 25 °C, 2 h, 72%, 81% brsm, 1.0:1.3 R/S; (p) K2CO3 (10.0 equiv.), MeOH, 25 °C, 2 h, 30% 35, 40% 7. :1 dr; (m) 33 (1.8 equiv.), n-BuLi (1.6 equiv.), THF, 0 °C, 30 min, then 19, THF, 0 °C, <5 min, 58%, 80% brsm, 2:1 E/Z; (n) TBAF (2.0 equiv.), THF, 0 °C, 5 min, 81%; (o) BDSB (1.2 equiv.), MeCN, 25 °C, 2 h, 72%, 81% brsm, 1.0:1.3 R/S; (p) K2CO3 (10.0 equiv.), MeOH, 25 °C, 2 h, 30% 35, 40% 7. | ||
As shown in Table 1, attempted cyclization and ring-expansion of 23 with BDSB using the conditions we have typically deployed for such events (EtNO2, –78 °C, 10 min or MeNO2, –20 °C, 10 min, entries 1 and 2) did afford the desired ring system (19) as the predominant diastereomer (∼5:1), but only in low yield (∼30% brsm).6,7 Unfortunately, higher temperatures (0 °C, entry 3) diminished both yield and diastereoselectivity. We then tested CH2Cl2 as part of standard solvent screening, but were disappointed to find that at −20 °C, the reaction was even less effective than with our standard solvents. Pleasingly, though, performing the reaction at 0 °C in CH2Cl2 (entry 5) led to a complete, better yielding reaction (49%) with superior diastereoselectivity (7.0:1); that yield was slightly reduced when the reaction was conducted on larger scale. Critically, we observed no α-bromination of the aldehyde within 23, though such reactions have been observed in other contexts.15 As further evidence for the uniqueness of the developed solution and the power of BDSB to effect this cyclization/ring-expansion chemistry, attempts to use NBS in HFIP, convenient and readily executed conditions recently reported to effect bromenium-induced polyene cyclizations with equal facility as BDSB,16 did not effect any conversion in this process, even at reflux. Additionally, attempts to cyclize acetate-protected and free hydroxyl analogues in lieu of the aldehyde group within 23 failed to produce the desired 8-membered ring. Finally, lactone 22 (synthesis shown in ESI†) also did not participate in the desired BDSB-induced cyclization, suggesting it does not have the conformational flexibility needed for intramolecular attack by the Boc group. Interestingly, when the same DFT-based conformational analysis described earlier was performed on models of the acetate-protected and free hydroxyl analogues of 23, their dihedral angles (ϕ = 132.22° and ϕ = 134.32°, respectively – see ESI† for details) were closer to that of 23 than 22, but still of lower value. Whether that result suggests that a threshold angle exists for success is unclear, as conformational flexibility may not be the only important difference between 22 and 23. Nevertheless, given that the successful aldehyde case does have the highest dihedral angle observed, we believe that the low-level DFT conformational analysis presented here could potentially be predictive in assessing the order of potential precursors to test for other variants of the cyclization/ring-expansion process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Temperature (°C) |
19:32 |
Yield (%) |
| a Reactions were performed using 0.06 mmol of 23; all reactions using BDSB were conducted for 10 min total. | |||||
| 1 | BDSB | EtNO2 | −78 | 4.9:1 |
∼30 b.r.s.m. |
| 2 | BDSB | MeNO2 | −20 | 5.3:1 |
∼30 b.r.s.m. |
| 3 | BDSB | MeNO2 | 0 | 1.3:1 |
20% |
| 4 | BDSB | CH2Cl2 | −20 | N/A | <10% conv. |
| 5 | BDSB | CH2Cl2 | 0 | 7.0:1 |
49% |
| 6 | NBS, morpholine | HFIP | 0 to reflux | N/A | N.R. |

With the desired cyclization/ring-expansion effected, Wittig olefination of the aldehyde within 19 using the ylide derived from 33 yielded an inseparable mixture of E- and Z-enynes 34 (∼2:1) in 47% yield following TBAF deprotection.17 Initial removal of the TBS-protecting group to form a lactol prior to olefination and efforts to effect Horner–Wadsworth–Emmons variants did not prove more stereoselective. Pressing forward, 34 was then converted into laurendecumallene B (7) and its allene epimer (35) in an isolated 1.3:1 ratio through a second BDSB-induced cyclization and terminating carbonate cleavage. Key to the success of this bromenium-induced event was the use of MeCN as solvent, with other solvents such as EtNO2 and MeNO2 at various reaction temperatures affording inferior yields (see ESI section for a full screening table†). Although previous studies have indicated that the chirality of bromoallene formation can be solvent dependent,18 we did not observe similar trends here based on the range of solvents probed. All reactions gave a similar mixture of products (based on crude NMR analysis) irrespective of the starting enyne ratio. Of note, this event is the first successful report of BDSB reactivity in MeCN, with the outcome also being superior to TBCO, the typical reagent of choice for these transformations.5c,17,19 Furthermore, it is the first example of BDSB successfully forming a bromoallene from a free alcohol/enyne precursor. We previously attempted such a reaction in our synthesis of laurallene;6c however, only decomposition was observed in that case, likely because of the more reactive alkene present on that 8-membered ring precursor.
Pleasingly, all spectral data (1H and 13C NMR) for synthetic 7 matched that as reported by Wang and co-workers, noting the observed optical rotation was higher ([α]D24 + 117.4, c = 0.33 in CHCl3), but of the same rotational direction as the natural material ([α]D25 + 60.6, c = 0.33 in CHCl3).9 Extensive efforts were made to crystallize either 7 or its analogues to gain additional and definitive structural proof;20 however, these efforts were unsuccessful. Nevertheless, based on Lowe's rule and analysis performed by the Kim group, we are confident in the stereochemical assignment of the bromoallene moiety as drawn.5i,21 Our faith in the chirality about the exocyclic red-starred carbon derives from the array of tetrahydrofuran-containing alkenes probed to date through our core 8-membered ring formation process, many confirmed by X-ray analysis, with that stereochemistry encoded by the alkene geometry of the cyclization precursor. To the best of our knowledge, such a bromine disposition relative to the cis-hydrogen atoms neighboring the ether ring fusion (as found in 2 and 3, cf.Scheme 1), is unknown within the class.22
Conclusions
In conclusion, the first total synthesis of laurendecumallene B (7) has been achieved in 21 linear steps from (S)-(−)-glycidol. As a result, its absolute and relative stereochemical assignments have been confirmed, with one of those centers being unique. Critically, due to its global stereochemical and functional group array, a highly specific variant of our general cyclization/ring-expansion proved necessary to achieve success, with BDSB ultimately being essential for the incorporation of both bromine atoms. Of note, molecular modeling of potential oxonium intermediates was predictive of the type of structural unit needed for ultimate success, suggesting a tool of value for future applications of this design. Finally, with this work leading to the eighth different natural product generated to date with diverse functional groups and chiral centers of differential absolute configuration, it further affirms the value of our process in forging 8-membered bromoether rings in a general fashion.Author contributions
S. A. S., Y.-A. Z., and C. A. T. conceived the project. S. A. S directed the research, and all authors composed the manuscript and the ESI section.† C. A. T. executed the total synthesis and Y.-A. Z. provided significant intellectual contribution for route design and NMR analysis of key intermediates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Antoni Jurkiewicz and Dr C. Jin Qin for assistance with NMR and mass spectrometry, respectively. Support for the calculations performed was completed in part with resources provided by the University of Chicago's Research Computing Center. Financial support for this work came from the University of Chicago and the National Institutes of Health (T32 GM08720, Predoctoral Training Program in Chemistry and Biology, graduate fellowship to C. A. T.).Notes and references
- (a) T. Wanke, A. C. Philippus, G. A. Zatelli, L. F. O. Vieira, C. Lhullier and M. Falkenberg, Rev. Bras. Farmacogn., 2015, 25, 569 CrossRef CAS; (b) B.-G. Wang, J. B. Gloer, N.-Y. Ji and J.-C. Zhao, Chem. Rev., 2013, 113, 3632 CrossRef CAS PubMed.
- A. Fukuzawa, E. Kurosawa and T. Irie, Tetrahedron Lett., 1972, 13, 3 CrossRef.
- (a) A. G. González, J. D. Martín, V. S. Martín, M. Norte, R. Pérez, J. Z. Ruano, S. A. Drexler and J. Clardy, Tetrahedron, 1982, 38, 1009 CrossRef; (b) M. Noite, A. G. Gonzalez, F. Cataldo, M. L. Rodríguez and I. Brito, Tetrahedron, 1991, 47, 9411 CrossRef; (c) X.-D. Li, F.-P. Miao, K. Li and N.-Y. Ji, Fitoterapia, 2012, 83, 518 CrossRef CAS PubMed; (d) A. Fukuzawa and E. Kurosawa, Tetrahedron Lett., 1979, 20, 2797 CrossRef.
- (a) D. J. Kennedy, I. A. Selby, H. J. Cowe, P. J. Cox and R. H. Thomson, J. Chem. Soc., Chem. Commun., 1984, 153 RSC; (b) M. Suzuki, S. Nakano, Y. Takahashi, T. Abe, M. Masuda, H. Takahashi and K. Kobayashi, J. Nat. Prod., 2002, 65, 801 CrossRef CAS PubMed.
- For syntheses of natural products 1–6, see: (a) B. Kim, M. Lee, M. J. Kim, H. Lee, S. Kim, D. Kim, M. Koh, S. B. Park and K. J. Shin, J. Am. Chem. Soc., 2008, 130, 16807 CrossRef CAS PubMed; (b) H. S. Kim, T. Kim, J. Ahn, H. Yun, C. Lim, J. Jang, J. Sim, H. An, Y.-J. Surh, J. Lee and Y.-G. Suh, J. Org. Chem., 2018, 83, 1997 CrossRef CAS PubMed; (c) M. T. Crimmins and E. A. Tabet, J. Am. Chem. Soc., 2000, 122, 5473 CrossRef CAS; (d) T. Saitoh, T. Suzuki, M. Sugimoto, H. Hagiwara and T. Hoshi, Tetrahedron Lett., 2003, 44, 3175 CrossRef CAS; (e) M. Sasaki, A. Hashimoto, K. Tanaka, M. Kawahata, K. Yamaguchi and K. Takeda, Org. Lett., 2008, 10, 1803 CrossRef CAS PubMed; (f) M. Sasaki, K. Oyamada and K. Takeda, J. Org. Chem., 2010, 75, 3941 CrossRef CAS PubMed; (g) M. J. Kim, T.-i. Sohn, D. Kim and R. S. Paton, J. Am. Chem. Soc., 2012, 134, 20178 CrossRef CAS PubMed; (h) F. Yoshimura, T. Okada and K. Tanino, Org. Lett., 2019, 21, 559 CrossRef CAS PubMed; (i) T.-i. Sohn, D. Kim and R. S. Paton, Chem.–Eur. J., 2015, 21, 15988 CrossRef CAS PubMedFor the synthesis of a natural product closely related to 6, see: (j) J. Park, B. Kim, H. Kim, S. Kim and D. Kim, Angew. Chem., Int. Ed., 2007, 46, 4726 CrossRef PubMed.
- (a) S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898 CrossRef CAS PubMed; (b) S. A. Snyder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714 CrossRef CAS PubMed; (c) Y.-A. Zhang, N. Yaw and S. A. Snyder, J. Am. Chem. Soc., 2019, 141, 7776 CrossRef CAS PubMed.
- S. A. Snyder and D. S. Treitler, Angew. Chem., Int. Ed., 2009, 48, 7899 CrossRef CAS PubMed.
- (a) H. S. Sam Chan, Q. N. N. Nguyen, R. S. Paton and J. W. Burton, J. Am. Chem. Soc., 2019, 141, 15951 CrossRef CAS PubMedFor selected examples of other oxonium-based processes, see: (b) D. C. Braddock, Org. Lett., 2006, 8, 6055 CrossRef CAS PubMed; (c) M. Sugimoto, T. Suzuki, H. Hagiwara and T. Hoshi, Tetrahedron Lett., 2007, 48, 1109 CrossRef CAS; (d) B. Kim, M. Lee, M. J. Kim, H. Lee, S. Kim, D. Kim, M. Koh, S. B. Park and K. J. Shin, J. Am. Chem. Soc., 2008, 130, 16807 CrossRef CAS PubMed; (e) D. C. Braddock, D. S. Millan, Y. Pérez-Fuertes, R. H. Pouwer, R. N. Sheppard, S. Solanki and A. J. P. White, J. Org. Chem., 2009, 74, 1835 CrossRef CAS PubMed; (f) K. J. Bonney, D. C. Braddock, A. J. P. White and M. Yaqoob, J. Org. Chem., 2011, 76, 97 CrossRef CAS PubMed; (g) B. S. Dyson, J. W. Burton, T.-i. Sohn, B. Kim, H. Bae and D. Kim, J. Am. Chem. Soc., 2012, 134, 11781 CrossRef CAS PubMed; (h) S. Keshipeddy, I. Martínez, B. F. Castillo II, M. D. Morton and A. R. Howell, J. Org. Chem., 2012, 77, 7883 CrossRef CAS PubMed; (i) K. J. Bonney and D. C. Braddock, J. Org. Chem., 2012, 77, 9574 CrossRef CAS PubMed; (j) D. C. Braddock and D.-T. Sbircea, Chem. Commun., 2014, 50, 12691 RSC; (k) M. T. Taylor and J. M. Fox, Tetrahedron Lett., 2015, 56, 3560 CrossRef CAS PubMedR. Lin, L. Cao and F. G. West, Org. Lett., 2017, 19, 552 CrossRef CAS PubMed.
- N.-Y. Ji, X.-M. Li, K. Li and B.-G. Wang, J. Nat. Prod., 2007, 70, 1499 CrossRef CAS PubMed.
- Y. Yoshimitsu, S. Inuki, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2013, 15, 3046 CrossRef CAS PubMed.
- (a) O.-Y. Jeon and E. M. Carreira, Org. Lett., 2010, 12, 1772 CrossRef CAS PubMed; (b) C. Bonini, M. Campaniello, L. Chiummiento and V. Videtta, Tetrahedron, 2008, 64, 8766 CrossRef CAS; (c) L. Wang, P. Li and D. Menche, Angew. Chem., Int. Ed., 2010, 49, 9270 CrossRef CAS PubMed; (d) L. Wang and D. Menche, Angew. Chem., Int. Ed., 2012, 51, 9425 CrossRef CAS PubMed; (e) G. Stork and S. D. Rychnovsky, J. Am. Chem. Soc., 1987, 109, 1565 CrossRef CAS.
- K. C. Nicolaou, T. V. Koftis, S. Vyskocil, G. Petrovic, W. Tang, M. O. Frederick, D. Y. K. Chen, Y. Li, T. Ling and Y. M. A. Yamada, J. Am. Chem. Soc., 2006, 128, 2859 CrossRef CAS PubMed.
- A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 17, 1295 CrossRef.
- S. J. Danishefsky, M. P. DeNinno, G. B. Phillips, R. E. Zelle and P. A. Lartey, Tetrahedron, 1986, 42, 2809 CrossRef CAS.
- M. Shen, M. Kretschmer, Z. G. Brill and S. A. Snyder, Org. Lett., 2016, 18, 5018 CrossRef CAS PubMed.
- A. M. Arnold, A. Pöthig, M. Drees and T. Gulder, J. Am. Chem. Soc., 2018, 140, 4344 CrossRef CAS PubMed.
- P. A. Evans, V. S. Murthy, J. D. Roseman and A. L. Rheingold, Angew. Chem., Int. Ed., 1999, 38, 3175 CrossRef CAS PubMed.
- C. Sabot, D. Bérard and S. Canesi, Org. Lett., 2008, 10, 4629 CrossRef CAS PubMed.
- J. Ishihara, Y. Shimada, N. Kanoh, Y. Takasugi, A. Fukuzawa and A. Murai, Tetrahedron, 1997, 53, 8371 CrossRef CAS.
- The diol moiety of laurendecumallene B (7) and its epimer (35) were derivatized with the following protecting groups in attempts to grow a single crystal suitable for X-ray crystallographic analysis: p-bromobenzoate, ferrocene carboxylate, 4-nitrobenzoate, and 3,5-dinitrobenzoate. The latter three analogues were all solids; however, none afforded suitable single crystals despite our efforts.
- G. Lowe, Chem. Commun., 1965, 411 RSC.
- Fujii and Ohno proposed a structure for 7 with the opposite bromine chirality (ref. 10); while their 1H NMR spectra for the final material, which is acknowledged to be only a tentative assignment, is indeed quite close to that of the natural product, there are some subtle differences. See the ESI section† for NMR comparisons..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06116a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |