
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton-coupled electron transfer across benzimidazole bridges in bioinspired proton wires†
Emmanuel
Odella‡
 a,
S. Jimena
Mora‡
a,
Brian L.
Wadsworth‡
a,
Joshua J.
Goings‡
b,
Miguel A.
Gervaldo
c,
Leonides E.
Sereno
c,
Thomas L.
Groy
a,
Devens
Gust
a,
Thomas A.
Moore
a,
Gary F.
Moore
*a,
Sharon
Hammes-Schiffer
*b and
Ana L.
Moore
*a
a,
S. Jimena
Mora‡
a,
Brian L.
Wadsworth‡
a,
Joshua J.
Goings‡
b,
Miguel A.
Gervaldo
c,
Leonides E.
Sereno
c,
Thomas L.
Groy
a,
Devens
Gust
a,
Thomas A.
Moore
a,
Gary F.
Moore
*a,
Sharon
Hammes-Schiffer
*b and
Ana L.
Moore
*a
aSchool of Molecular Sciences, Arizona State University, Tempe, Arizona 85287-1604, USA. E-mail: gary.f.moore@asu.edu; amoore@asu.edu
bDepartment of Chemistry, Yale University, New Haven, Connecticut 06520-8107, USA. E-mail: sharon.hammes-schiffer@yale.edu
cDepartamento de Química, Facultad de Ciencias Exactas, Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Agencia Postal No 3, 5800 Río Cuarto, Córdoba, Argentina
First published on 20th March 2020
Abstract
Designing molecular platforms for controlling proton and electron movement in artificial photosynthetic systems is crucial to efficient catalysis and solar energy conversion. The transfer of both protons and electrons during a reaction is known as proton-coupled electron transfer (PCET) and is used by nature in myriad ways to provide low overpotential pathways for redox reactions and redox leveling, as well as to generate bioenergetic proton currents. Herein, we describe theoretical and electrochemical studies of a series of bioinspired benzimidazole-phenol (BIP) derivatives and a series of dibenzimidazole-phenol (BI2P) analogs with each series bearing the same set of terminal proton-accepting (TPA) groups. The set of TPAs spans more than 6 pKa units. These compounds have been designed to explore the role of the bridging benzimidazole(s) in a one-electron oxidation process coupled to intramolecular proton translocation across either two (the BIP series) or three (the BI2P series) acid/base sites. These molecular constructs feature an electrochemically active phenol connected to the TPA group through a benzimidazole-based bridge, which together with the phenol and TPA group form a covalent framework supporting a Grotthuss-type hydrogen-bonded network. Infrared spectroelectrochemistry demonstrates that upon oxidation of the phenol, protons translocate across this well-defined hydrogen-bonded network to a TPA group. The experimental data show the benzimidazole bridges are non-innocent participants in the PCET process in that the addition of each benzimidazole unit lowers the redox potential of the phenoxyl radical/phenol couple by 60 mV, regardless of the nature of the TPA group. Using a series of hypothetical thermodynamic steps, density functional theory calculations correctly predicted the dependence of the redox potential of the phenoxyl radical/phenol couple on the nature of the final protonated species and provided insight into the thermodynamic role of dibenzimidazole units in the PCET process. This information is crucial for developing molecular “dry proton wires” with these moieties, which can transfer protons via a Grotthuss-type mechanism over long distances without the intervention of water molecules.
Introduction
Energy transfer, electron transfer, and proton transfer processes are choreographed by a light-driven water-plastoquinone oxidoreductase enzyme better known as photosystem II (PSII) in all water oxidizing photosynthetic organisms. Understanding how these processes are linked and coupled to one another is one of the key principles for designing efficient artificial photosynthetic systems.1–3 In PSII, photochemical events begin with either direct absorption of light by several chlorophyll a molecules, known collectively as P680, or the absorption of light by antenna pigments followed by energy transfer to P680. The excited singlet state of P680 (P680*) then undergoes rapid electron transfer to reduce a nearby chlorophyll (Chl), forming a charge separated pair, P680˙+–Chl˙−. In a series of electron transfer steps, the strongly reducing Chl˙− species reduces first a pheophytin and subsequently plastoquinone A and then B. The highly oxidizing P680˙+ species, the other half of the charge separated pair, is reduced by electron transfer from the tyrosine Z (Tyrz) residue. This redox event occurs with concomitant transfer of the phenolic proton to its hydrogen-bonded partner, His190, in a proton-coupled electron transfer (PCET) process leading to formation of a neutral tyrosine radical (Tyrz˙).4–6 Subsequently, Tyrz˙ advances the oxidation state of the oxygen evolving complex (OEC) by electron transfer from the Mn4O5Ca OEC and recovers its proton, presumably from His190.PCET reactions are also involved in the water oxidation chemistry that occurs during advancement of the OEC's oxidation state, which results in redox leveling and is central to bioenergetics.7 Coupled to electron transfer from the Mn4O5Ca complex to Tyrz˙, protons are extracted from substrate water by the Mn4O5Ca catalytic complex. These protons ultimately go to the lumen of the thylakoid membrane, most likely being transferred along Grotthuss-type proton wires.8 Overall the process involves transfer of four electrons and four protons to accomplish the oxidation of two water molecules. Although atomic level mechanistic details are not fully understood, there is general agreement that proton transfer along the “wires” to the lumen is coupled to the oxidation process and is part of the bioenergetic electrical system generating proton-motive force (PMF) and driving ATP synthesis.9
The thermodynamics of these processes are crucial to the control of proton activity in catalysis and the generation of proton currents involved in bioenergetics. The relatively high redox potential required for water splitting is delivered by Tyrz˙, which seems to require participation in a local hydrogen-bond network. Model studies indicate that losing the phenolic hydrogen-bonded proton in the Tyrz–His190 pair to the surroundings would result in a redox couple (Tyrz˙/Tyrz− instead of Tyrz˙/Tyrz) with a midpoint potential (E1/2) that is insufficient to oxidize water (and would likely render the redox process irreversible).10,11 At the other limit, if the phenolic proton remains on the Tyrz residue and is not transferred to its hydrogen-bonded partner His190, the oxidation of Tyrz and formation of Tyrz˙+ by P680˙+ would be thermodynamically uphill.10,11 To keep the redox potential of Tyrz in the appropriate range, nature employs the reaction pathway alluded to above, involving hydrogen-bonded assemblies where both electrons and protons are transferred in a PCET process.9,12–15
The PCET concept has been extensively studied in connection with chemical and biochemical catalysis, bioenergetics, and Grotthuss-type proton wires and has inspired many researchers to build artificial constructs to mimic the Tyrz–His190 function.16–21 Among them, a benzimidazole-phenol (BIP, containing an unsubstituted benzimidazole group, see structure in Scheme 1, ESI Page S5†), where the phenol moiety mimics Tyrz and the benzimidazole mimics His190, provides a minimalist structure demonstrating PCET.11,22–25
In our previous work, the amino-substituted BIP (4, see Fig. 1) featured a concerted one-electron, two-proton transfer (E2PT) process upon electrochemical oxidation of the phenol (see Fig. 2).22 The PCET process in this genre of compounds is concerted in that no thermodynamically stable intermediate was found. This designation is further supported by the agreement between the calculated redox potential for the E2PT process and the experimentally measured redox potential. However, PCET in these systems may be asynchronous on the femtosecond timescale, although such asynchronicity does not impact the electrochemical measurements. As a consequence of transferring the proton to the terminal proton-accepting (TPA) group in 4, the potential of the phenoxyl radical/phenol redox couple is ∼300 mV lower than that measured in experiments using BIP. This drop in the E1/2 would restrict its use as a redox mediator in a water splitting process at near neutral pH. Theoretical calculations predicted that BIPs bearing substituents on the 7-position of the benzimidazole with lower pKa's than the amino group would have E1/2 values closer to the ∼0.95 V vs. SCE associated with the one-electron one-proton transfer (E1PT) process characteristic of the BIP phenoxyl radical/phenol redox couple.22 Indeed, BIP compounds substituted with N-phenylimines as a TPA group did have midpoint potentials near 1.00 V vs. SCE.26 Moreover, the ratio of E2PT to E1PT products following oxidation of the phenol could be modulated by the nature of the substituents at the para-position of the N-phenylimine moiety. Density functional theory (DFT) calculations and infrared spectroelectrochemistry (IRSEC) results showed that with stronger electron-donating substituents the proton of the benzimidazole is transferred to the imine group upon phenol oxidation, leading to a net proton movement of ∼6.4 Å along the hydrogen-bond network.26
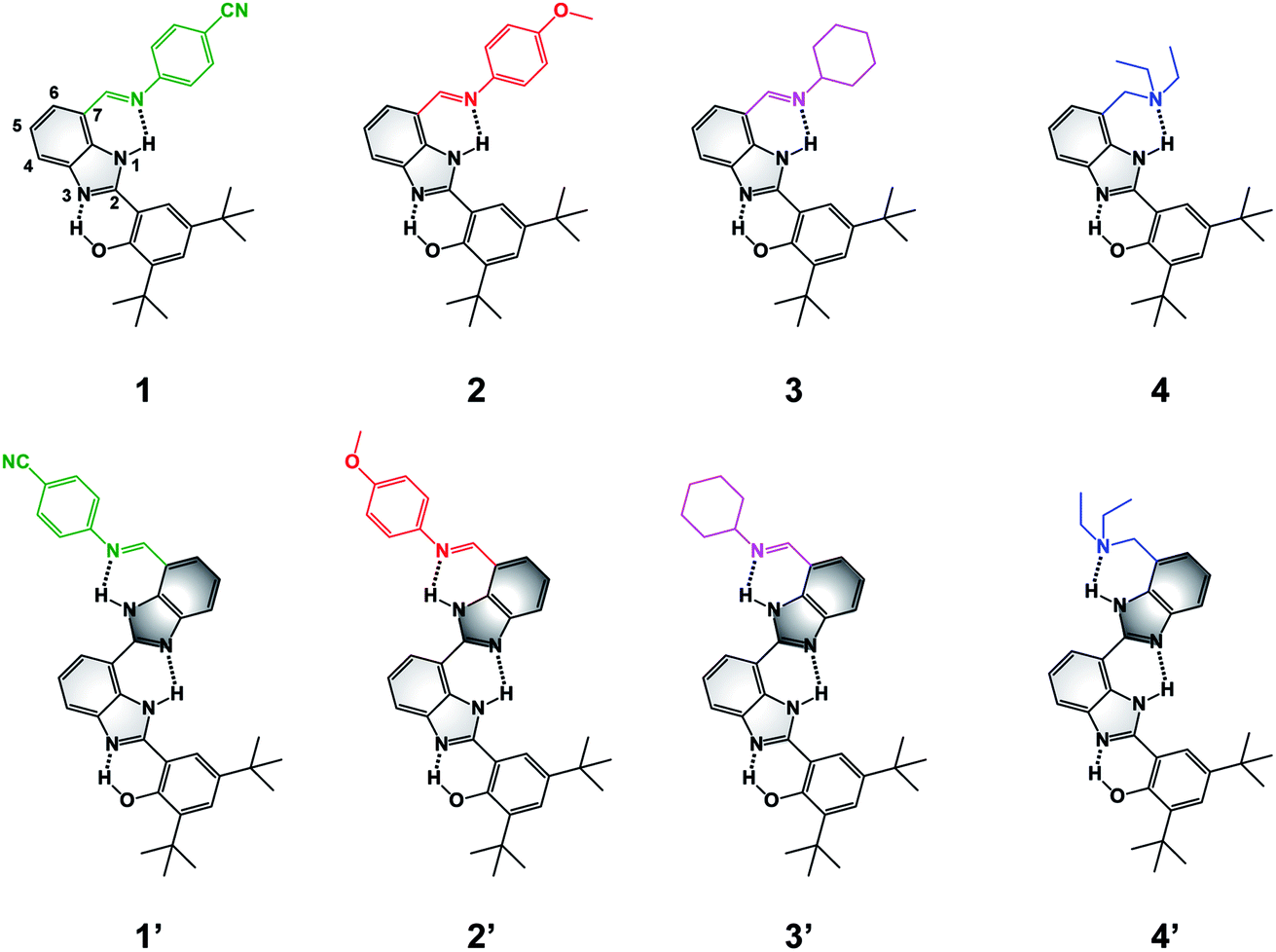 | ||
| Fig. 1 Molecular structures of BIP (1–4) and BI2P (1′–4′) constructs bearing different terminal proton-accepting groups (color-coded as in Fig. 4–6). The numbering system is shown in 1, and the incorporation of a second benzimidazole to form dibenzimidazole based bridges between the phenol moiety and the terminal proton-accepting group are shown in dark grey in 1′–4′. | ||
To extend the proton translocation, new constructs bearing an additional benzimidazole moiety, which generates a dibenzimidazole bridge and increases the spatial separation between the oxidation center (the phenol moiety) and the TPA group, have been synthesized (see Fig. 1). These molecules were designed to investigate the control of phenol redox potentials in amino and imine-substituted BIP systems,22,26 and to further explore the thermodynamics of proton transfer across a dibenzimidazole bridge unit (see Fig. 2). Fig. 1 shows compounds 1, 2, 3 and 4, which are BIPs substituted at the 7-position with para-cyano-N-phenylimine, para-methoxy-N-phenylimine, N-cyclohexylimine, and an exocyclic diethylamino group, respectively. By including a second benzimidazole, the analogous compounds, BI2Ps 1′–4′ bearing the same TPA groups as in 1–4, were synthesized.
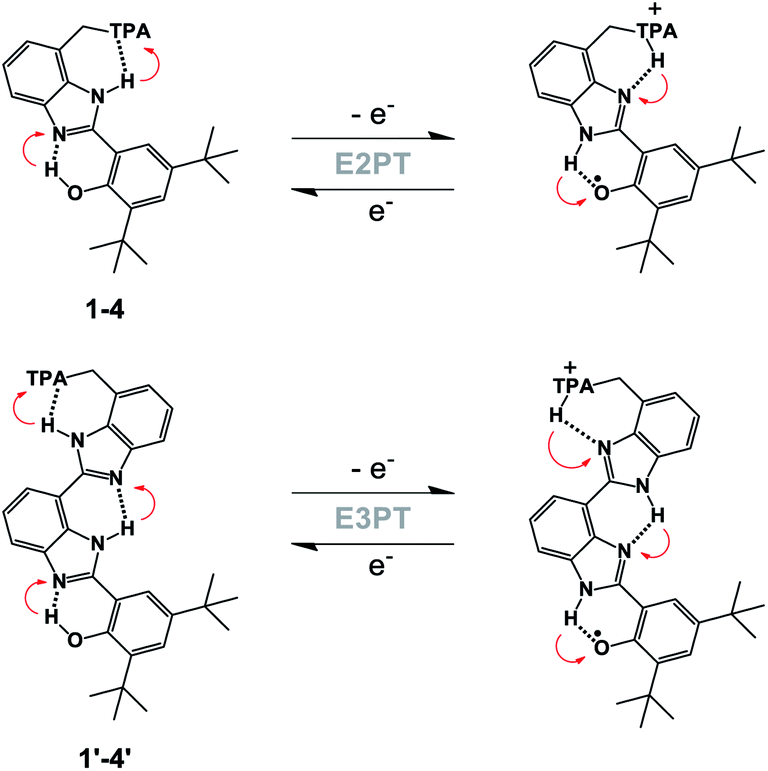 | ||
| Fig. 2 Generic structure of BIP derivatives 1–4 (top), and BI2P analogs 1′–4′ (bottom) showing the electrochemical oxidation of the phenol coupled with two (E2PT) or three (E3PT) proton transfers. Red arrows indicate proton movement for the forward and reverse processes. | ||
This work provides insights regarding structure–function relationships governing the influence of hydrogen-bond networks involving phenols, bridging benzimidazoles and TPAs on the E1/2 of the phenol redox process, which initiates the fully reversible PCET reaction. This is a requirement for the design of efficient artificial proton wires able to conduct protons over distances of ∼30 Å, necessary for the construction of functional artificial biological membranes.
Results and discussion
Synthesis and structural characterization
Model compounds 1–3 were obtained by reaction of a derivative of BIP containing a formyl group at the 7-position (BIP–CHO) with the corresponding aromatic or aliphatic primary amino derivatives in the presence of a catalytic amount of pyrrolidine.26,27 Compound 4 was obtained by reduction of the tertiary amide group in the 7-position of BIP (BIP–CONEt2) following a procedure previously described.22 The second benzimidazole moiety of the BI2P family was introduced by condensation of BIP–CHO with methyl 2,3-diaminobenzoate followed by conversion of the ester group to the aldehyde (BI2P–CHO), the precursor of compounds 1′–4′. The complete synthetic route, conditions, and structural characterization are described in the ESI (Pages S4–S16†).The spatial arrangement of the proton donor–acceptor systems and the strength of the hydrogen bonds connecting them are important design parameters for the success of intramolecular PCET reactions.28–30 In this regard, the crystal structures of both 4 and 2′ indicate the presence of the intramolecular hydrogen bonds in these molecules (see Fig. 3, CCDC deposition no. 1968269 and 1968270, respectively). In the case of 4, the O1–N1 distance between the phenolic O and the proximal N of the benzimidazole is 2.59 Å, and the N2–N3 distance between the NH of the benzimidazole and the N of the TPA (exocyclic diethylamine) is 2.85 Å. Those distances remain basically constant in 2′; the O1–N1 distance between the phenol O and the proximal N of the first benzimidazole unit is 2.59 Å, and the N4–N5 distance between the N of the second benzimidazole and the imine N of the TPA is 2.84 Å. The two benzimidazoles are also connected through a hydrogen bond with the corresponding N2–N3 distance of 2.80 Å. The X-ray crystallographic data also reflects a small dihedral angle between the phenol and the benzimidazole moiety in 4 and 2′, with angles of 8.0° and 3.6°, respectively. Disregarding the crystal packing effects, these small dihedral angles indicate that the extent of conjugation between the phenol and benzimidazole is essentially the same in 4 and 2′. Similarly, small dihedral angles in related compounds bearing benzimidazole-phenol moieties have been observed.26,31 The optimized structures determined from DFT calculations are in good agreement with the crystallographic data of 4 and 2′, and some selected bond lengths and dihedral angles are summarized in Tables S23 and S24.†
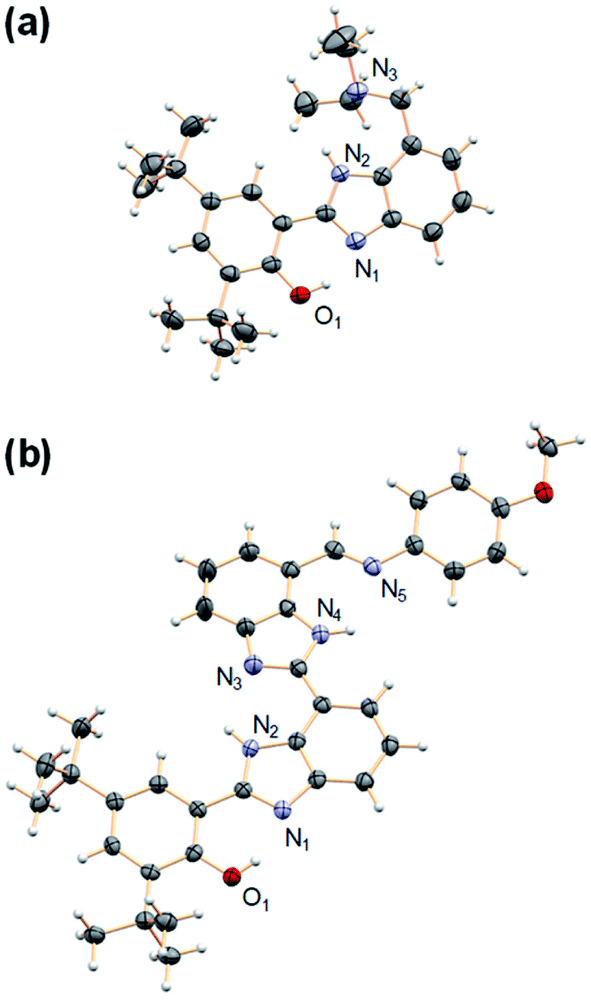 | ||
| Fig. 3 Crystal structures of (a) 4 and (b) 2′. Carbon atoms are shown in gray, oxygens in red, nitrogens in light violet, and hydrogens in white. The heteroatoms involved in hydrogen bonds are labeled. Thermal ellipsoids are drawn at the 50% probability level. A molecule of DCM was found in the crystal unit cell (see ESI†) of 2′ and has been omitted for clarity. | ||
In agreement with the crystal structure data indicating a partially conjugated and extended π system provided by the dibenzimidazole in 2′, a red shift in the absorption spectrum of 2′ as compared to 2 is observed. In fact, a red shift between all the BIP molecules (1–4) and the analogous BI2P molecules (1′–4′) is observed (Fig. S11†). This change in electronic structure as the partially conjugated framework is extended contributes to the redox potential lowering as the Grotthuss-type wire is extended (vide infra and analysis on Page S62†).
In solution strong hydrogen bonds in 4 have been previously demonstrated22 and, in the case of 2′, evidence for strong hydrogen bonds is clear from the relatively downfield phenolic proton resonances observed in the 400 MHz 1H NMR spectra (see Table 1 and structural characterization in the ESI). In CDCl3 solution, the chemical shift of the phenolic proton appears at 13.40 ppm, and the chemical shifts of both benzimidazole NH groups are clearly resolved. The signal corresponding to the distal NH proton (second benzimidazole) forming the hydrogen bond with the imine N (N4H⋯N5 in Fig. 3b) is assigned to the resonance at 11.83 ppm. By extension, the NH proton connecting the two benzimidazole moieties through a hydrogen bond (N2H⋯N3 in Fig. 3b) is assigned to the resonance at 12.12 ppm.
| Compound | δ OH (ppm) | δ NH (ppm)a | δ NH (ppm)b |
|---|---|---|---|
| a NH involved in the internal hydrogen bond between benzimidazoles (N2H⋯N3 in Fig. 3b). b NH involved in the internal hydrogen bond between the benzimidazole and the terminal proton-accepting group (either imine, such as N4H⋯N5 in Fig. 3b, or exocyclic amine, such as N2H⋯N3 in Fig. 3a). c Data from ref. 26. d Data from ref. 27. e Data from ref. 22. f Broad signal presumably overlapped with the NH signal assigned to the internal hydrogen bond between benzimidazoles (N2H⋯N3 in Fig. 3b). | |||
| 1 | 13.19c | — | 11.48c |
| 2 | 13.30c | — | 11.84c |
| 3 | 13.30d | — | 11.87d |
| 4 | 13.45e | — | 11.17e |
| 1′ | 13.36 | 12.07 | 11.54 |
| 2′ | 13.40 | 12.12 | 11.83 |
| 3′ | 13.42d | 12.15d | 11.90d |
| 4′ | 13.43 | 12.20 | ∼12.20f |
In the case of compound 1′, the –CN group does not affect the strength of the internal hydrogen bond connecting both benzimidazoles (N2H⋯N3), and this chemical shift is basically the same as that observed for the analogous internal hydrogen bond in 2′. However, the chemical shift of the NH proton hydrogen bonded with the imine N (N4H⋯N5) is upfield of the corresponding resonance observed in 2′, suggesting that this hydrogen bond is weaker due to the decrease in electron density on the imine N resulting from the electron-withdrawing effect of the –CN group.32 The opposite effect is observed in 3′, where the more basic cyclohexylimine group strengthens the corresponding hydrogen bond.27
A general trend in the chemical shifts of protons involved in hydrogen bonds in the BIPs (1–4) and BI2Ps (1′–4′) is revealed by inspection of the 1H NMR data (see Table 1). There is a slight downfield chemical shift, which generally is associated with stronger hydrogen bonds27,31,33 of the NH proton hydrogen bonded to the TPA group in BI2P compounds compared to the analogous BIPs. Within the 1′–4′ series, evidence of the strength of the hydrogen bonds is also indicated by the frequency of the benzimidazole NH IR stretching mode (νNH). A progressive shift in the νNH to lower frequencies is observed across the BI2P series (see Fig. S12†), consistent with the trend observed in the 1H NMR. This behavior supports the increase in the hydrogen-bond network strength due to the increase in the basicity of the TPA.
The existence of isomers resulting from 1,3-tautomerism is seen in some benzimidazole-containing compounds.34 Indeed, depending on both the hydrogen-bond ability of the solvent and the strength of the hydrogen bond between the phenol and the imidazole N, compounds 1–4 may have additional isomers resulting from 1,3-tautomerism and rotation around the bond linking the benzimidazole and the phenolic moiety.22,26 In the case of 1–4, only one isomer has the internal hydrogen-bond network necessary to achieve two intramolecular proton transfers upon phenol oxidation. Because compounds 1′–4′ have an additional benzimidazole unit, the number of possible isomers in solution increases relative to 1–4. For 1′ and its precursors (BI2P–COOCH3, BI2P–CH2OH and BI2P–CHO, see synthesis and NMR characterization in the ESI†), the presence of at least two isomers in CDCl3 was detected, and one of them lacks the hydrogen-bond network necessary for the two-proton intramolecular translocations. On the contrary, the 1H NMR spectra of compounds 2′–4′ clearly show the existence of essentially one isomer in non-polar solvents such as CDCl3, suggesting that a more basic group (N-phenylimine p-substituted with a strong electron donating group such as –OCH3, cyclohexylimine, or tertiary amine) strengthens the terminal hydrogen bond and thereby favors the conformation where the hydrogen-bond chain is unbroken across the molecule.27
Electrochemical studies
Fig. 4 shows cyclic voltammograms (CVs) recorded using BI2Ps 1′–4′ and their analogous BIPs 1–4. For compounds 1–4, data relevant to the chemical and electrochemical reversibility of the phenoxyl radical/phenol redox couples are included in Table 2. In 0.5 M tetrabutylammonium hexafluorophosphate (TBAPF6) acetonitrile solutions containing 1–4, the cathodic to anodic peak intensity ratios (ic/ia), a measure of chemical reversibility, are ≥0.65 at a scan rate of 100 mV s−1 (see ESI†), and the peak-to-peak separations (ΔEp), a measure of electrochemical reversibility, are 0.07 V for 1–3 and 0.10 V for 4. In the BI2P series, the ΔEp is 0.11 for 1′, 2′ and 4′, and 0.10 for 3′.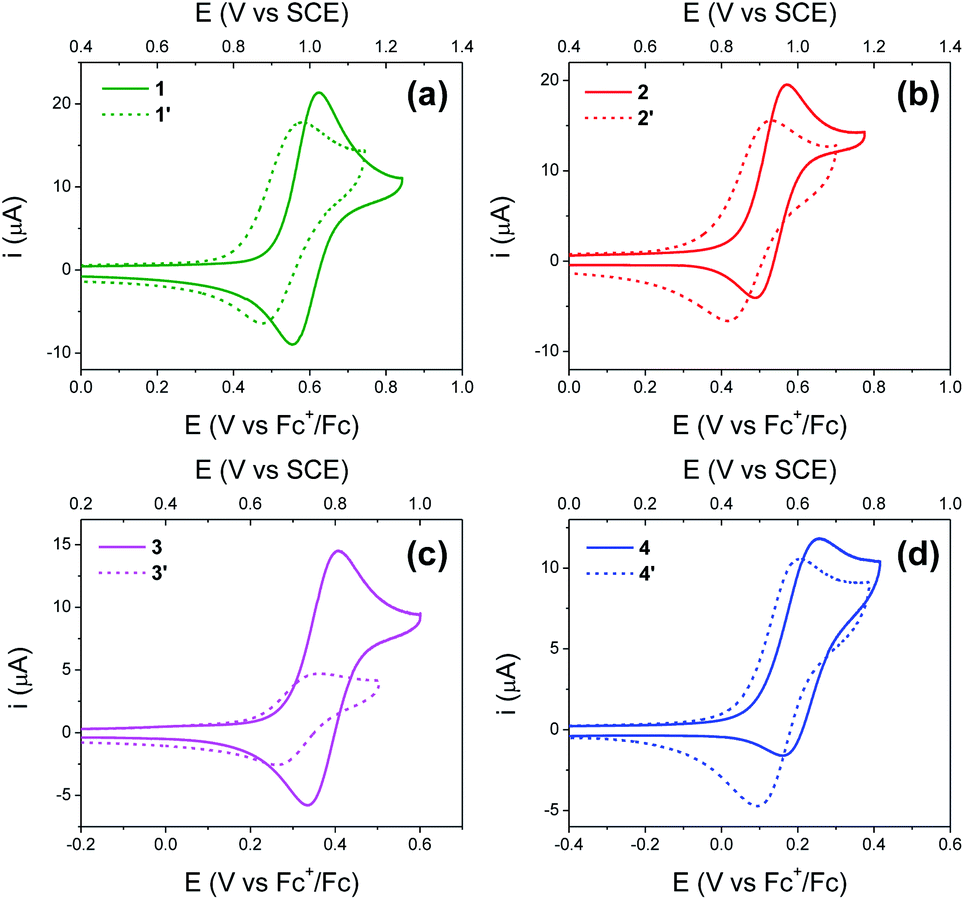 | ||
| Fig. 4 (a–d) Cyclic voltammograms of BI2Ps 1′–4′ (dashed lines). The cyclic voltammograms of analogous BIPs 1–4 (solid lines) are included for comparison. Concentration: 1 mM of the indicated compound, 0.5 M TBAPF6 supporting electrolyte in dry acetonitrile. WE: glassy carbon. Pseudo RE: Ag wire (ferrocene as internal reference). CE: Pt wire. Scan rate, 100 mV s−1. Compound 3′ has low solubility in acetonitrile. | ||
| Compound | CalculatedaE1/2 (V vs. SCE) | ExperimentalbE1/2 (V vs. SCE) | ΔEp (V) | i c/ia |
|---|---|---|---|---|
| a Each computed redox potential assumes the maximum number of intramolecular proton transfers. The redox potentials corresponding to the intermediates that would arise from only some of these proton transfers are shown in Table S25. b The potential of the pseudoreference electrode was determined using the ferrocenium/ferrocene redox couple as an internal standard and adjusting to the saturated calomel electrode (SCE) scale (with E1/2 taken to be 0.40 V vs. SCE in acetonitrile).35 c Values from ref. 26. d Reference potential for all compounds in this table; agrees by construction. | ||||
| 1 | 1.00 | 0.99c | 0.07c | 0.76c |
| 2 | 0.93d | 0.93c | 0.07c | 0.65c |
| 3 | 0.77 | 0.77 | 0.07 | 0.83 |
| 4 | 0.61 | 0.61 | 0.10 | 0.67 |
| 1′ | 0.91 | 0.93 | 0.11 | 0.88 |
| 2′ | 0.77 | 0.87 | 0.11 | 0.98 |
| 3′ | 0.72 | 0.71 | 0.10 | 1.00 |
| 4′ | 0.55 | 0.55 | 0.11 | 0.98 |
Experimental and calculated E1/2 values for the phenoxyl radical/phenol couple, as well as ΔEp's, are summarized in Table 2. Each E1/2 was calculated assuming that all possible intramolecular proton transfer events occur, i.e., two protons transfer in 1–4, and three protons transfer in 1′–4′.
For BIPs 1–4, E1/2 values of the phenoxyl radical/phenol redox couples were taken as the average of the anodic and cathodic peak potentials, yielding values of 0.99, 0.93, 0.77 and 0.61 V vs. SCE, respectively.22,26 In the case of the BI2P family, the same trend in the E1/2 to lower values with increasing basicity of the TPAs was observed. The reduction of the E1/2 across the BI2P series follows the same trend as the hydrogen-bond strength caused by the nature of the TPA group. Compound 1′ has the weakest hydrogen-bond network in the BI2P series, consistent with the chemical-shift values of the protons attached to the heteroatoms (NHs and OH, Table 1), and the phenol is relatively more difficult to oxidize (highest E1/2). The opposite effect is observed in the case of 4′, which exhibits the strongest hydrogen-bond network in the BI2Ps series, and the phenol is easier to oxidize. In our previous work, we observed a clear relationship between the electronic effect of the imine substituents (–CN and –OCH3 groups in compounds 1 and 2, respectively) and the E1/2 of the phenol at ∼12 Å distance from the substituent.26 In the case of 1′ and 2′, the effect of the substituent on the E1/2 is still present, even though the distance between the phenol group and the substituent is even larger (∼16 Å).
As illustrated in Fig. 5, the E1/2 of each compound in the BI2P series (1′–4′) is 60 mV lower than in each analogous compound in the BIP series (1–4). In other words, this 60 mV shift is a consequence of the added benzimidazole and is independent of the TPA group. A similar 60 mV shift following each successive addition of a bridging benzimidazole was previously reported for a series of BIPs having the same TPA group but containing either one, two, or three bridging benzimidazole groups making up the Grotthuss-type proton wire.27 Thus, in that case the E1/2 is 120 mV lower for the BIP featuring three benzimidazole groups than the E1/2 of the BIP featuring one benzimidazole group.27
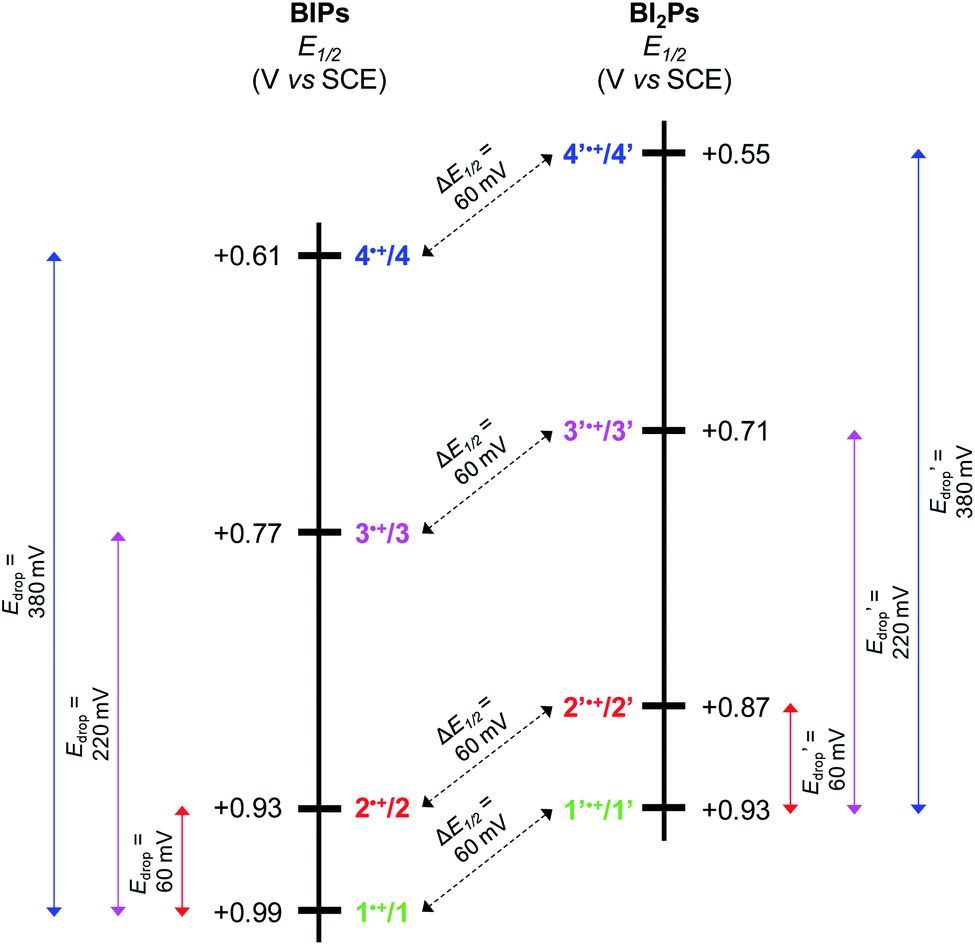 | ||
| Fig. 5 Diagram showing the experimentally determined E1/2 for BIP derivatives (1–4) and BI2P analogs (1′–4′). The colored arrows show that the drop in E1/2 (Edrop) for the different TPAs is the same in both series. The dashed black arrows indicate the decrease in E1/2 (ΔE1/2) due to the addition of one benzimidazole unit, which is 60 mV regardless of the TPA. | ||
In Fig. 5 we show that the ΔE1/2 shift of 60 mV does not depend on the TPA group. Therefore, it must be a property of the electronic structure of the linked benzimidazole groups and their partial conjugation with the phenol. Fig. 5 also shows the changes in redox potential of the phenoxyl radical/phenol couple within the 1–4 or 1′–4′ series (see Edrop and  indicated by colored arrows). These Edrop and
indicated by colored arrows). These Edrop and 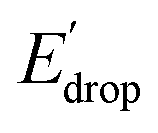 values can be predicted using the pKa values associated with the respective TPA groups, which are the same in both series. Thus, Edrop and
values can be predicted using the pKa values associated with the respective TPA groups, which are the same in both series. Thus, Edrop and 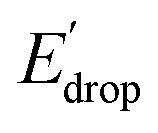 do not depend on the number of benzimidazole units comprising the bridge and their partial conjugation with the phenol. For example, the value of Edrop or
do not depend on the number of benzimidazole units comprising the bridge and their partial conjugation with the phenol. For example, the value of Edrop or 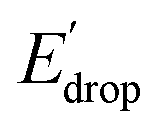 resulting from changing the TPAs between 4 and 1 or 4′ and 1′ is 380 mV, qualitatively in agreement with the ΔpKa in acetonitrile of a tertiary amine and a benzimidazole substituted with an electron-withdrawing group.36
resulting from changing the TPAs between 4 and 1 or 4′ and 1′ is 380 mV, qualitatively in agreement with the ΔpKa in acetonitrile of a tertiary amine and a benzimidazole substituted with an electron-withdrawing group.36
In our theoretical treatment, the change in the redox potential due to incorporation of a second benzimidazole and different TPAs can be expressed by the thermodynamics of the overall PCET process (see analysis Page S62†). The calculations show that upon oxidation of the phenol, each benzimidazole and TPA contributes a term corresponding to ΔG < 0 for proton transfer. In other words, each successive proton transfer reaction is exoergic and therefore lowers the overall E1/2 of the phenoxyl radical/phenol redox couple (see Table S25†).
Infrared spectroelectrochemistry
In addition to the close agreement between the experimental and calculated E1/2, the use of infrared spectroelectrochemistry (IRSEC) further corroborates proton translocation in these systems. If the movement of protons to the TPA upon phenol oxidation is inhibited in 1′–4′, formation of E1PT or E2PT products would be evidenced by the appearance of the characteristic bands at ∼1556 cm−1 and ∼3320 cm−1, which are assigned to the NH in-plane bending vibration and the NH stretching modes of the benzimidazolium cation, respectively.22,26 Conversely, if the electrochemical oxidation of the phenol drives intramolecular proton transfer to the TPA, a characteristic band at ∼1650 cm−1 associated with the coupling between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching and the CNH+ bending modes of the protonated imine26,27 is expected for BI2Ps having the imine linkage as a TPA group (1′–3′). In the case of 4′, a determination of the different oxidized species formed upon electrochemical oxidation is enabled by analysis of the changes in the absorbance associated with NH vibrational modes before and after protonation (see discussion below).22
N stretching and the CNH+ bending modes of the protonated imine26,27 is expected for BI2Ps having the imine linkage as a TPA group (1′–3′). In the case of 4′, a determination of the different oxidized species formed upon electrochemical oxidation is enabled by analysis of the changes in the absorbance associated with NH vibrational modes before and after protonation (see discussion below).22
Fig. 6a and c show IRSEC data (in the 1700–1400 cm−1 region) collected using solutions of compounds 2′ and 4′, respectively. The most significant and characteristic changes in absorbance upon increasing the polarization potential are indicated by upward and downward arrows. As seen in the case of 2′ (Fig. 6a), the bands at 1442 cm−1, 1505 cm−1, 1531 cm−1, 1610 cm−1, and 1625 cm−1 progressively decrease, and new bands at 1570 cm−1 and 1653 cm−1 appear. Among them, the bands at ∼1442 cm−1 and ∼1570 cm−1 (observed for both 2′ and 4′) are attributed to CC ring stretching modes. These bands show similar changes in absorbance intensity upon increasing the potential of polarization as previously observed for other electrochemically generated phenoxyl radicals.37 Formation of the E3PT product upon oxidation of 2′ is indicated by the decrease of the band at 1625 cm−1 assigned to the CN stretching of the imine group,38 the absence of the NH in-plane bending vibration mode of the benzimidazolium ion at ∼1556 cm−1,22 and the increase of the band at 1653 cm−1 attributed to the protonated imine group.26,27 In the high frequency region (Fig. 6b), the absence of a typical broad band at ∼3320 cm−1 assigned to the NH stretching mode of the benzimidazolium ion22,26,27 also indicates that the PCET process does not give rise to either the E1PT or E2PT species as the final products, and the proton transfer to the TPA (forming the E3PT product) is the only species detected following electrochemical oxidation of 2′.
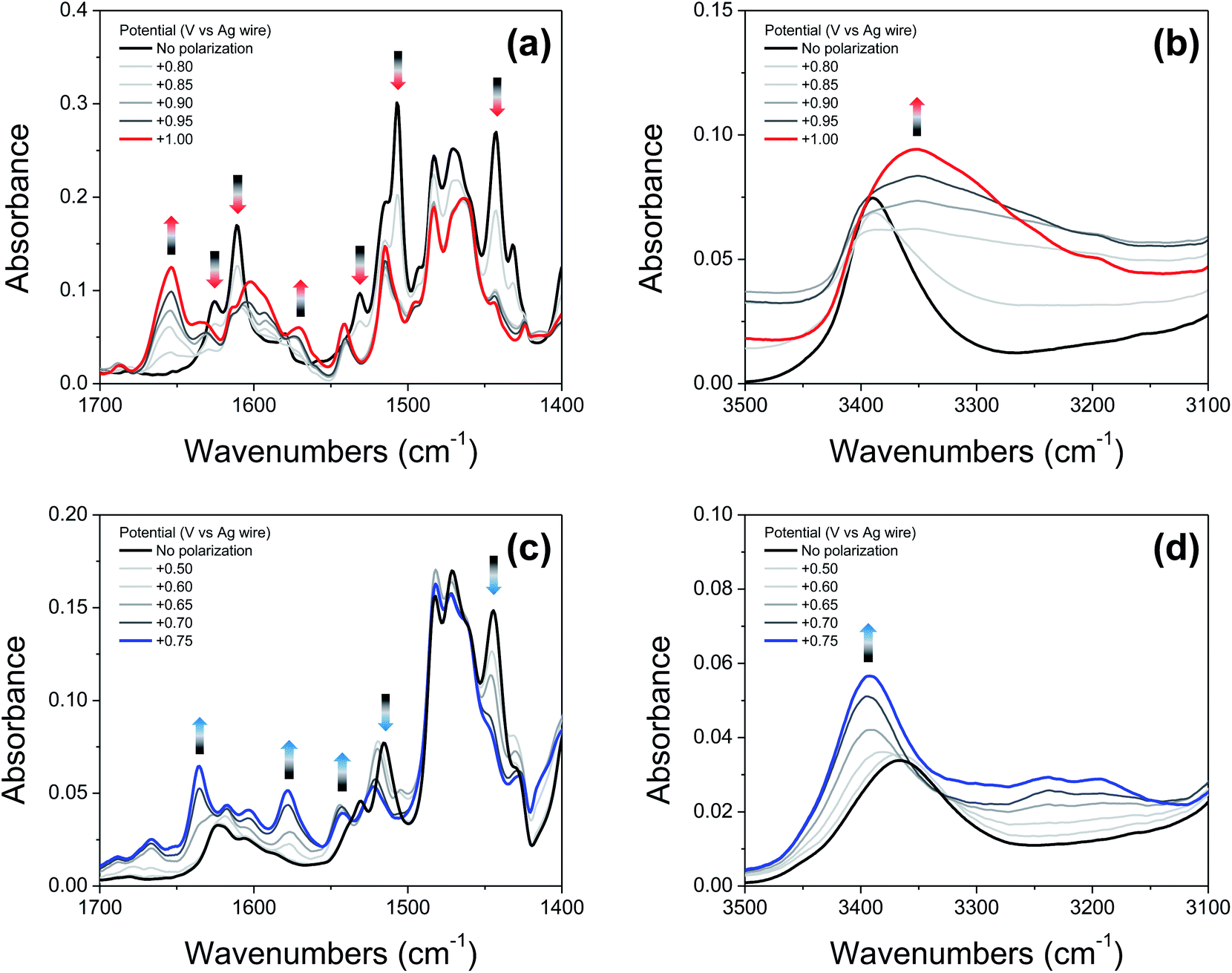 | ||
| Fig. 6 (a and c): IRSEC spectra of 2′ and 4′, respectively, (19 mM solutions) polarized in 50 mV or 100 mV steps in the 1700–1400 cm−1 region. (b and d): IRSEC spectra of 2′ and 4′, respectively, under the same conditions in the 3500–3100 cm−1 region. Solvent: DCM, 0.1 M TBAPF6. Some of the characteristic bands showing changes upon electro-oxidation are indicated with upward and downward arrows. | ||
Based on the agreement between theoretical calculations and experimental values (see Table 2) for 4′, translocating three protons upon phenol oxidation leads to the most thermodynamically favorable oxidized state (the E3PT product). For the neutral species, the bands at 1530 cm−1 and 1515 cm−1 are assigned to a vibration including the NH in-plane bending of the NH hydrogen bonded with the exocyclic amine and the NH connecting both benzimdazole moieties, respectively. In the case of the analogous BIP 4, the band at 1515 cm−1 (associated with the NH in-plane bending of the NH hydrogen bonded with the exocyclic amine) decreases and a new band arises at 1529 cm−1 upon electro-oxidative conditions.22 For these compounds, control experiments together with DFT modeling were used to explain these changes in terms of a concerted or nearly concerted two-proton translocation leading to the protonation of both the exocyclic amine and the proximal N of the benzimidazole.22 Following electrochemical oxidation of 4′, a similar change in the pattern of the related bands is observed. In particular, the band at 1515 cm−1 decreases in intensity and a new band at 1522 cm−1 grows in, whereas the band at 1530 cm−1 is not apparent and a new band appears at ∼1542 cm−1, suggesting that both benzimidazole NHs have been transferred to their nearest basic sites (the second benzimidazole moiety and exocyclic amine) forming the E3PT product. As previously discussed for the imine 2′, the absence of the characteristic NH in-plane bending band (∼1556 cm−1) of the benzimidazolium ion confirms non-detectable levels of E1PT and E2PT products following electrochemical oxidation of 4′. This conclusion is also supported by the absence of the characteristically strong NH stretching mode of the benzimidazolium ion (at ∼3320 cm−1).22,26 Upon phenol oxidation, the intensity of the νNH increases and a shift from 3366 cm−1 to 3392 cm−1 is observed (Fig. 6d), suggesting that the hydrogen-bond network weakens in the oxidized form of 4′. This weakening is ascribed to a disruption of the hydrogen bond due to protonation of the tertiary amine.22 Moreover, all assignments and changes observed for both molecules (2′ and 4′) are supported by DFT calculations (see ESI, Fig. S19 and Table S26†).
The previously published IRSEC of 3′ indicated the formation of the E3PT product unequivocally,27 while the IRSEC of 1′ shows the formation of the E3PT product, although in low yield (see ESI, Fig. S13†). In this case, the formation of the E2PT product is slightly favored theoretically, but the experimental IRSEC data is inconclusive concerning the formation of such a product.
Kinetic isotopic effect
Deuterium kinetic isotope effect values have been determined electrochemically and are shown in Table S22.† The KIE values for 1, 2 and 4 have been previously reported,22,26 and the values obtained for 2′–4′ are similar (within experimental error) to those of their analogous BIPs with only one benzimidazole bridge. The experimental values obtained for 4 and other related BIPs agree with the values obtained theoretically, assuming a concerted mechanism.22,26 However, the experimental error and the uncertainty inherent in the calculated values precluded the extraction of mechanistic information. The KIE values obtained for 1–4 are relatively small (i.e., 0.9–1.5) but are in agreement with previously reported values for related systems.28,29,31,33 These small values have been explained theoretically in terms of dominant contributions from excited vibronic states with significant overlap integrals between the reactant and product proton vibrational wave functions.22 Similarly, small KIE values were obtained for the more extended 2′–4′ systems.Conclusions
Using both experimental and theoretical techniques, we have characterized a PCET process in which the translocation of protons across a benzimidazole or a dibenzimidazole proton wire to a TPA is initiated by the electrochemical oxidation of the phenol. In contrast to our previous report, in which different length proton wires using the same TPA were studied, herein several different TPAs were used with the same benzimidazole and dibenzimidazole wires.From the experimental observations summarized in Fig. 5, it is clear that the Edrop values do not depend on the addition of a second benzimidazole to the bridge but rather are governed by the basicity of the TPA, and that the ΔE1/2 shift does not depend on the structure or basicity of the TPA. Therefore, we associate the 60 mV ΔE1/2 shift with the expansion of the partially conjugated system due to the addition of the second benzimidazole to the bridge.
Within the DFT error of ∼100 mV, our theoretical model reliably predicts the increasingly negative Gibbs free energy change indicated by the decreasing E1/2 for oxidation of the phenol with increasing basicity of the TPA and addition of a second benzimidazole. However, theoretically it is difficult to parse the origin of the changes in E1/2 into separate contributions from changing the intrinsic nature of the TPA and extending the π-electronic structure upon addition of the second benzimidazole.
The structural framework supporting the hydrogen-bond network may play several roles in these systems. By extending the framework, the benzimidazole/dibenzimidazole bridge plays a key role in supporting the hydrogen-bond network and thereby providing a reversible pathway for the protons, resulting in essentially electrochemically and chemically reversible redox behavior.27 The observation that the equilibrium electrochemical oxidation of the phenol decreases by up to 440 mV (4′vs.1) as a function of the TPA and changes in the extended π system suggests that the coupling provided by PCET during the electron and proton transfer processes occurs over ∼11 Å. Moreover, in our previous report this coupling extended over a hydrogen-bond network that spanned ∼16 Å.27
Looking to the future, PCET in a construct that would include the generation of biomimetic proton currents, in conjunction with chemical reversibility, must provide a low overpotential pathway for redox processes driving proton currents. A low overpotential, chemically reversible pathway coupling redox processes to proton activities is characteristic of myriad biochemical processes. For example, these processes include the efficient generation and maintenance of PMF and its dissipation in the performance of biochemical work, and proton management at catalytic sites such as those involved in water oxidation as well as CO2 and O2 reduction. Our combination of theoretical and experimental approaches to understanding and expanding PCET will enable the design of efficient artificial proton wires conducting proton currents over considerably longer molecular distances. This basic knowledge is necessary to develop a new generation of artificial photosynthetic systems and to re-engineer natural photosynthesis to improve its efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-FG02-03ER15393. The theoretical portion of this research was supported as part of the Center for Molecular Electrocatalysis, an Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences.Notes and references
- C. Tommos and G. T. Babcock, Biochim. Biophys. Acta, Bioenerg., 2000, 1458, 199–219 CrossRef CAS.
- B. A. Barry and G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7099–7103 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J.-R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- C. Carra, N. Iordanova and S. Hammes-Schiffer, J. Am. Chem. Soc., 2003, 125, 10429–10436 CrossRef CAS PubMed.
- M. Amin, L. Vogt, W. Szejgis, S. Vassiliev, G. W. Brudvig, D. Bruce and M. R. Gunner, J. Phys. Chem. B, 2015, 119, 7366–7377 CrossRef CAS PubMed.
- K. Saito, A. William Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley Blackwell, Oxford, UK, Hoboken, NJ, USA, Second edn, 2014 Search PubMed.
- S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2018, 51, 445–453 CrossRef CAS PubMed.
- G. F. Moore, M. Hambourger, G. Kodis, W. Michl, D. Gust, T. A. Moore and A. L. Moore, J. Phys. Chem. B, 2010, 114, 14450–14457 CrossRef CAS PubMed.
- F. Rappaport, A. Boussac, D. A. Force, J. Peloquin, M. Brynda, M. Sugiura, S. Un, R. D. Britt and B. A. Diner, J. Am. Chem. Soc., 2009, 131, 4425–4433 CrossRef CAS.
- J. M. Keough, D. L. Jenson, A. N. Zuniga and B. A. Barry, J. Am. Chem. Soc., 2011, 133, 11084–11087 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. Pettersson Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-Schiffer and J. M. Mayer, Science, 2019, 364, 471–475 CrossRef CAS PubMed.
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- J. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Chem. Rev., 2003, 103, 2167–2202 CrossRef CAS PubMed.
- I. Chaves, R. Pokorny, M. Byrdin, N. Hoang, T. Ritz, K. Brettel, L.-O. Essen, G. T. J. van der Horst, A. Batschauer and M. Ahmad, Annu. Rev. Plant Biol., 2011, 62, 335–364 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10920–10925 CrossRef CAS PubMed.
- M. R. A. Blomberg, Biochemistry, 2016, 55, 489–500 CrossRef CAS PubMed.
- T. Mathes, I. H. M. van Stokkum, M. Stierl and J. T. M. Kennis, J. Biol. Chem., 2012, 287, 31725–31738 CrossRef CAS.
- M. T. Huynh, S. J. Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A.-L. Teillout, C. W. Machan, C. P. Kubiak, D. Gust, T. A. Moore, S. Hammes-Schiffer and A. L. Moore, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466–10467 CrossRef CAS PubMed.
- T. A. Moore, D. Gust, S. Hatlevig, A. L. Moore, L. R. Makings, P. J. Pesski, F. C. De Schryver, M. van Der Auweraer, D. Lexa, R. V. Bensasson and M. Rougée, Isr. J. Chem., 1988, 28, 87–95 CrossRef CAS.
- S. Jimena Mora, D. A. Heredia, E. Odella, U. Vrudhula, D. Gust, T. A. Moore and A. L. Moore, J. Porphyrins Phthalocyanines, 2019, 23, 1–10 CrossRef.
- E. Odella, S. J. Mora, B. L. Wadsworth, M. T. Huynh, J. J. Goings, P. A. Liddell, T. L. Groy, M. Gervaldo, L. E. Sereno, D. Gust, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, J. Am. Chem. Soc., 2018, 140, 15450–15460 CrossRef CAS PubMed.
- E. Odella, B. L. Wadsworth, S. J. Mora, J. J. Goings, M. T. Huynh, D. Gust, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, J. Am. Chem. Soc., 2019, 141, 14057–14061 CrossRef CAS.
- S. D. Glover, G. A. Parada, T. F. Markle, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2017, 139, 2090–2101 CrossRef CAS PubMed.
- M.-T. Zhang, T. Irebo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 13224–13227 CrossRef CAS PubMed.
- T. F. Markle, I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2011, 133, 17341–17352 CrossRef CAS PubMed.
- I. J. Rhile, T. F. Markle, H. Nagao, A. G. DiPasquale, O. P. Lam, M. A. Lockwood, K. Rotter and J. M. Mayer, J. Am. Chem. Soc., 2006, 128, 6075–6088 CrossRef CAS PubMed.
- L. E. Khoo, Spectrochim. Acta, Part A, 1979, 35, 993–995 CrossRef.
- T. F. Markle, I. J. Rhile, A. G. DiPasquale and J. M. Mayer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8185–8190 CrossRef CAS PubMed.
- M. Forés, M. Duran, M. Solà, M. Orozco and F. J. Luque, J. Phys. Chem. A, 1999, 103, 4525–4532 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 2019, 6735–6748 CrossRef CAS.
- R. D. Webster, Electrochem. Commun., 2003, 5, 6–11 CrossRef CAS.
- C.-T. Cao, Y. Bi and C. Cao, Spectrochim. Acta, Part A, 2016, 163, 96–101 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1968269 and 1968270. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06010c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |