
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gd(III)–Pt(IV) theranostic contrast agents for tandem MR imaging and chemotherapy†
Casey J.
Adams
 and
Thomas J.
Meade
*
and
Thomas J.
Meade
*
Departments of Chemistry, Molecular Biosciences, Neurobiology, and Radiology, Northwestern University, Evanston, Illinois 60208, USA. E-mail: tmeade@northwestern.edu
First published on 28th January 2020
Abstract
Pt(IV) prodrugs have emerged as versatile therapeutics for addressing issues regarding off-target toxicity and the chemoresistance of classic Pt(II) drugs such as cisplatin and carboplatin. There is significant potential for Pt(IV) complexes to be used as theranostic agents, yet there are currently no reported examples of Gd(III)–Pt(IV) agents for simultaneous MR imaging and chemotherapy. Here we report the synthesis, characterization, and in vitro efficacy of two Gd(III)–Pt(IV) agents, GP1 and GP2. Both agents are water soluble and stable under extracellularly relevant conditions but are reduced under intracellular conditions. Both are cytotoxic in multiple cancer cell lines, cell permeable, and significantly enhance the T1-weighted MR contrast of multiple cell lines. Thus, GP1 and GP2 are promising agents for tandem MR imaging and chemotherapy and provide a versatile platform through which future Gd(III)–Pt(IV) agents can be developed.
Introduction
For decades, Pt(II) chemotherapeutics have been fundamental tools for the treatment of solid tumors.1–3 Although there have been extensive efforts to develop new Pt-based chemotherapeutics, there are currently only three approved by the FDA: cisplatin, carboplatin, and oxaliplatin.4–7 All FDA approved Pt chemotherapeutics are Pt(II) square planar complexes that cross-link DNA, ultimately leading to apoptosis in fast-dividing cells.1,8 This mechanism of action makes them highly effective at treating solid tumors. Even so, they have significant off-target toxicity that can result in a number of serious side effects including renal failure, hearing loss, and myelosuppression.1–5 Pt(II) chemotherapeutics are also susceptible to chemoresistance. This stems from multiple mechanisms, including decreased drug accumulation, cytosolic sequestration, and resistance to DNA damage.8–12 Despite issues with toxicity and chemoresistance, Pt(II) complexes are used in nearly 50% of all chemotherapy regimens, making them some of the most widely used cancer drugs available.13A number of approaches have been attempted to mitigate off-target toxicity and chemoresistance of Pt(II) drugs. This includes using tumor-targeting groups, nanoconstructs, and selective release mechanisms.14–16 In recent years, Pt(IV) prodrugs have become a promising approach to alleviate off-target toxicity and reduce chemoresistance.14–19 Pt(IV) complexes are octahedral and inert compared to their Pt(II) square planar analogues. In an oxidizing extracellular environment Pt(IV) complexes remain inert. They can be reduced intracellularly to Pt(II), triggering the dissociation of a reactive Pt(II) drug from its axial ligands.15–19 A variety of groups can be incorporated as axial ligands to allow for tumor targeting, combination therapy, bioimaging, and controlled reduction of the Pt(IV).20–31 Sessler and coworkers were the first to demonstrate how Gd(III) complexes can be used synergistically with Pt(IV) prodrugs. Their Gd(III)-texaphyrin complexes have been used to increase tumor localization and mediate the reduction of Pt(IV) to Pt(II).30,31 However, we believe there is a significant opportunity to investigate Gd(III)–Pt(IV) mixed metal complexes for applications as theranostic agents.
Several examples of theranostic Pt(IV) prodrug complexes for dual chemotherapy and fluorescence imaging have been reported.24–27 Some of these probes can provide important information regarding drug delivery and subsequent reduction of the Pt(IV) complex in vitro. However, in vivo fluorescence imaging has limited clinical utility.32,33 The primary modality for imaging tumors is MRI, which allows for whole body, non-invasive imaging with excellent soft tissue contrast and spatial resolution.32–36 While a few examples of Gd(III)–Pt(II) theranostics have appeared,37–39 to our knowledge there are no reported examples of Pt(IV) prodrugs containing a Gd(III) complex for contrast-enhanced MR imaging either in vitro or in vivo.
Here, we describe two Gd(III)–Pt(IV) theranostic agents that were synthesized by coupling a Gd(III) MR contrast agent axially to cisplatin and carboplatin-based Pt(IV) complexes. These agents are water soluble, cell permeable, and oxidatively stable, but are reduced under biologically relevant intracellular conditions to release the toxic Pt(II) payload and the Gd(III) MR agent (Scheme 1). These complexes are designed for intracellular contrast enhancement of cancer cells, whereas typical Gd(III) contrast agents are limited to the extracellular space surrounding tumors.34,40 This Gd(III)–Pt(IV) platform possesses a second axial site that can be used to couple targeting groups for tumor specificity, drugs to combat chemoresistance, or fluorophores for multimodal imaging and validation.
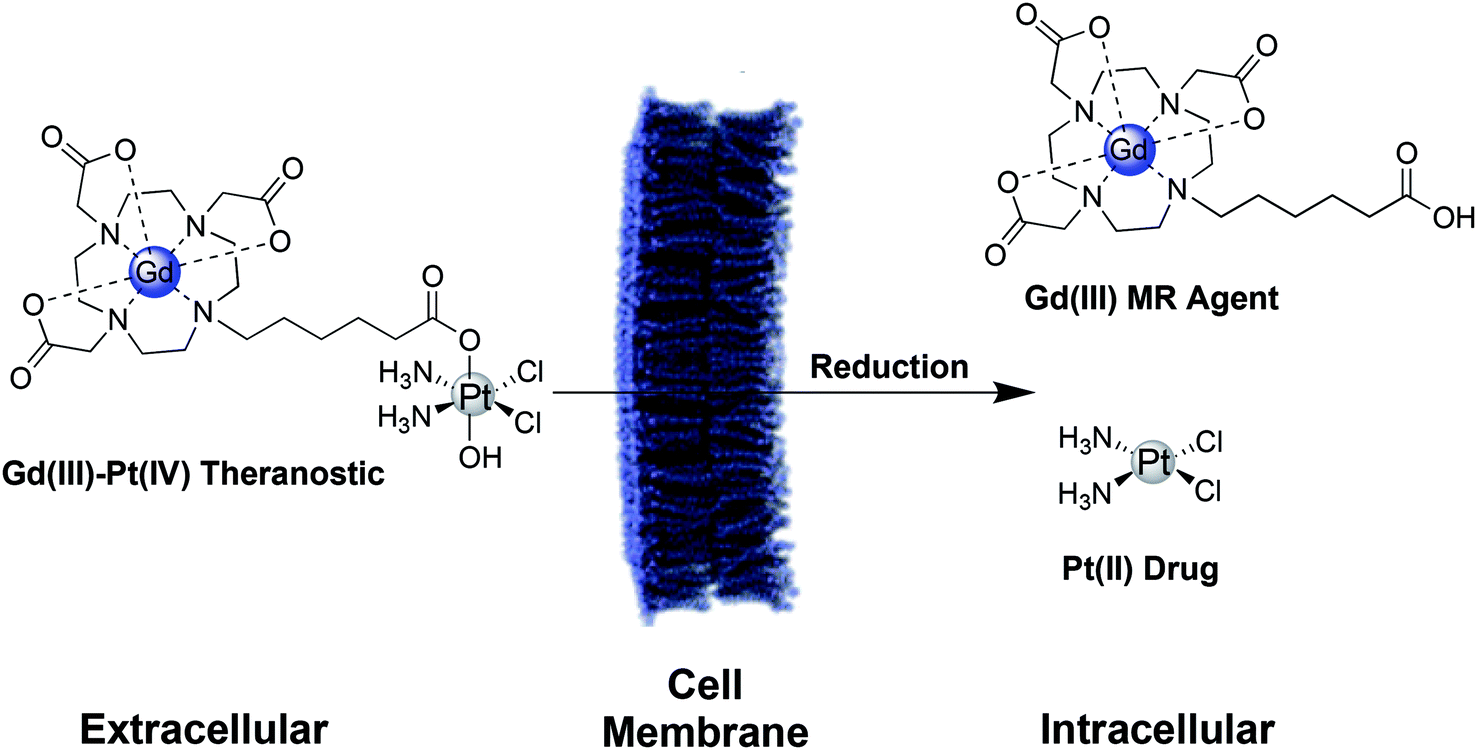 | ||
| Scheme 1 Schematic of a Gd(III)–Pt(IV) theranostic agent. In the oxidizing extracellular environment, the agent remains in the Pt(IV) oxidation state.15 Upon entering the cell, the agent is reduced to Pt(II), simultaneously releasing the therapeutic Pt(II) drug and a Gd(III) MR contrast agent. | ||
Results and discussion
Synthesis and purification of the agents
Two Gd(III)–Pt(IV) agents, GP1 and GP2 were synthesized by coupling a Gd(III) complex, 1, with Pt(IV) complexes 2 and 3 respectively (Fig. 1A). Complexes 1, 2, and 3 were all synthesized following literature protocols.41–43 The structures of GP1 and GP2 are found in Fig. 1A. Both agents were purified by preparatory HPLC, characterized by HPLC-MS (see ESI†) and analyzed by ICP-MS to ensure the Gd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratio was 1:1.
:Pt ratio was 1:1.
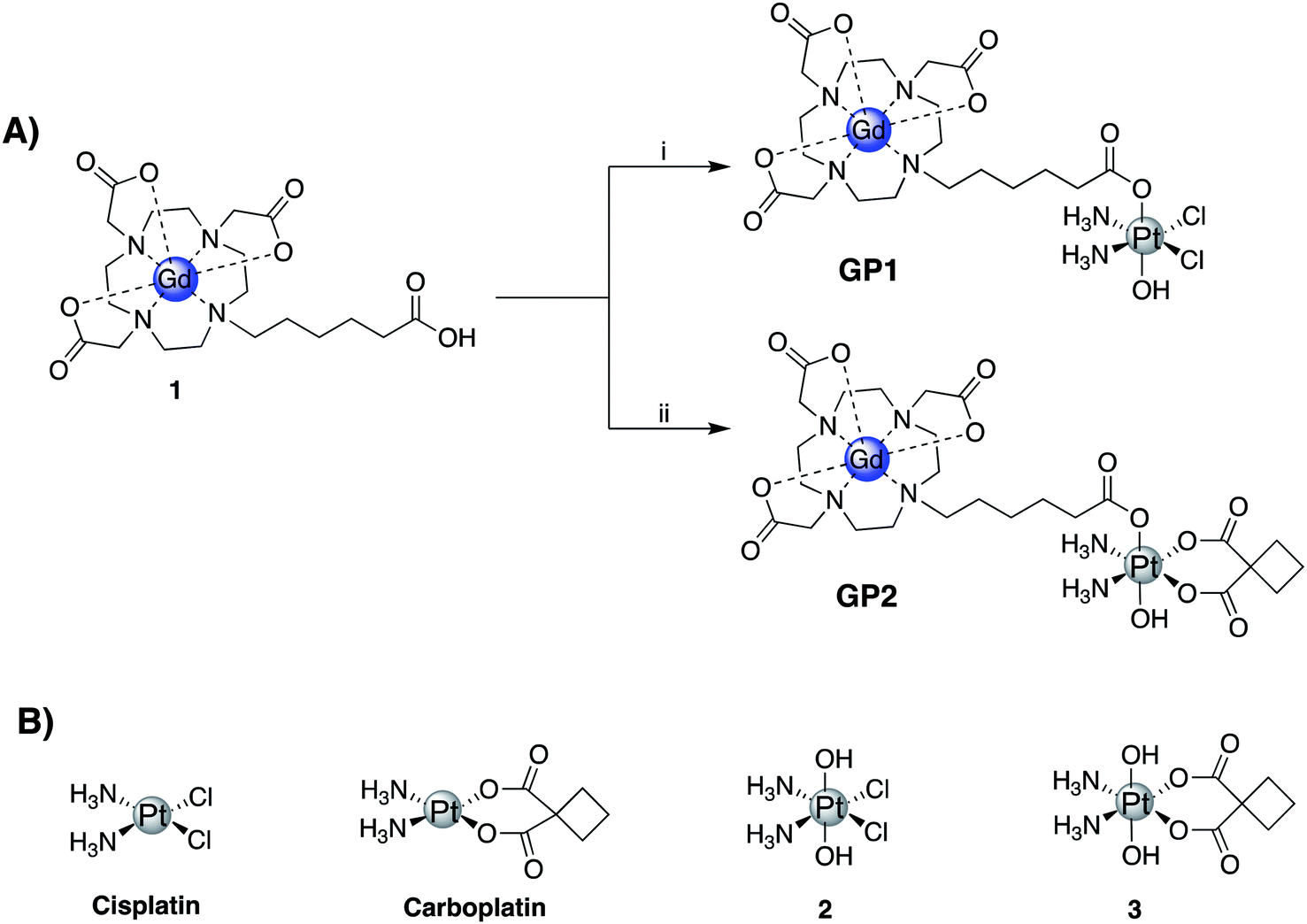 | ||
| Fig. 1 (A) Synthetic scheme of GP1 and GP2: (i) TBTU (1 equiv.), triethylamine (1 equiv.), 2 (1.5 equiv.), DMSO, 45 °C, 12 h, 44%; (ii) TBTU (1 equiv.), triethylamine (1 equiv.), 3 (1.5 equiv.), DMSO, 45 °C, 12 h, 56%. (B) Structures of cisplatin, carboplatin, and the corresponding Pt(IV) complexes 2 and 3. | ||
Relaxivity measurements
The T1 relaxivity (r1) and T2 relaxivity (r2) of 1, GP1, and GP2 were measured at both low (1.41 T) and high (7 T) magnetic field strength in phosphate-buffered saline (PBS) to quantify how well they behave as MR contrast agents. Relaxivity values are summarized in Table 1. At 1.41 T, 1 had an r1 of 4.1 mM−1 s−1 and an r2 of 4.6 mM−1 s−1. At 7 T, the r1 had a slight increase while the r2 significantly increased, as expected at high field. The relaxivity values for 1 are well within the range of what is expected for a small molecule Gd(III) complex.34 At 1.41 T, GP1 and GP2 had an r1 of 7.0 and 8.8 mM−1 s1 and r2 of 7.5 and 10.7 mM−1 s−1 respectively. At 7 T, there was little change in r1 for both agents, while both r2 values increased, as expected.| Agent | r 1 (mM−1 s−1) | r 2 (mM−1 s−1) | ||
|---|---|---|---|---|
| 1.41 T | 7 T | 1.41 T | 7 T | |
| 1 | 4.1 | 4.7 | 4.6 | 6.9 |
| GP1 | 7.0 | 7.1 | 7.5 | 10.7 |
| GP2 | 8.8 | 8.5 | 9.2 | 12.4 |
Compared to 1, both GP1 and GP2 had significantly increased relaxivity (both r1 and r2). This is possibly due to an increase in the rotational correlation time (τR) or a change in the inner sphere hydration number (q) of Gd(III). The differences in relaxivity of GP1 and GP2 compared to 1 offers the possibility of monitoring reduction of the agents by MR at both high and low field strength. To test this, GP1, GP2, and 1 were incubated in 5 mM GSH in PBS and the relaxivity was measured (see ESI†). The relaxivity of each converged to the same value (r1 of 4.4, 4.5, and 4.6 mM−1 s−1 for 1, GP1, and GP2 respectively), indicating the same Gd(III) species was formed after reduction. The significant change in r1 upon reduction of GP1 and GP2 theoretically could be used to monitor intracellular reduction by MR. However, further testing needs to be done to determine if these agents are suitable for this application.
Stability and reduction of the Gd(III)–Pt(IV) complexes
Though most Pt(IV) complexes (see 2 and 3 in Fig. 1) are insoluble in most solvents, both GP1 and GP2 are readily soluble in aqueous solutions. To ensure both agents remain stable in aqueous media under various conditions, they were dissolved in PBS, two types of cell culture media (MEM and RPMI-1640), pH 5 H2O, and porcine live esterase (PLE) in PBS and monitored over time by HPLC-MS.Fig. 2A and B shows that over 48 hours, GP1 and GP2 both remained completely stable in PBS and pH 5 H2O. This suggests that both agents can remain intact even in the most acidic conditions in cells, such as in lysosomes (pH 4.5–5).44 Both agents also remained highly stable in MEM (≥92% intact) at 48 hours. GP1 and GP2 were mostly stable in the presence of PLE, and the slight decrease in stability that was observed (∼10%) occurred on a much slower timescale than the reduction of both agents by GSH (Fig. 2C and D). It is unlikely that esterase cleavage is a competing intracellular dissociation mechanism.
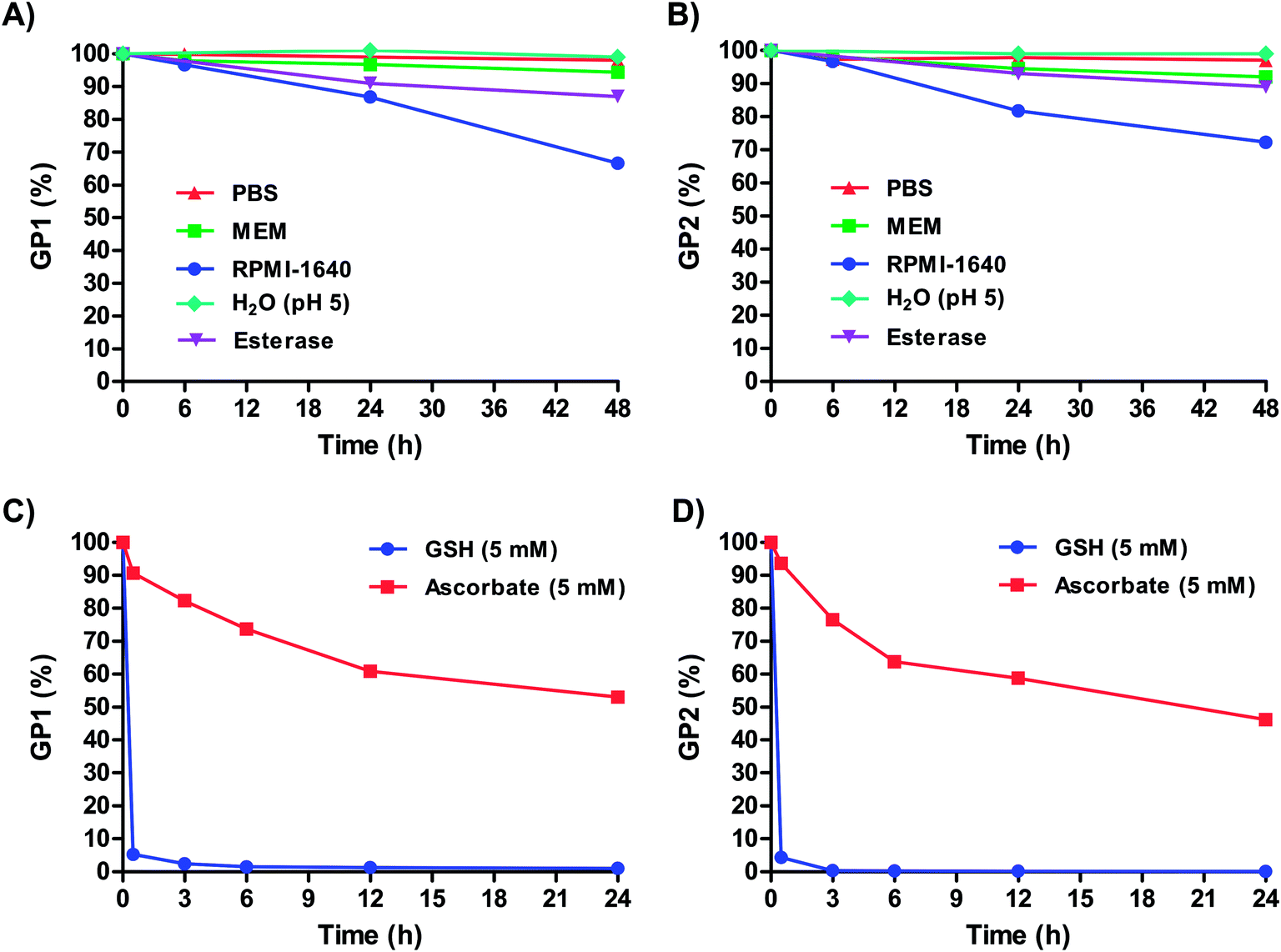 | ||
| Fig. 2 Stability of (A) GP1 and (B) GP2 in PBS, MEM, RPMI-1640, pH 5 H2O, and porcine liver esterase (PLE) in PBS. The observed partial reduction over long periods of time in RPMI-1640 is likely a result of glutathione (GSH) in the media. Both agents are highly stable in PBS, MEM, and pH 5 H2O over extended periods of time while PLE resulted in slight decrease in stability. Stability of (C) GP1 and (D) GP2 in 5 mM GSH and 5 mM ascorbate. GP1 and GP2 are both rapidly reduced by intracellular concentrations of GSH. | ||
In RPMI-1640, 33% of GP1 and 28% of GP2 were reduced to Pt(II) within 48 hours. Unlike MEM, which contains no glutathione (GSH), RPMI-1640 has 0.003 mM GSH. Though this is an extracellularly relevant concentration of GSH (0.002–0.02 mM),45,46 a minority of GP1 and GP2 are reduced over long periods of time. However, it is important to note that at 6 hours, both agents are ≥96% intact (i.e., they remain in the Pt(IV) oxidation state). In vivo, agent that does not get into cells within this time frame will likely be renally cleared before it is reduced. Therefore, within the time frame relevant for in vivo experiments, GP1 and GP2 stay intact and stable in extracellularly relevant conditions.
Under reducing conditions both agents rapidly converted to a monomeric Gd(III) complex and a Pt(II) drug. Each agent was incubated with 5 mM GSH and 5 mM ascorbate at 37 °C to mimic intracellular conditions. Fig. 2C and D shows that both agents are rapidly reduced by GSH at these concentrations (≥94% reduction in less than an hour). As expected, GP1 and GP2 are also reduced by ascorbate, albeit it at a much slower rate. This is not surprising, as ascorbate is a weaker reducing agent. It is clear from these results that under conditions similar to those inside cells, both agents are reduced, releasing a Gd(III) contrast agent and Pt(II) chemotherapeutic.
Cytotoxicity of the agents
To assess the cytotoxicity of GP1 and GP2in vitro, cell viability assays were performed to determine the IC50 concentrations in three cancer cell lines (A2780, HeLa, and MCF-7). Table 2 summarizes the IC50 concentrations of both agents compared to those of cisplatin and carboplatin. Cells were incubated with cisplatin and GP1 for 48 h and carboplatin and GP2 for 72 h to compensate for the inherent decreased potency of carboplatin compared to cisplatin. GP1 had an IC50 of 29.8 μM in A2780 cells, 49.3 μM in HeLa cells, and 113 μM in MCF-7 cells. For all cell lines, GP1 was less toxic than cisplatin but followed the same trend of highest toxicity in A2780 cells and least toxic in MCF-7 cells. GP1 releases cisplatin intracellularly, therefore the apparent difference in toxicity of the two is likely due to decreased cell permeability of GP1 caused by the Gd(III) complex. However, because GP1 is inert in the extracellular matrix, higher doses can be safely administered to account for the decreased cell permeability.| Complex | IC50 (μM) | ||
|---|---|---|---|
| A2780 | HeLa | MCF-7 | |
| Cisplatin | 7.6 ± 2.3 | 14.7 ± 1.2 | 22.4 ± 2.0 |
| Carboplatin | 15.3 ± 5.8 | 71.2 ± 4.9 | 124 ± 4 |
| GP1 | 29.8 ± 2.5 | 49.3 ± 1.3 | 113 ± 4 |
| GP2 | 55.0 ± 2.9 | 258 ± 5.0 | 382 ± 6 |
GP2 had an IC50 of 55.0 μM in A2780 cells, 258 μM in HeLa cells, and 382 μM in MCF-7 cells. The decreased toxicity of GP2 compared to GP1 is expected because carboplatin is known to be significantly less reactive than cisplatin. GP2 was similarly less toxic than carboplatin, but followed the same trend of highest toxicity in A2780 cells and lowest toxicity in MCF-7 cells. The decreased toxicity compared to carboplatin is again attributed to decreased cell permeability due to the presence of the Gd(III) complex.
These results demonstrate that GP1 and GP2 are cytotoxic in three different cancer cell lines. Both agents show similar trends in toxicity as their Pt(II) analogues, which suggests that upon entering the cells they are reduced and behave like typical Pt(II) chemotherapeutics.
Accumulation of the Gd(III)–Pt(IV) agents in cells
Accumulation of GP1 and GP2 in cells was measured by concentration-dependent uptake experiments in A2780 and HeLa cells. Individual concentration-dependent uptake graphs are found in the ESI.†Table 3 compares the accumulation of Gd and Pt in A2780 and HeLa cells that were incubated with GP1, GP2, cisplatin, and carboplatin near their respective IC50 concentrations. For comparison, both cell lines were dosed with complex 1 at similar concentrations as GP1 and GP2. The results of these uptake experiments demonstrate several important points. First, the accumulation of Gd in both cell lines for both agents is higher than what is typically considered the amount necessary for detection by MR (high μM concentrations).32,33 This suggests that GP1 and GP2 can significantly enhance intracellular MR contrast. Typical Gd(III) complexes alone are not cell permeable, therefore the Pt(IV) complexes likely make it possible for GP1 and GP2 to get into cells. This is evidenced by the significant increase in cellular uptake of GP1 and GP2 compared to complex 1 in both cell lines.| Complex | Cell accumulation (fmol per cell) | |||
|---|---|---|---|---|
| A2780 cells | HeLa cells | |||
| Gd | Pt | Gd | Pt | |
| GP1 | 2.5 ± 0.3 | 0.20 ± 0.03 | 5.7 ± 1.9 | 0.17 ± 0.01 |
| GP2 | 1.4 ± 0.2 | 0.38 ± 0.1 | 1.1 ± 0.2 | 0.30 ± 0.06 |
| 1 | 0.26 ± 0.02 | N/A | 0.21 ± 0.01 | N/A |
| Cisplatin | N/A | 0.21 ± 0.02 | N/A | 0.27 ± 0.02 |
| Carboplatin | N/A | 0.22 ± 0.02 | N/A | 0.60 ± 0.11 |
Second, intracellular Pt levels at the IC50 concentrations were similar between GP1 and cisplatin and GP2 and carboplatin. From a therapeutic standpoint, the agents behave like cisplatin and carboplatin once they enter cells and are reduced from Pt(IV) to Pt(II). These results support that the higher IC50 concentrations of GP1 and GP2 compared to cisplatin and carboplatin are caused by decreased cell-permeability, not a decrease in intracellular toxicity. It is clear that both GP1 and GP2 penetrate cells well enough to provide sufficient MR-Gd(III) concentrations for imaging and therapeutically relevant Pt concentrations in cells at low incubation concentrations.
Finally, there is a significant preferential accumulation of Gd compared to Pt from both agents in both cell lines. The difference in accumulation of the two is possible because they dissociate from one another after the intracellular reduction of Pt(IV) to Pt(II). For GP1, a 13-fold higher accumulation of Gd than Pt in A2780 cells and 34-fold higher Gd(III) accumulation in HeLa cells was observed. For GP2, Gd accumulation was 3.7-fold higher in both A2780 and HeLa cells. These results suggest that Gd is effluxed to a lesser extent than Pt. When GP1 and GP2 are reduced intracellularly, the cell impermeable charged Gd(III) complex is likely prevented from exiting the cells as readily as the cell permeable Pt(II) complexes. This is of particular consequence for the ability of GP1 and GP2 to act as MR contrast agents because higher cellular amounts of Gd(III) increase MR contrast.
Cell uptake of GP1 and GP2 was additionally measured in a time-dependent manner in A2780 cells. Fig. 3 demonstrates that the accumulation of both Gd and Pt from both agents is directly proportional to time. In these experiments, higher accumulation of Gd compared to Pt was observed at every timepoint. Initial uptake of GP1 and GP2 from 0–30 min was fast compared to uptake from 0.5–24 h. Fast uptake and intracellular reduction can explain why there was significantly more Gd than Pt even at the 30 min timepoint. For both agents, the Gd:Pt ratio in cells continues to increase over time, which is consistent with a higher rate of efflux of Pt. These results also suggest MR-relevant amounts of Gd from both agents accumulate in cells within a few hours. This is promising for the prospect of imaging within the time frame relevant for in vivo experiments.
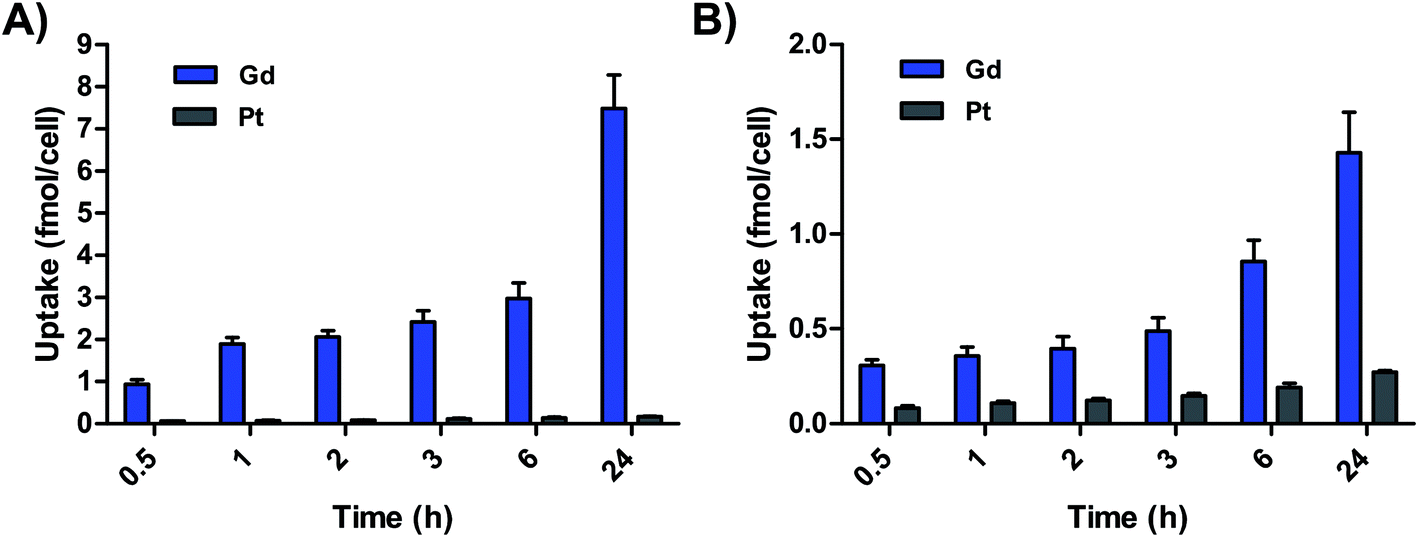 | ||
| Fig. 3 (A) Accumulation of Gd and Pt in A2780 cells incubated with 65 μM GP1 over time. (B) Accumulation of Gd and Pt in A2780 cells incubated with 62.5 μM GP2 over time. In both cases, accumulation of Gd was significantly higher than Pt. | ||
MR imaging of A2780 and HeLa cell pellets
To quantify the ability of GP1 and GP2 to enhance MR contrast intracellularly, MR cell pellet imaging experiments were performed. A2780 and HeLa cells were incubated with IC50 concentrations of GP1 and GP2 for 6 hours then imaged in a 7 T MR scanner. The relaxation rate (R1), which is defined as 1/T1 and is directly proportional to MR contrast, of each cell pellet was determined and compared to control cells that were untreated, incubated with 100 μM complex 1, or incubated with IC50 concentrations of cisplatin.Fig. 4 demonstrates that GP1 and GP2 significantly enhance the intracellular MR contrast of both A2780 and HeLa cells when dosed at concentrations near the IC50. GP1 increased the R1 by 36% in A2780 cells and 48% in HeLa cells compared to the untreated controls. GP2 increased the R1 by 26% in A2780 cells and 23% in HeLa cells. Treating cells with similar concentrations of complex 1 and IC50 concentrations of cisplatin resulted in minimal increases in R1. This supports that the observed contrast enhancement by GP1 and GP2 is a result of the agents effectively accumulating in cells, something 1 cannot do alone. Furthermore, any physiological changes in the cells caused by the presence of a Pt(II) drug have little effect on the MR contrast. Therefore, the contrast enhancement is a result of intracellular Gd(III).
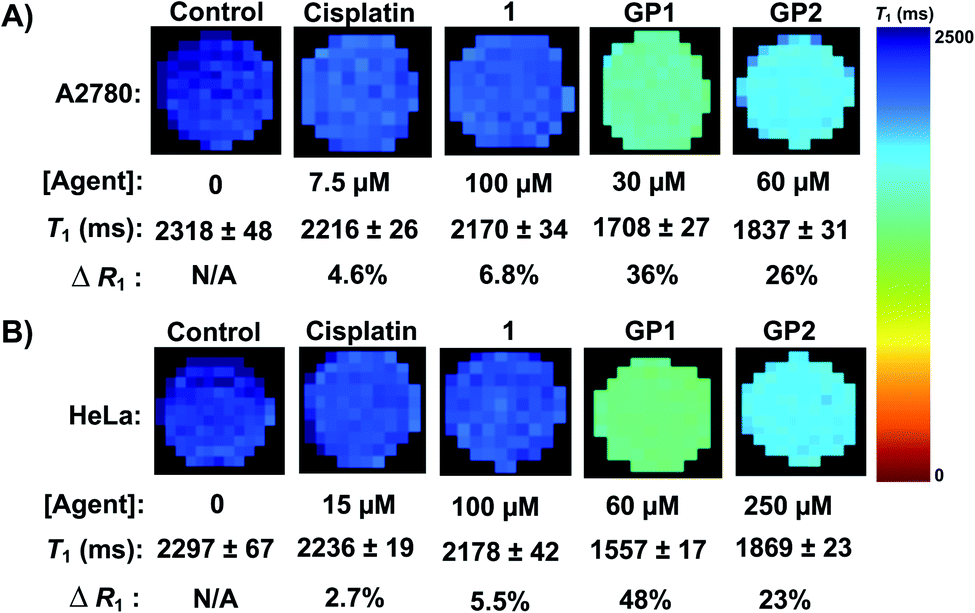 | ||
| Fig. 4 (A) MR imaging of A2780 cell pellets at 7 T. IC50 concentrations of GP1 and GP2 significantly enhanced the intracellular contrast of A2780 cells (increase in R1) compared to the untreated control cells while 1 and cisplatin had a minimal effect. (B) MR imaging of HeLa cell pellets at 7 T. IC50 concentrations of GP1 and GP2 significantly enhanced the intracellular contrast of HeLa cells compared to the untreated control cells while 1 and cisplatin had a minimal effect. | ||
Notably, the cell pellet experiments were performed within a time frame relevant for in vivo MR imaging (6 hours). These results are very promising for the prospect of using Gd(III)–Pt(IV) theranostics like GP1 and GP2 for in vivo MR imaging.
Conclusions
We have described the synthesis, characterization and cellular uptake of two new Gd(III)–Pt(IV) agents. GP1 and GP2 represent the first examples of Gd(III)–Pt(IV) agents that are simultaneously MR contrast agents and are reduced to provide chemotherapy. Of the two agents, GP1 is most promising as it exhibits greater cellular toxicity, higher intracellular accumulation of Gd(III) and better MR contrast enhancement in vitro. Future work will focus on demonstrating the in vivo efficacy of these agents for both imaging and treatment.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
C. Adams acknowledges support from the National Cancer Institute (NCI) Grant 1F31CA239426-01. MR imaging was performed with help from E. Waters at the Northwestern University Center for Advanced Molecular Imaging generously supported by NCI grant P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Metal analysis was performed with help from R. Sponenburg at the Northwestern University Quantitative Bio-element Imaging Center generously supported by NASA Ames Research Center NNA04CC36G.References
- S. Dasari and P. Bernard Tchounwou, Cisplatin in cancer therapy: Molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- B. Desoize and C. Madoulet, Particular aspects of platinum compounds used at present in cancer treatment, Critical Reviews in Oncology/Hematology, 2002, 42(3), 317–325 CrossRef PubMed.
- D. S. Goodsell, The Molecular Perspective: Cisplatin, Stem Cells, 2006, 24(3), 514–515 CrossRef PubMed.
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, The status of platinum anticancer drugs in the clinic and in clinical trials, Dalton Trans., 2010, 39(35), 8113–8127 RSC.
- J. J. Wilson and S. J. Lippard, Synthetic Methods for the Preparation of Platinum Anticancer Complexes, Chem. Rev., 2014, 114(8), 4470–4495 CrossRef CAS PubMed.
- N. P. Farrell, Multi-platinum anti-cancer agents. Substitution-inert compounds for tumor selectivity and new targets, Chem. Soc. Rev., 2015, 44(24), 8773–8785 RSC.
- Z. H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene, 2003, 22(47), 7265–7279 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harel-Bellan, M. Castedo and G. Kroemer, Systems biology of cisplatin resistance: past, present and future, Cell Death Dis., 2014, 5(5), e1257 CrossRef CAS PubMed.
- G. Giaccone, Clinical Perspectives on Platinum Resistance, Drugs, 2000, 59(4), 9–17 CrossRef CAS PubMed.
- B. Köberle, M. T. Tomicic, S. Usanova and B. Kaina, Cisplatin resistance: Preclinical findings and clinical implications, Biochim. Biophys. Acta, Rev. Cancer, 2010, 1806(2), 172–182 CrossRef PubMed.
- D.-W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes, Pharmacol. Rev., 2012, 64(3), 706–721 CrossRef CAS PubMed.
- T. C. Johnstone, G. Y. Park and S. J. Lippard, Understanding and improving platinum anticancer drugs-phenanthriplatin, Anticancer Res., 2014, 34(1), 471–476 CAS.
- R. J. Browning, P. J. T. Reardon, M. Parhizkar, R. B. Pedley, M. Edirisinghe, J. C. Knowles and E. Stride, Drug Delivery Strategies for Platinum-Based Chemotherapy, ACS Nano, 2017, 11(9), 8560–8578 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs, Chem. Rev., 2016, 116(5), 3436–3486 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Targeting and delivery of platinum-based anticancer drugs, Chem. Soc. Rev., 2013, 42(1), 202–224 RSC.
- X. Li, Y. Liu and H. Tian, Current Developments in Pt(IV) Prodrugs Conjugated with Bioactive Ligands, Bioinorg. Chem. Appl., 2018, 2018, 8276139 Search PubMed.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Platinum(IV) Prodrugs – A Step Closer to Ehrlich's Vision?, Eur. J. Inorg. Chem., 2017, 2017(12), 1596–1612 CrossRef CAS.
- Z. Wang, Z. Deng and G. Zhu, Emerging platinum(IV) prodrugs to combat cisplatin resistance: from isolated cancer cells to tumor microenvironment, Dalton Trans., 2019, 48(8), 2536–2544 RSC.
- D. Montagner, D. Tolan, E. Andriollo, V. Gandin and C. Marzano, A Pt(IV) Prodrug Combining Chlorambucil and Cisplatin: a Dual-Acting Weapon for Targeting DNA in Cancer Cells, Int. J. Mol. Sci., 2018, 19(12), 3775 CrossRef PubMed.
- H. Chen, X. Wang and S. Gou, A cisplatin-based platinum(IV) prodrug containing a glutathione s-transferase inhibitor to reverse cisplatin resistance in non-small cell lung cancer, J. Inorg. Biochem., 2019, 193, 133–142 CrossRef CAS PubMed.
- H. Huang, Y. Dong, Y. Zhang, D. Ru, Z. Wu, J. Zhang, M. SHen, Y. Duan and Y. Sun, GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer, Theranostics, 2019, 9(4), 1047–1065 CrossRef CAS PubMed.
- X. Qin, L. Fang, J. Zhao and S. Gou, Theranostic Pt(IV) Conjugate with Target Selectivity for Androgen Receptor, Inorg. Chem., 2018, 57(9), 5019–5029 CrossRef CAS PubMed.
- J. X. Ong, C. S. Q. Lim, H. V. Le and W. H. Ang, A Ratiometric Fluorescent Probe for Cisplatin: Investigating the Intracellular Reduction of Platinum(IV) Prodrug Complexes, Angew. Chem., Int. Ed., 2019, 58(1), 164–167 CrossRef CAS PubMed.
- Y. Song, K. Suntharalingam, J. S. Yeung, M. Royzen and S. J. Lippard, Synthesis and characterization of Pt(IV) fluorescein conjugates to investigate Pt(IV) intracellular transformations, Bioconjugate Chem., 2013, 24(10), 1733–1740 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, Modulation of Ligand Fluorescence by the Pt(II)/Pt(IV) Redox Couple, Inorg. Chim. Acta, 2012, 389, 77–84 CrossRef CAS PubMed.
- Y. Yuan, Y. Chen, B. Z. Tang and B. Liu, A targeted theranostic platinum(IV) prodrug containing a luminogen with aggregation-induced emission (AIE) characteristics for in situ monitoring of drug activation, Chem. Commun., 2014, 50(29), 3868–3870 RSC.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Diazido Mixed-Amine Platinum(IV) Anticancer Complexes Activatable by Visible-Light Form Novel DNA Adducts, Chem.–Eur. J., 2013, 19(29), 9578–9591 CrossRef CAS PubMed.
- J. Kasparkova, H. Kostrhunova, O. Novakova, R. Křikavová, J. Vančo, Z. Trávníček and V. Brabec, A Photoactivatable Platinum(IV) Complex Targeting Genomic DNA and Histone Deacetylases, Angew. Chem., Int. Ed., 2015, 54(48), 14478–14482 CrossRef CAS PubMed.
- G. Thiabaud, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Photoinduced Reduction of PtIV within an Anti-Proliferative PtIV–Texaphyrin Conjugate, Chem.–Eur. J., 2014, 20(29), 8942–8947 CrossRef CAS.
- G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Activation of Platinum(IV) Prodrugs By Motexafin Gadolinium as a Redox Mediator, Angew. Chem., Int. Ed. Engl., 2016, 55(41), 12626–12631 CrossRef CAS PubMed.
- M. L. James and S. S. Gambhir, A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications, Physiol. Rev., 2012, 92(2), 897–965 CrossRef CAS PubMed.
- M. A. Pysz, S. S. Gambhir and J. K. Willmann, Molecular imaging: current status and emerging strategies, Clin. Radiol., 2010, 65(7), 500–516 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119(2), 957–1057 CrossRef CAS PubMed.
- N. Rammohan, K. W. MacRenaris, L. K. Moore, G. Parigi, D. J. Mastarone, L. M. Manus, L. M. Lilley, A. T. Preslar, E. A. Waters, A. Filicko, C. Luchinat, D. Ho and T. J. Meade, Nanodiamond-Gadolinium(III) Aggregates for Tracking Cancer Growth In Vivo at High Field, Nano Lett., 2016, 16(12), 7551–7564 CrossRef CAS PubMed.
- R. J. Holbrook, N. Rammohan, M. W. Rotz, K. W. MacRenaris, A. T. Preslar and T. J. Meade, Gd(III)-Dithiolane Gold Nanoparticles for T1-Weighted Magnetic Resonance Imaging of the Pancreas, Nano Lett., 2016, 16(5), 3202–3209 CrossRef CAS PubMed.
- Z. Zhu, X. Wang, T. Li, S. Aime, P. J. Sadler and Z. Guo, Platinum(II)–Gadolinium(III) Complexes as Potential Single-Molecular Theranostic Agents for Cancer Treatment, Angew. Chem., Int. Ed., 2014, 53(48), 13225–13228 CrossRef CAS PubMed.
- B. Wu, X.-Q. Li, T. Huang, S.-T. Lu, B. Wan, R.-F. Liao, Y.-S. Li, A. Baidya, Q.-Y. Long and H.-B. Xu, MRI-guided tumor chemo-photodynamic therapy with Gd/Pt bifunctionalized porphyrin, Biomater. Sci., 2017, 5(9), 1746–1750 RSC.
- O. J. Stacey, A. J. Amoroso, J. A. Platts, P. N. Horton, S. J. Coles, D. Lloyd, C. F. Williams, A. J. Hayes, J. J. Dunsford and S. J. A. Pope, Water soluble, cyclometalated Pt(II)–Ln(III) conjugates towards novel bimodal imaging agents, Chem. Commun., 2015, 51(61), 12305–12308 RSC.
- H. Li and T. J. Meade, Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances, J. Am. Chem. Soc., 2019, 141(43), 17025–17041 CrossRef CAS PubMed.
- N. Rammohan, K. W. MacRenaris, L. K. Moore, G. Parigi, D. J. Mastarone, L. M. Manus, L. M. Lilley, A. T. Preslar, E. A. Waters, A. Filicko, C. Luchinat, D. Ho and T. J. Meade, Nanodiamond–Gadolinium(III) Aggregates for Tracking Cancer Growth In Vivo at High Field, Nano Lett., 2016, 16(12), 7551–7564 CrossRef CAS PubMed.
- H. P. Varbanov, S. M. Valiahdi, C. R. Kowol, M. A. Jakupec, M. Galanski and B. K. Keppler, Novel tetracarboxylatoplatinum(IV) complexes as carboplatin prodrugs, Dalton Trans., 2012, 41(47), 14404–14415 RSC.
- M. D. Hall, C. T. Dillon, M. Zhang, P. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, The cellular distribution and oxidation state of platinum(II) and platinum(IV) antitumour complexes in cancer cells, JBIC, J. Biol. Inorg. Chem., 2003, 8(7), 726–732 CrossRef CAS PubMed.
- K. Coen, R. S. Flannagan, S. Baron, L. R. Carraro-Lacroix, D. Wang, W. Vermeire, C. Michiels, S. Munck, V. Baert, S. Sugita, F. Wuytack, P. R. Hiesinger, S. Grinstein and W. Annaert, Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells, J. Cell Biol., 2012, 198(1), 23–35 CrossRef CAS PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Glutathione: overview of its protective roles, measurement, and biosynthesis, Mol. Aspects Med., 2009, 30(1–2), 1–12 CrossRef CAS PubMed.
- D. Tolan, V. Gandin, L. Morrison, A. El-Nahas, C. Marzano, D. Montagner and A. Erxleben, Oxidative Stress Induced by Pt(IV) Pro-drugs Based on the Cisplatin Scaffold and Indole Carboxylic Acids in Axial Position, Sci. Rep., 2016, 6, 29367 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05937g |
| This journal is © The Royal Society of Chemistry 2020 |