
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar water splitting over Rh0.5Cr1.5O3-loaded AgTaO3 of a valence-band-controlled metal oxide photocatalyst†
Kenta
Watanabe
a,
Akihide
Iwase
 ab and
Akihiko
Kudo
*ab
ab and
Akihiko
Kudo
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.tus.ac.jp
bPhotocatalysis International Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda-shi, Chiba-ken 278-8510, Japan
First published on 28th January 2020
Abstract
Improvement of water splitting performance of AgTaO3 (BG 3.4 eV) of a valence-band-controlled photocatalyst was examined. Survey of cocatalysts revealed that a Rh0.5Cr1.5O3 cocatalyst was much more effective than Cr2O3, RuO2, NiO and Pt for water splitting into H2 and O2 in a stoichiometric amount. The optimum loading amount of the Rh0.5Cr1.5O3 cocatalyst was 0.2 wt%. The apparent quantum yield (AQY) at 340 nm of the optimized Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 photocatalyst reached to about 40%. Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 gave a solar to hydrogen conversion efficiency (STH) of 0.13% for photocatalytic water splitting under simulated sunlight irradiation. Bubbles of gasses evolved by the solar water splitting were visually observed under atmospheric pressure at room temperature.
Introduction
Photocatalytic solar water splitting is an important reaction because it has the potential for solving resources, energy and environmental issues. Hence, many researchers have aimed to develop water splitting systems of a one-step photoexcitation type and a two-step photoexcitation type (Z-scheme) with visible-light-driven powdered photocatalysts.1–6 The simplicity of the one-step photoexcitation type will be an advantage in practical use. Domen and co-workers have reported that (oxy)nitride and oxysulfide photocatalysts such as GaN–ZnO,7,8 ZnGeN2–ZnO,9,10 LaMg1/3Ta2/3O2N,11–13 TaON,14 CaTaO2N,15 Ta3N5 (ref. 16) and Y2Ti2O5S2 (ref. 17) show activities for one-step photoexcitation type water splitting under visible light irradiation. We have also reported that Rh,Sb-codoped SrTiO3 of a metal oxide photocatalyst shows the water splitting activity under visible light irradiation by loading of an IrO2 cocatalyst.18 Although above visible-light-responsive photocatalysts split water under sunlight irradiation, their solar energy conversion efficiencies (solar to hydrogen conversion efficiency, STH) do not reach the level for practical use due to low apparent quantum yields (AQY). In terms of the high AQY, NiO-loaded La-doped NaTaO3 (ref. 19) (BG 4.1 eV), Rh0.5Cr1.5O3-loaded Zn,Ca-codoped Ga2O3 (ref. 20) (BG 4.6 eV) and MoOy,RhCrOx-coloaded Al-doped SrTiO3 (ref. 21) (BG 3.2 eV) show the AQYs of 56% at 270 nm, 71% at 254 nm and 69% at 365 nm, for photocatalytic water splitting under UV irradiation, respectively. However, only Al-doped SrTiO3 can respond to sunlight (λ > 300 nm) with STH of 0.4%.22 Thus, the development of sunlight-driven photocatalysts with one-step photoexcitation for water splitting is an important research topic.We have reported that a AgTaO3 photocatalyst (BG 3.4 eV) shows water splitting activity under UV irradiation by loading of a NiO cocatalyst.23 The characteristics of AgTaO3 is the valence band maximums formed by Ag 4d orbitals which are located above the bands formed by O 2p orbitals, and hence the band gap of AgTaO3 is relatively narrow among the metal oxides containing Ta. AgTaO3 is expected to respond to sunlight judging from the band gap of 3.4 eV, while the water splitting under sunlight irradiation has not been achieved yet. Moreover, the crystal structure of perovskite for AgTaO3 is the same as that for SrTiO3 and NaTaO3 photocatalysts with high activities for water splitting. These backgrounds motivate us to investigate solar water splitting using the AgTaO3 photocatalyst.
A cocatalyst is one of the most effective factors for improvement of the photocatalytic activity. Actually, the water splitting activity of AgTaO3 is drastically improved by loading of a NiO cocatalyst which is widely used in water splitting over metal oxides.23 Recently, Rh0.5Cr1.5O3 has also arisen as a potential candidate cocatalyst for water splitting over metal oxides.20,21 In the present study, we investigated the loading effect of various cocatalysts on water splitting over AgTaO3. Solar water splitting was also demonstrated using the AgTaO3 with the optimized cocatalyst.
Experimental
Preparation of photocatalysts
AgTaO3 was synthesized by a solid-state reaction using Ag2O (Kanto Chemical; 99.0%) and Ta2O5 (Rare Metallic; 99.99%) as starting materials. The starting materials were mixed in an alumina mortar in a ratio of Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ta = 1 + x:1 (x = 0, 0.03, 0.05, 0.07). The mixture was calcined in air at 1173–1373 K for 5–20 h in an alumina crucible. The calcined materials were washed with 1 mol L−1 of an aqueous HNO3 solution in ultrasonication for 1 h in order to remove Ag metals deposited on the surface of AgTaO3.
:Ta = 1 + x:1 (x = 0, 0.03, 0.05, 0.07). The mixture was calcined in air at 1173–1373 K for 5–20 h in an alumina crucible. The calcined materials were washed with 1 mol L−1 of an aqueous HNO3 solution in ultrasonication for 1 h in order to remove Ag metals deposited on the surface of AgTaO3.
A Pt cocatalyst was loaded in situ by a photodeposition method using an aqueous H2PtCl6 (Tanaka Kikinzoku; 37.7 wt% as Pt) solution. NiO,23 RuO2 (ref. 24) and Cr2O3 (ref. 25) of cocatalysts were loaded on AgTaO3 photocatalysts by an impregnation method using Ni(NO3)2·6H2O (Wako Pure Chemical; 98.0%), Ru3(CO)12 (Aldrich; 99%), Cr(NO3)3·9H2O (Kanto Chemical; 98.0–103.0%). AgTaO3 powder was suspended in aqueous solutions dissolving Ni(NO3)2 or Cr(NO3)3, and an acetone solution dissolving Ru3(CO)12 in porcelain crucibles. The AgTaO3-suspended solutions were dried up on a water bath and subsequently calcined in an electric furnace with conditions of 543 K – 1 h for NiO, 673 K – 2 h for RuO2 and 623 K – 1 h for Cr2O3. Rh0.5Cr1.5O3 (ref. 25) cocatalyst was loaded by a simple impregnation method using Cr(NO3)3·9H2O and Rh(NO3)3 (Kanto Chemical; >80.0% as anhydrous). AgTaO3-suspended aqueous solution containing both Rh(NO3)3 and Cr(NO3)3 was dried up and subsequently calcined at 623 K for 1 h. It is reported that Rh0.5Cr1.5O3 of a mixed oxide is naturally formed on a photocatalyst by this process.26 The loading amount of Rh0.5Cr1.5O3 was controlled by adjusting the amount of starting materials.
Characterization
The crystal structure of the synthesized AgTaO3 was identified by powder X-ray diffraction (Rigaku; MiniFlex600). The elemental composition of the synthesized AgTaO3 was measured by an X-ray fluorescence spectrometer (Rigaku; NEX DE). A Diffuse reflectance spectrum was measured using a UV-vis-NIR spectrometer with an integrating sphere (Jasco; UbeatV-570) and was converted from reflection to absorbance mode by the Kubelka–Munk method. Morphologies of photocatalyst particles and Rh0.5Cr1.5O3-cocatalysts were observed using a scanning electron microscope (JEOL; JSM-7400F) and a transmission electron microscope (JEOL; JEM-2100F). A chemical state of Rh in Rh0.5Cr1.5O3-loaded AgTaO3 was analyzed using an X-ray photoelectron spectroscopy (JEOL; JPS-9010MC).Photocatalytic reaction
Photocatalytic water splitting was carried out in a gas-closed-circulation system. Photocatalyst powder (0.3 g) was dispersed in distilled water (120 mL) in a top-irradiation cell with a Pyrex window. A 300 W Xe-arc lamp (PerkinElmer; Cermax-PE300BF) and a solar simulator (Asahi spectra; HAL-320) were employed as a light source. An inner-irradiation reaction cell made of quartz equipped with a 400 W high-pressure Hg lamp (SEN; HL400EH-5) was also used for photocatalytic water splitting in order to compare with the activity of NiO/NaTaO3:La.19 Amounts of evolved H2 and O2 gases were determined with a gas chromatograph (Shimadzu; GC-8A, MS-5A, Ar carrier gas, TCD). Turnover number (TON) of the amount of reacted electrons/holes to the molar quantity of AgTaO3 was estimated using the eqn (1).| [TON] = [the molar quantity of reacted electrons]/[the molar quantity of AgTaO3] = [(the amount of evolved H2) × 2/mol]/[the molar quantity of AgTaO3/mol] | (1) |
Apparent quantum yields (AQY) were measured using a 300 W Xe-arc lamp (Asahi Spectra; MAX-302) with band-pass filters (Asahi Spectra). The photon flux of the monochromatic light through the long-pass filters was measured using a silicon diode head (OPHIR; PD300-UV head and NOVA display). An AQY and a solar to hydrogen conversion efficiency (STH) were estimated using the following eqn (2) and (3).
| [AQY%] = 100 × [the number of reacted electrons or holes]/[the number of incident photons] = 100 × [(the number of evolved H2 molecules) × 2]/[the number of incident photons] | (2) |
| [STH%] = 100 × ([ΔG0(H2O)/kJ mol−1] × [rate of H2 evolution/μmol h−1])/([irradiation time/s] × [solar energy (AM1.5G)/mW cm−2] × [irradiation area/cm2]) = (237 × [rate of H2 evolution/μmol h−1])/(3600 × 100 × 25) × 100 | (3) |
Results and discussion
XRD measurement confirmed that trigonal AgTaO3 with a perovskite structure was successfully synthesized in a single phase as previously reported (Fig. S1†).23 The peaks due to metallic Ag were not observed in XRD patterns of AgTaO3 both before and after washing with an aqueous HNO3 solution. However, the absorption due to the surface plasmonic resonance of Ag was observed in diffuse reflectance spectra of the AgTaO3 before and after the washing (Fig. S2†). The absorption due to the surface plasmonic resonance after the washing was a little bit smaller than that before the washing. Therefore, metallic Ag on the surface would be removed by the washing but the amount of removed Ag was quite small. Actually, atomic ratios of Ag to Ta in the AgTaO3 after the washing were calculated to be 1.00 and 0.99 from XPS and XRF measurements, respectively. These results indicate that the ratios of Ag to Ta on the surface and in the bulk were almost stoichiometric even if after the washing. The absorption edge of AgTaO3 was around 380 nm in a diffuse reflectance spectrum (Fig. S3†), indicating 3.4 eV of the band gap. Scanning electron microscope observation revealed that the size of AgTaO3 particles was from several hundreds nm to several μm (Fig. S4†).Table 1 shows the activities for photocatalytic water splitting over AgTaO3 loaded with various cocatalysts under UV irradiation. Non-loaded AgTaO3 produced only a small amount of H2 without O2 evolution. In other words, water splitting did not proceed. In contrast, all AgTaO3 loaded with various cocatalysts produced both H2 and O2. However, the amount of evolved H2 was more than a stoichiometric amount when NiO, RuO2 and Pt were loaded. When Cr2O3 and Rh0.5Cr1.5O3 were loaded, the stoichiometric amounts of H2 and O2 evolved, indicating that ideal water splitting proceeded. In particular, the activity of AgTaO3 was greatly improved by loading of the Rh0.5Cr1.5O3 cocatalyst. This result is appropriate because Rh0.5Cr1.5O3 is known as an effective cocatalyst for photocatalytic water splitting. It is reported that Cr2O3 does not work as a cocatalyst for photocatalytic water splitting but Ag coloaded with Cr works as a cocatalyst.25 In the present case, only Cr2O3-loaded AgTaO3 showed the water splitting activity. This is probably because a small amount of metallic Ag still remained on the surface of AgTaO3 even after washing with an aqueous HNO3 solution and a Ag–Cr cocatalyst was formed by loading of Cr2O3 as an effective cocatalyst. We optimized the loading amount of the Rh0.5Cr1.5O3 cocatalyst and synthesis conditions of AgTaO3. The water splitting activity of AgTaO3 slightly depended on synthesis conditions and 1273 K – 15 h was the best condition for the activity (Table S1†). In addition, the activity was insensitive for addition of an excess amount of Ag. On the other hand, the activity much depended on the loading amount of Rh0.5Cr1.5O3 cocatalyst and 0.2 wt% was optimum (Table 1). The optimized Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 stably and continuously produced H2 and O2 using a Xe lamp (Fig. 1).
| Cocatalyst (wt%) | Loading method | Activity/μmol h−1 | |
|---|---|---|---|
| H2 | O2 | ||
| a Photocatalyst: 0.3 g, reactant solution: distilled water (120 mL), cell: top-irradiation cell with a Pyrex window, light source: 300 W Xe-arc lamp (λ > 300 nm). | |||
| None | — | 0.5 | 0 |
| Rh0.5Cr1.5O3 (0.05) | Impregnation (623 K – 1 h) | 56 | 28 |
| Rh0.5Cr1.5O3 (0.1) | Impregnation (623 K – 1 h) | 318 | 162 |
| Rh0.5Cr1.5O3 (0.2) | Impregnation (623 K – 1 h) | 400 | 192 |
| Rh0.5Cr1.5O3 (0.3) | Impregnation (623 K – 1 h) | 217 | 111 |
| Cr2O3 (0.13) | Impregnation (623 K – 1 h) | 1.8 | 0.9 |
| RuO2 (0.2) | Impregnation (673 K – 2 h) | 1.6 | 0.5 |
| NiO (0.2) | Impregnation (543 K – 1 h) | 18 | 4 |
| Pt (0.2) | Photodeposition (in situ) | 45 | 4 |
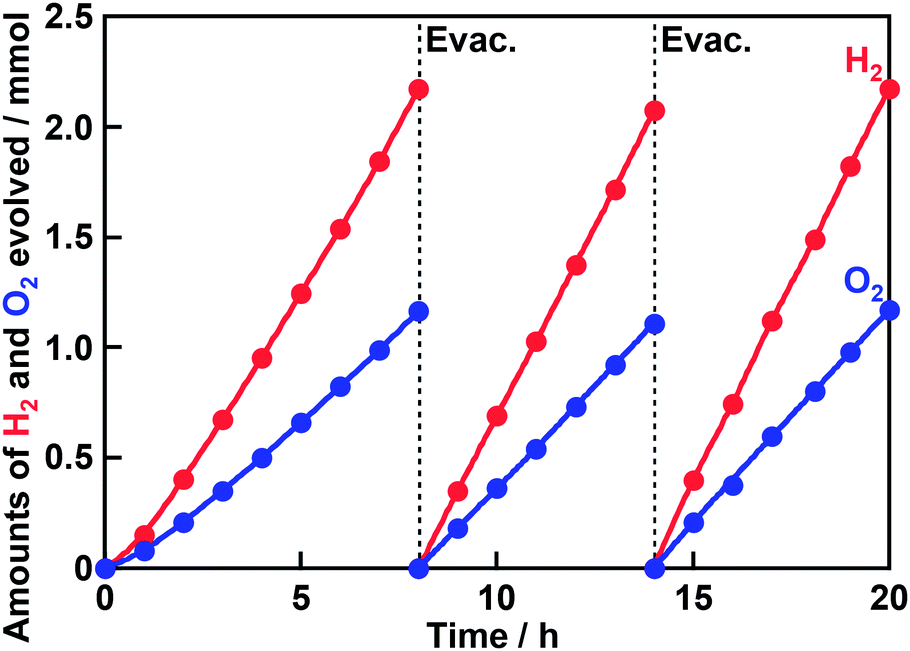 | ||
| Fig. 1 Photocatalytic water splitting over Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3. AgTaO3 was synthesized by a solid state reaction at 1273 K for 15 h without excess Ag. Rh0.5Cr1.5O3 was loaded by an impregnation method at 623 K for 1 h. Photocatalyst: 0.3 g, reactant solution: distilled water (120 mL), cell: top-irradiation cell with a Pyrex window, light source: 300 W Xe-arc lamp (λ > 300 nm). | ||
Chemical and physical states of the loaded Rh0.5Cr1.5O3 in Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 before and after water splitting were characterized using a transmission electron microscope (TEM) and an X-ray photoelectron spectroscopy (XPS) because the water splitting activity of AgTaO3 was greatly improved by loading a Rh0.5Cr1.5O3 cocatalyst. It was confirmed by TEM that Rh0.5Cr1.5O3 particles with a diameter of about 10–20 nm were loaded on AgTaO3 (Fig. 2), as observed for GaN–ZnO.26 In XPS spectra, the binding energy of the Rh 3d5/2 peak for Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was 309.0 eV (Fig. S5†) which is close to not that for Rh2O3 (308.3 eV) but that for Rh0.5Cr1.5O3 (308.9 eV).26 The slightly positive shift in the binding energy was also observed for the Rh0.5Cr1.5O3 loaded on GaN–ZnO (309.1 eV).26 These results indicate that the loaded Rh0.5Cr1.5O3 on AgTaO3 in the present study was similar to that on GaN–ZnO. Additionally, the TEM image of Rh0.5Cr1.5O3 cocatalysts did not change so much after photocatalytic water splitting. On the other hand, in the XPS spectra, the Rh 3d5/2 peak for Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 after photocatalytic water splitting was broadened to lower binding energy than that before photocatalytic water splitting. This observation indicated that the photocatalyst was activated by partial reduction of Rh in loaded Rh0.5Cr1.5O3 cocatalyst to the metallic form during the induction period in Fig. 1.
 | ||
| Fig. 2 TEM images of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 (a) before and (b) after photocatalytic water splitting under UV irradiation using a 300 W Xe-arc lamp. AgTaO3 was synthesized by a solid state reaction at 1273 K for 15 h without excess Ag. Rh0.5Cr1.5O3 was loaded by an impregnation method at 623 K for 1 h. | ||
An action spectrum of water splitting on the optimized Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was measured, as shown in Fig. 3. The maximum AQY was 38% at 340 nm. As far as we know, the AQY is the highest among one-step photoexcitation type water splitting using metal oxide photocatalysts with valence bands consisting of metal cation orbitals. In order to compare the activity of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 with that of highly active NiO/NaTaO3:La, photocatalytic water splitting over Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was carried out in an inner-irradiation reaction cell made of quartz equipped with a 400 W high pressure Hg-lamp using 1 g of photocatalyst powder suspended in 340 mL of distilled water, as shown in Fig. 4. Evolution rates of H2 and O2 were 20 mmol h−1 and 10 mmol h−1, respectively. The activity of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was similar to that of NiO/NaTaO3:La.19 This reaction catalytically proceeded because TON was 47. Thus, Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 interestingly showed the high activity for water splitting even without doping unlike La-doped NaTaO3, Zn,Ca-codoped Ga2O3, and Al-doped SrTiO3, suggesting that AgTaO3 itself has good potential for water splitting. One possible explanation about the potential of AgTaO3 will be the distortion of its crystal structure. The Ta–O–Ta bond angle of AgTaO3 (164 degree) is very similar to that of NaTaO3 (163 degree).23 The similarity in the lattice distortion between AgTaO3 and NaTaO3 causes a similar property of a conduction band of AgTaO3 to a highly active NaTaO3. Therefore, mobility of photogenerated electrons and a reduction potential of water reduction of AgTaO3 should be similar to those of NaTaO3. Additionally, the valence band of NaTaO3 is formed by only O 2p orbitals, whereas that of AgTaO3 is formed by hybridized orbitals between Ag 4d and O 2p.23 Therefore, photogenerated holes in AgTaO3 could migrate more easily than those in NaTaO3. These positive factors in AgTaO3 gave high photocatalytic activity.
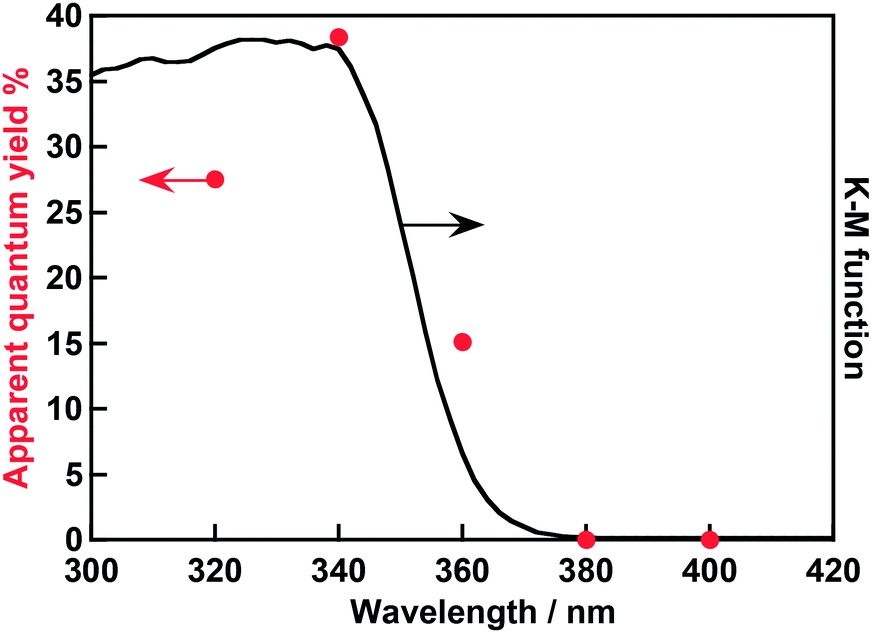 | ||
| Fig. 3 An action spectrum of water splitting over Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 and a diffuse reflectance spectrum of AgTaO3 prepared by a solid state reaction at 1273 K for 15 h without excess Ag. Rh0.5Cr1.5O3 was loaded by an impregnation method at 623 K for 1 h. Photocatalyst: 0.3 g, reactant solution: distilled water (120 mL), cell: top-irradiation cell with a Pyrex window, light source: 300 W Xe-arc lamp with band-pass filters. | ||
 | ||
| Fig. 4 Photocatalytic water splitting over Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 under UV irradiation. Photocatalyst: 1 g, reactant solution: distilled water (340 mL), cell: inner-irradiation cell made of quartz, light source: 400 W high pressure Hg-lamp. AgTaO3 was synthesized by a solid state reaction at 1273 K for 15 h without excess Ag. Rh0.5Cr1.5O3 was loaded by an impregnation method at 623 K for 1 h. | ||
Water splitting proceeded over the Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 photocatalyst even under simulated sunlight irradiation, as shown in Fig. 5. H2 and O2 were steadily evolved with the rates of 486 mL m−2 h−1 and 247 mL m−2 h−1, respectively. The STH was estimated to be 0.13%. Additionally, we were able to watch evolved bubbles when Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 on the bottom of the reaction cell was irradiated with simulated sunlight under 1 atm at room temperature. The STH of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 is lower than those of TiO2/CoOOH/RhCrOx/SrTiO3:Al (STH = 0.4%)22 and SrTiO3:Rh,La-Au-BiVO4:Mo photocatalyst sheet (STH = 1.1%),27 whereas it is higher than those of Z-schematic water splitting using SrTiO3:Rh of a H2-evolving photocatalyst, BiVO4 of an O2-evolving photocatalyst, and ionic electron mediators such as Fe3+/2+ and [Co(bpy)3]3+/2+.28,29 Thus, we successfully achieved highly efficient one-step photoexcitation type solar water splitting using Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 of a valence-band-controlled metal oxide photocatalyst.
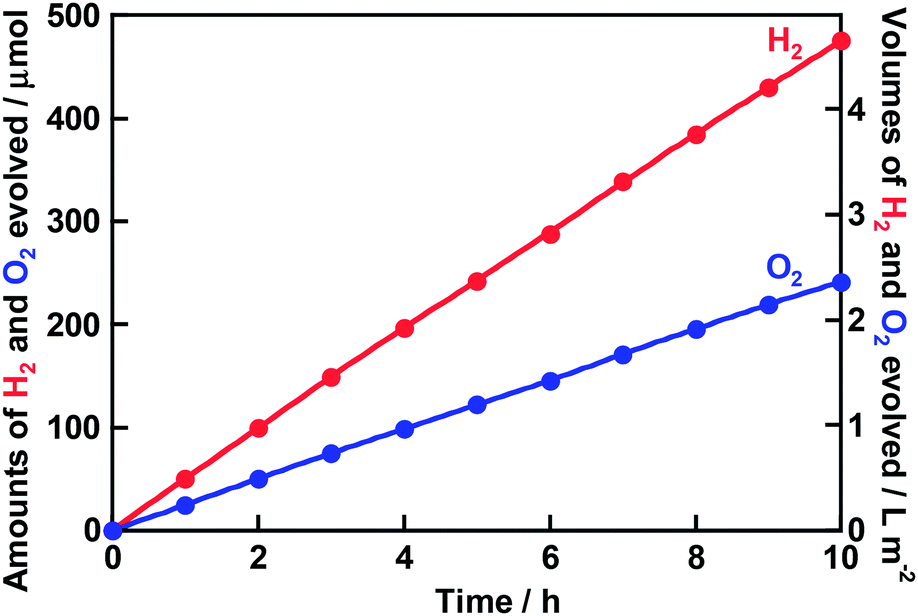 | ||
| Fig. 5 Photocatalytic solar water splitting over Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3. AgTaO3 was synthesized by a solid state reaction at 1273 K for 15 h without excess Ag. Rh0.5Cr1.5O3 was loaded by an impregnation method at 623 K for 1 h. Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was activated by a 300 W Xe-arc lamp in distilled water for 20 h before this reaction (Fig. 1) in order to eliminate the induction period early. Photocatalyst: 0.3 g, reactant solution: distilled water (120 mL), cell: top-irradiation cell with a Pyrex window, light source: a solar simulator with an AM1.5-filter (100 mW cm−2), irradiation area: 25 cm2. | ||
Conclusions
Rh0.5Cr1.5O3(0.2 wt%)-loaded AgTaO3 has arisen as a photocatalyst for solar water splitting in a suspension system. The AQY of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was about 40% at 340 nm. The activity of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 was similar to that of NiO/NaTaO3:La under the same experimental condition, using 1 g of photocatalyst powder suspended in 340 mL of distilled water, in an inner-irradiation cell made of quartz equipped with a 400 W high pressure Hg-lamp. AgTaO3 seems to have good potential for water splitting because Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 showed the high AQY even without doping of elements. Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 also showed the photocatalytic activity for water splitting under simulated sunlight irradiation with the STH of 0.13%. The AQY and STH of Rh0.5Cr1.5O3(0.2 wt%)/AgTaO3 were the highest for one-step photoexcitation type water splitting using a valence-band-controlled metal oxide photocatalyst. Thus, it was clarified that a metal oxide photocatalyst with a valence band consisting of metal cation orbitals rather than O 2p could be utilized as a photocatalyst for highly efficient water splitting. The knowledge is expected to be applied to visible-light-driven photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 17H06433 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)” and 17H0127. The authors gratefully thank Dr T. Ichihashi for assistance with TEM measurements.Notes and references
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- K. Maeda and K. Domen, Bull. Chem. Soc. Jpn., 2016, 89, 627–648 CrossRef CAS.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042–1048 CrossRef CAS.
- K. Takanabe, T. Uzawa, X. Wang, K. Maeda, M. Katayama, J. Kubota, A. Kudo and K. Domen, Dalton Trans., 2009, 10055–10062 RSC.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- C. Pan, T. Takata and K. Domen, Chem.–Eur. J., 2016, 22, 1854–1862 CrossRef CAS PubMed.
- C. Pan, T. Takata, K. Kumamoto, T. Minegishi, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, J. Mater. Chem. A, 2016, 4, 4544–4552 RSC.
- K. Maeda, D. Lu and K. Domen, Chem.–Eur. J., 2013, 19, 4986–4991 CrossRef CAS.
- J. Xu, C. Pan, T. Takata and K. Domen, Chem. Commun., 2015, 51, 7191–7194 RSC.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- H. T. Chiang, H. Lyu, T. Hisatomi, Y. Goto, T. Takata, M. Katayama, T. Minegishi and K. Domen, ACS Catal., 2018, 8, 2782–2788 CrossRef.
- H. Lyu, T. Hisatomi, Y. Goto, M. Yoshida, T. Higashi, M. Katayama, T. Takata, T. Minegishi, H. Nishiyama, T. Yamada, Y. Sakata, K. Asakura and K. Domen, Chem. Sci., 2019, 10, 3196–3201 RSC.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441–12447 CrossRef CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 RSC.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303–308 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753–13758 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05909a |
| This journal is © The Royal Society of Chemistry 2020 |