
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA programmable chemical switch based on triggerable Michael acceptors†
Jiaming
Zhuang
 *a,
Bo
Zhao
a,
Xiangxi
Meng
d,
Jessica D.
Schiffman
bd,
Sarah L.
Perry
bd,
Richard W.
Vachet
abc and
S.
Thayumanavan
*abc
*a,
Bo
Zhao
a,
Xiangxi
Meng
d,
Jessica D.
Schiffman
bd,
Sarah L.
Perry
bd,
Richard W.
Vachet
abc and
S.
Thayumanavan
*abc
aDepartment of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: jzhuang@chem.umass.edu; thai@chem.umass.edu
bCenter for Bioactive Delivery, Institute for Applied Life Science, University of Massachusetts, Amherst, Massachusetts 01003, USA
cMolecular and Cellular Biology Program, University of Massachusetts, Amherst, Massachusetts 01003, USA
dDepartment of Chemical Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA
First published on 10th January 2020
Abstract
Developing an engineerable chemical reaction that is triggerable for simultaneous chemical bond formation and cleavage by external cues offers tunability and orthogonality which is highly desired in many biological and materials applications. Here, we present a chemical switch that concurrently captures these features in response to chemically and biologically abundant and important cues, viz., thiols and amines. This thiol/amine-triggerable chemical switch is based on a Triggerable Michael Acceptor (TMAc) which bears good leaving groups at its β-position. The acceptor undergoes a “trigger-to-release” process where thiol/amine addition triggers cascaded release of leaving groups and generates a less activated acceptor. The newly generated TMAc can be further reversed to liberate the original thiol/amine by a second nucleophile trigger through a “trigger-to-reverse” process. Within the small molecular volume of the switch, we have shown five locations that can be engineered to achieve tunable “trigger-to-release” kinetics and tailored reversibility. The potential of the engineerable bonding/debonding capability of the chemical switch is demonstrated by applications in cysteine-selective and reversible protein modification, universal self-immolative linkers, and orthogonally addressable hydrogels.
Introduction
As two of the most abundant and important nucleophiles that are widely involved in many chemical and biological processes, thiol and amine moieties have drawn tremendous research interest.1–3 The significance of these reactions has been found in a variety of applications, ranging from drug discovery to protein modification, drug delivery, and materials preparation.4–12 Highlighted by the thiol–X click reaction and activated ester based amidations, considerable efforts have been devoted to developing efficient C–S/C–N bond formation chemistry with these functionalities.13–23 On the other hand, triggered debonding reactions with high fidelity to either produce thiol/amine or use them as an external cue to liberate other molecular identities under mild conditions are very few, though their importance is well recognized.24–29 Triggerable debonding chemistry can be broadly classified into two categories: (i) trigger-to-reverse and (ii) trigger-to-release. The former approach is based on reversible reactions, where a coupling product can be chemically, thermally or mechanically decoupled and reversed to regenerate the original starting materials.26,30 The reversibility and dynamic nature of the reversible reaction offer unique access to introduce interesting features such as malleability, self-healing, shape-memory adaptability, stress relaxation and responsiveness into new functional materials and even the opportunity to regulate the function of biomolecules.25,31–34 In comparison, the trigger-to-release strategy uses an efficient bond formation reaction to trigger the cascaded cleavage of preinstalled functionalities of interest.35 The ability to incorporate various releasable functionalities with this approach has great potential in the design of cleavable traceless linkers for antibody drug conjugates, neuromodulator delivery strategies, triggered catalysis and bioimaging.35–40 Thiol/amine associated reactions that concurrently capture the key advantageous features of trigger-to-release and trigger-to-reverse processes are not well-studied. Combining these features offers the opportunity to bring about new functions in areas that are independently represented by both these strategies. Moreover, many of the reported reactions in both of the strategies lack the structural handles to instill programmable reaction kinetics and customizable reversibility. For example, water-soluble allylsulfones have been used to execute double Michael addition reactions, where the disulfide bonds in proteins can be replaced with a functional handle.41 This modification capability has been used further to introduce biorthogonal functionalities onto protein surfaces.42 To fully realize the potential of these types of modifications in a much larger variety of applications, the reactivity of these functional groups with other common bio-nucleophiles and their reversibility need to be fully understood. Herein, we present a class of Triggerable Michael Acceptors (TMAcs) that not only exhibit the combination of advantages of both trigger-to-reverse and trigger-to-release processes with programmable reactivity and reversibility, but also offer the capability for both chemical bond formation and cleavage as presented in Scheme 1. Considering the indispensable roles played by these nucleophiles, we envision that developing reactions featuring the above capabilities for thiols and amines will allow us to build a molecular portfolio based on a single platform for a variety of chemical, biological and materials applications. In addition to demonstrating the unprecedented structural control over the two processes, we also showcase the utility of the reaction as a bonding and debonding chemical switch in cysteine selective and reversible protein modification, universal self-immolative linkers, and preparation of orthogonally addressable hydrogels.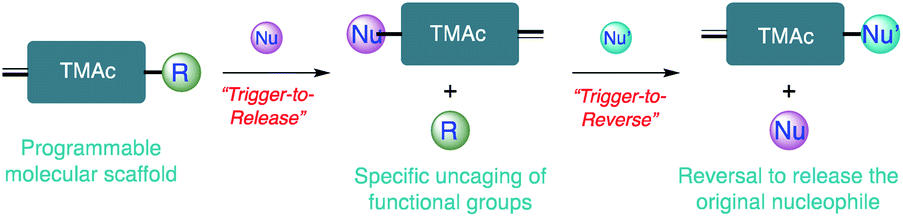 | ||
| Scheme 1 Schematic representation of a scaffold with programmable features in the trigger-to-release and trigger-to-reverse processes. | ||
Results and discussion
Design of an engineerable chemical switch for bonding and debonding
We have designed a chemical switch that can undergo a trigger-to-release process and a structure-dependent trigger-to-reverse process in the presence of certain nucleophiles. The general structure of the TMAc switch is represented by A1 in Fig. 1A. The molecule is simply an activated Michael acceptor, bearing a good leaving group (X) at the β-position. We envisioned that the nucleophilic addition of N1 to A1, which is already favored, can be significantly enhanced due to the elimination of the leaving group; the subsequent generation of a less activated TMAc, A2, should result in a relatively stable product. We envisaged several advantages of this very simple, but underexplored reaction: (i) the leaving group ‘X’ can be used to mask and then provide triggered release of a variety of functional groups, with implications in the activated release of a drug molecule or a sensing reporter (trigger-to-release); (ii) the product A2 can be structurally customized to be either inert or susceptible to a stronger nucleophile N2 that causes the release of the originally conjugated nucleophile N1 (trigger-to-reverse); and (iii) within a relatively small molecular volume, there exist five different locations (R1–R4, and X) that impact on the kinetics of these reactions, thus offering a fundamental understanding of the structural factors that underlie these two processes.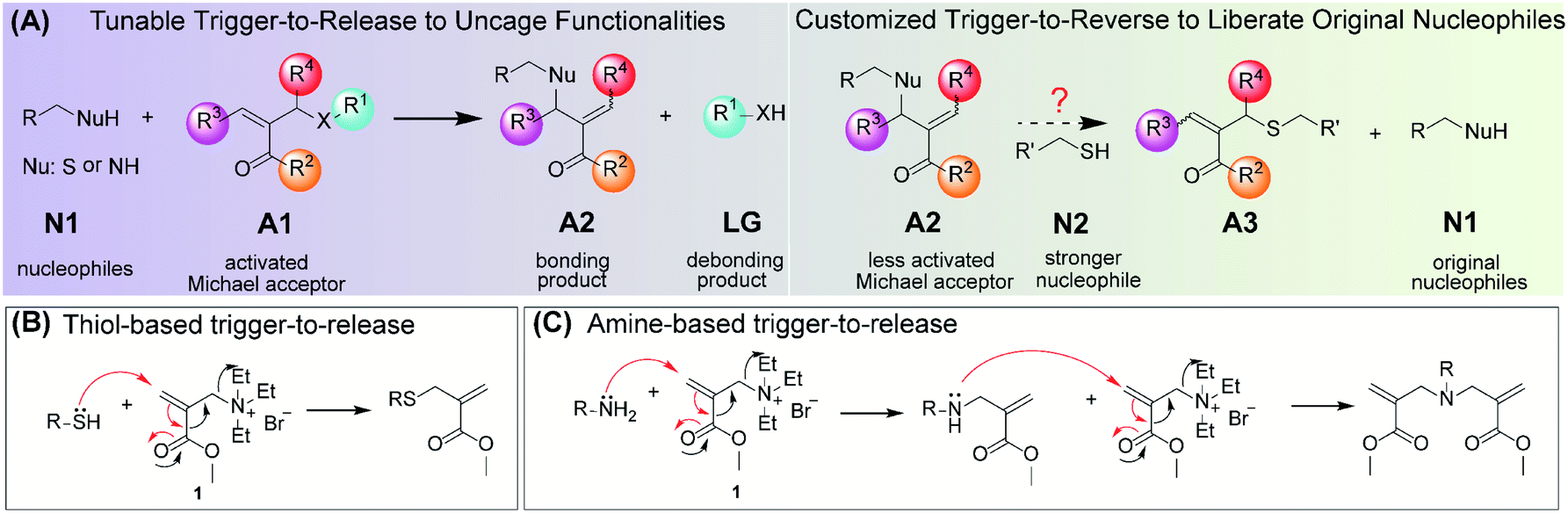 | ||
| Fig. 1 Chemical switch for bonding and debonding using Triggerable Michael Acceptors (TMAc). (A) Schematic illustration of the trigger-to-release process as a universal strategy for uncaging of functional groups (left) and the trigger-to-reverse process, the reversibility of which can be structurally customized (right). (B) Proposed thiol-based trigger-to-release. (C) Proposed amine-based trigger-to-release. | ||
For the initial test of the reaction hypothesis, molecule 1, an acrylate with a quaternary ammonium leaving group, was synthesized and evaluated for its reaction with two nucleophiles, 2-(2-methoxyethoxy)ethanethiol and benzylamine. 1H NMR showed that the reaction of 1 with the thiol (0.5 equiv.) is completed in 3 minutes (Fig. S1†) and that the product is indeed due to the Michael-type addition (Fig. 1B and S2†). Interestingly, the reaction of 1 with benzylamine was much slower at pH 7.4, where the reaction took ∼50 minutes for completion. This is attributed to the weaker nucleophilicity of amines under these conditions. Also, the product here is a dialkylated product with no discernible monoalkylated product (Fig. S3 and S4†), likely because the secondary amine product of the first step is a much better nucleophile than the primary benzylamine. These differences in the kinetics and the nature of products formed from thiol and amine nucleophiles provided us with an additional handle to understand the structural factors that could influence the behavior of the molecular switch.
Trigger-to-release process with tunable reaction kinetics
To understand factors that could influence trigger-to-release kinetics, we systematically varied the R1, R2, R3, R4 and X groups on A1 (Fig. 2A). The stoichiometry between A1 and the nucleophiles was maintained as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all experiments. Similarly, reaction conditions such as solvent, concentration and pH were identical within a comparison group (color-coded group in Fig. 2). However, these conditions might be different between two different comparison groups, either because of solubilization issues associated with a specific set of A1 derivatives or due to the need to slow down the reaction in order to achieve measurable kinetics. For this reason, whenever needed, we also carried out the reaction of at least one molecule under two different reaction conditions to bridge reaction rates in two comparison groups. Reaction kinetics were measured by NMR, and then the half-life of the thiol or amine was extrapolated from the kinetics curve (Fig. S5†) to compare the influence of structural factors on A1 reactivity. First, the thiol-addition reactions for 1–5 with aliphatic and aromatic ammonium leaving groups were studied (Fig. 2B, S5B and H†). The reactions were completed within just a few minutes for molecules 1–3, where the leaving group was an aliphatic ammonium moiety. However, the reaction was much slower for 4 and 5, which contain aromatic ammonium leaving groups. This difference is attributed to the fact that the positive charge in aromatic ammonium ions is more stabilized by delocalization. By extension, 5 would be less reactive than 4, as the electron-donating dimethylamino group can better stabilize the charge. However, we observed that the reaction of 5 was faster than that of 4. The observation is likely due to the basicity of the released DMAP molecule, which accelerates the reaction in MeOH-d4 where the pH of the reaction mixture could not be maintained. This assertion is supported by the drastic rate enhancement observed in the presence of extrinsic DMAP in the 4/thiol reaction (Fig. S6†). To further investigate this, we analyzed the reactivity of 4 and 5 towards benzylamine, where the pH of the solution was maintained in deuterated MeOH/buffer mixed media (Fig. 2C and S5E†). Indeed, the reactivity of 4 toward the amine is higher than that of 5 here. The influence of the leaving group was observed to be similar in both thiol and amine conjugation. Additionally, the reaction kinetics of Michael addition can be significantly influenced by the polarity of the solvent because of the stabilization of the developing transition state. To test this hypothesis, thiol-based trigger-to-release reactions of 1, 4, and 8 were carried out in both methanol and buffer solution. As shown in Fig. S7,† the kinetics in buffer solution is much faster than that in methanol except for compound 1, in which the reactivity is too high to be distinguished in the two solvents. In all these reactions, we were also concerned about any potential solvent-dependent interferences from the air oxidation of thiols. To understand this, oxidation of thiols in both methanol and buffer was monitored by NMR. We did not see any discernible changes in the stability of thiols over the period of this study, which indicates that thiol oxidation has negligible influence on the obtained Michael addition kinetics (Fig. S105 and S106†). Overall, this study provided the initial indication that the leaving group can significantly influence the kinetics of the trigger-to-release process.
:1 in all experiments. Similarly, reaction conditions such as solvent, concentration and pH were identical within a comparison group (color-coded group in Fig. 2). However, these conditions might be different between two different comparison groups, either because of solubilization issues associated with a specific set of A1 derivatives or due to the need to slow down the reaction in order to achieve measurable kinetics. For this reason, whenever needed, we also carried out the reaction of at least one molecule under two different reaction conditions to bridge reaction rates in two comparison groups. Reaction kinetics were measured by NMR, and then the half-life of the thiol or amine was extrapolated from the kinetics curve (Fig. S5†) to compare the influence of structural factors on A1 reactivity. First, the thiol-addition reactions for 1–5 with aliphatic and aromatic ammonium leaving groups were studied (Fig. 2B, S5B and H†). The reactions were completed within just a few minutes for molecules 1–3, where the leaving group was an aliphatic ammonium moiety. However, the reaction was much slower for 4 and 5, which contain aromatic ammonium leaving groups. This difference is attributed to the fact that the positive charge in aromatic ammonium ions is more stabilized by delocalization. By extension, 5 would be less reactive than 4, as the electron-donating dimethylamino group can better stabilize the charge. However, we observed that the reaction of 5 was faster than that of 4. The observation is likely due to the basicity of the released DMAP molecule, which accelerates the reaction in MeOH-d4 where the pH of the reaction mixture could not be maintained. This assertion is supported by the drastic rate enhancement observed in the presence of extrinsic DMAP in the 4/thiol reaction (Fig. S6†). To further investigate this, we analyzed the reactivity of 4 and 5 towards benzylamine, where the pH of the solution was maintained in deuterated MeOH/buffer mixed media (Fig. 2C and S5E†). Indeed, the reactivity of 4 toward the amine is higher than that of 5 here. The influence of the leaving group was observed to be similar in both thiol and amine conjugation. Additionally, the reaction kinetics of Michael addition can be significantly influenced by the polarity of the solvent because of the stabilization of the developing transition state. To test this hypothesis, thiol-based trigger-to-release reactions of 1, 4, and 8 were carried out in both methanol and buffer solution. As shown in Fig. S7,† the kinetics in buffer solution is much faster than that in methanol except for compound 1, in which the reactivity is too high to be distinguished in the two solvents. In all these reactions, we were also concerned about any potential solvent-dependent interferences from the air oxidation of thiols. To understand this, oxidation of thiols in both methanol and buffer was monitored by NMR. We did not see any discernible changes in the stability of thiols over the period of this study, which indicates that thiol oxidation has negligible influence on the obtained Michael addition kinetics (Fig. S105 and S106†). Overall, this study provided the initial indication that the leaving group can significantly influence the kinetics of the trigger-to-release process.
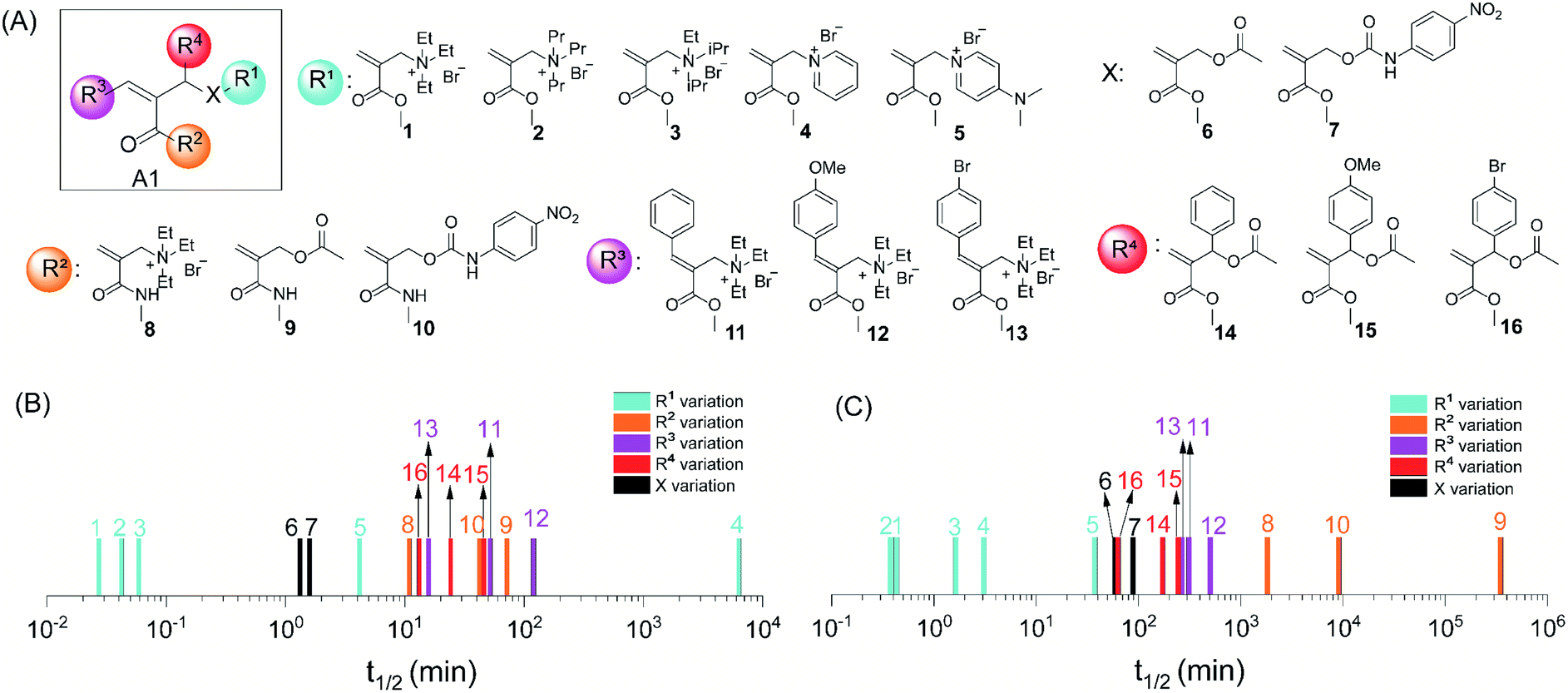 | ||
| Fig. 2 Structure–property relationship investigation of trigger-to-release kinetics. (A) Structural variations of A1. (B) Substitution effect on thiol-based trigger-to-release kinetics described using the half-life of thiol. (C) Substitution effect on amine-based trigger-to-release kinetics described using the half-life of amine. Note: the half-life of the thiol and amine was extrapolated from the kinetics curves obtained by NMR (Fig. S5†). The reaction conditions were identical within a comparison group (color-coded) and can be found in Fig. S5.† | ||
To further test the diversity of leaving groups, we varied the leaving group (X) from quaternary ammonium (1) to carboxylate (6) to carbamate moieties (7), where the released product would be a tertiary amine, carboxylic acid or a primary amine, respectively. For direct comparison, reactions of 1, 6 and 7 with a thiol were performed in the mixed MeOH-d4/buffer solution. The reaction for 1 was too fast to distinguish reactivity difference between pure MeOH-d4 and the mixed solvent within the detectable time scale (Fig. S7†). Indeed, both with the thiol and amine addition reactions, the relative reactivities correlate (1 > 6 ≥ 7) with the leaving ability of ‘X’ (Fig. 2B, C, S5C, F and I†). Considering the electronic influence on reactivity, we hypothesized next that the reactivity can be altered by varying the electrophilicity through variations in the R2 moiety. Accordingly, we synthesized acrylamide molecules 8, 9, and 10 as the weak electron acceptor analogs of 1, 6, and 7, respectively. Indeed, the acrylamides were found to be considerably slower than the corresponding acrylates (Fig. 2B, C, S5C, F and I†).
Next, the influence of R3 substitution was investigated using 11–13, the reactivities of which were substantially lower compared to that of the unsubstituted olefin 1 (Fig. 2B, C and S5D and G†). In addition to the steric barrier to nucleophilic attack, the extended conjugation could also deactivate the double bond. In such a scenario, an electron-donating substituent should make the molecule less reactive. Indeed, we found the reactivity order here to be bromophenyl > phenyl > methoxylphenyl, for both thiol and amine reactions. Interestingly, only a mono-addition product was observed for the latter, which also supports the steric-based inhibition of this reaction (Fig. S8, S82, S83, S86, S87, S90 and S91†). When substituted phenyl groups are incorporated onto the allylic position as R4 substituents, an electron-withdrawing bromo moiety was found to accelerate the reaction (Fig. 2B, C, S5D and G†). This observation was taken to suggest that the reaction goes through an enolate intermediate, and the step involving the leaving group has an early transition state structure with a developing negative charge at the benzylic position.
Engineering the capability of the trigger-to-reverse process to liberate original nucleophiles
Both reversible and irreversible conjugation reactions have implications and the preference depends on the specific applications.31,43–45 In this context, we evaluated the structural factors that dictate the reversibility of trigger-to-release products. Possible products from thiol and amine-based trigger-to-release reactions with A1 molecules are shown in Fig. 3A. Reversibility, i.e. the release of the originally conjugated nucleophile, was studied in the presence of a second nucleophile by following the generation of the original thiol and amine by time-dependent NMR measurement.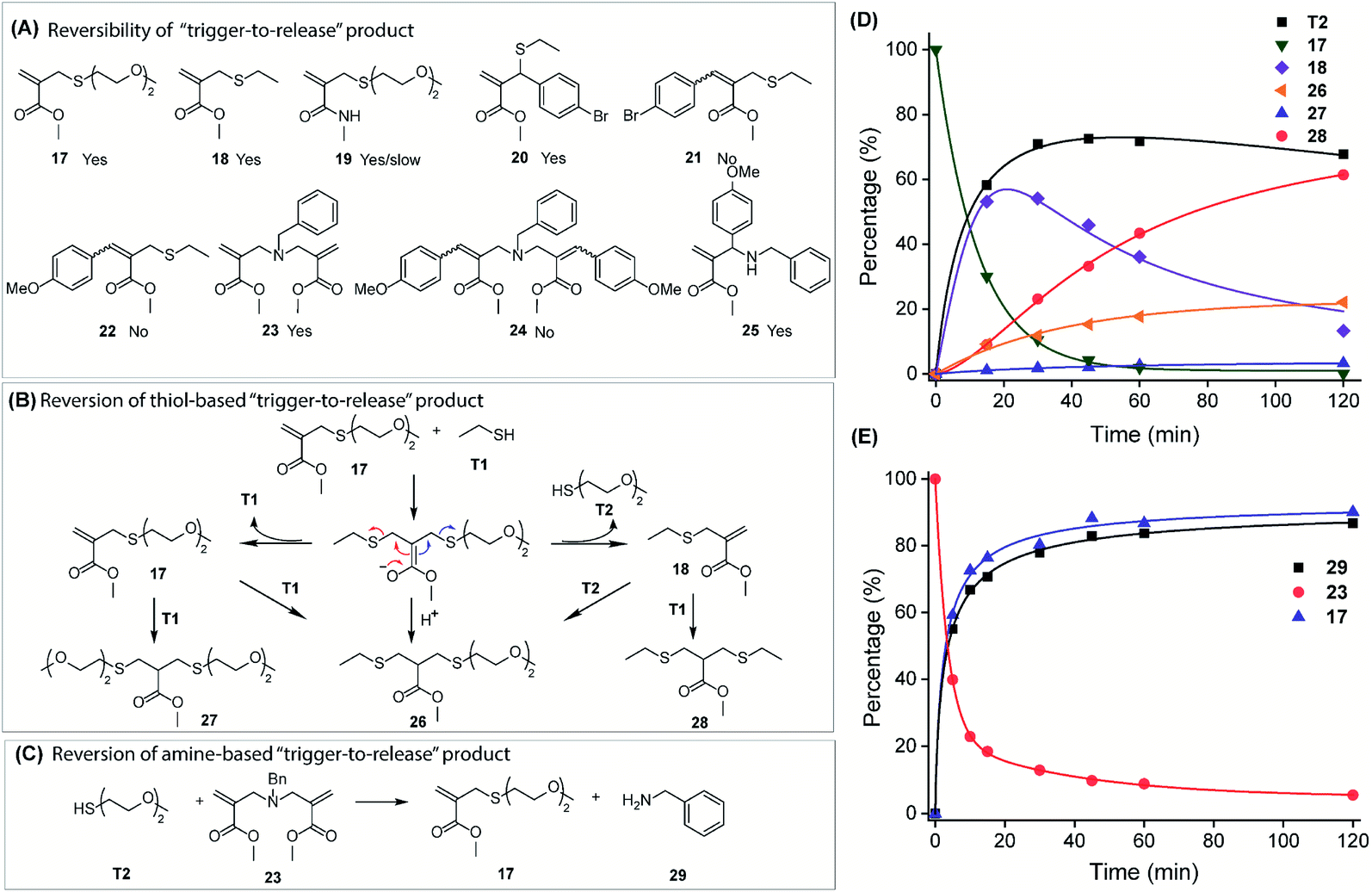 | ||
| Fig. 3 Customization of the reversibility of the trigger-to-release product. (A) Structures of thiol- and amine-based trigger-to-release products (occurrence of triggered reversion is indicated by “Yes”). (B) Triggered reversion of the thiol-based trigger-to-release product, 17. [T1]/[17] = 8. (C) Triggered reversion of the amine-based trigger-to-release product, 23. [T2]/[23] = 2. (D) Percentage of existing species in the course of the trigger-to-reverse process of 17. (E) Percentage of existing species in the course of the trigger-to-reverse process of 23. | ||
First, when 17 was treated with eight equivalents of ethanethiol (T1), generation of 18 was expected along with the released diethyleneglycolthiol (T2) (Fig. 3B and D). Interestingly, concurrent formation of other species, 26–28, was also observed, which can be understood based on the Michael addition–elimination mechanism (Fig. S9 and S10†). Solely from the deconjugation perspective, 70% of the originally conjugated T2 was released in this reaction. The lack of complete release of T2 is attributed to its reaction with 18 to generate 26. The fact that ethanethiol (T1) also reacts with 18 to form 28, relative to 26, suggests that the product distribution is simply determined by the ratio of T1 and T2 nucleophiles. This is understandable as the reaction is at equilibrium and this assertion was confirmed by decreasing the amount of T1, where an increase in the percentage of 26 was observed (Fig. S11†).
Similarly, the thiol triggered reversal of the amine-conjugated product, 23, was also investigated (Fig. 3C). More than 80% of benzylamine (29) was released in the presence of just 2 equivalents of T2 (Fig. 3E and S12†). The rather clean conversion, even with low amounts of thiol, is attributed to the lack of nucleophilicity of the protonated benzylamine at pH 7.4. Further studies on thiol-triggered reversal of other compounds show that 18, 19, 20, and 25 can also be reversed to release the original thiol or amine (Fig. S13–S16 and S20†). Interestingly, though a thiol-triggered deconjugate of 19 was observed, the reaction rate was very slow, which can be explained by the decreased electrophilicity of the double bond due to the conjugation effect of amides. This inspired us to further take advantage of the structural tunability of A1 to engineer the reversibility of the thiol/amine trigger-to-release reaction. We hypothesized that thiol-triggered reversal of the conjugate reaction will be profoundly suppressed by a phenyl substitution on the double bond, because the substitution not only reduces its electrophilicity, but also increases the steric hindrance of nucleophilic addition. To test this rationale, compounds 21, 22, and 24 were synthesized by conjugating ethane thiol or benzyl amine to the corresponding A1 molecules and were subjected to a thiol-triggered deconjugation reaction. Indeed, 21, 22, and 24 were all inert to the thiol and generation of original nucleophiles (ethane thiol or benzyl amine) was not observed (Fig. S17–S19†). Additionally, the trigger-to-reverse process is also promoted by the polarity of the reaction medium suggested by the observation of the accelerated reaction in phosphate buffer compared to that in pure MeOD (Fig. S107 and S108†). Overall, the results above demonstrate that the capability of reversing both thiol and amine conjugate reactions can be conveniently engineered by simply varying the A1 structure.
Chemoselective and reversible protein modification by trigger-to-release and trigger-to-reverse processes
As a demonstration of versatility, we investigated the chemical modification of proteins as it provides a powerful tool for expanding their functions and profiling activity.46 Here, we envision that the structural tunability of the TMAc can provide an opportunity to tailor its trigger-to-release reactivity in order to selectively react with thiol for cysteine-selective protein modification. Considering the delicate nature of proteins, modification reactions must be fast and free of organic solvents. The ammonium version of A1 is both reactive and water soluble and therefore was investigated for the modification. Specifically, acrylamide 8 exhibits clearly distinguishable reactivity between thiols and amines in aqueous medium (Fig. S21†), and it is far less reactive than ester 1. Both molecules were tested for the modification of β-lactoglobulin B (βLGb), which has one cysteine and fifteen lysine residues. As illustrated in Fig. 4A, we envisaged that modifications on multiple nucleophilic residues would occur with the highly reactive 1, while 8 would selectively modify cysteine. βLGb was fully labelled within 5 and 40 minutes, when treated with an excess of 1 and 8, respectively, as discerned from the disappearance of the unmodified protein peaks in ESI-MS (Fig. 4B and S22†). The mass shift in the concurrently appearing new peaks corresponds to one to four modifications in βLGb, when treated with 1. Multiple modifications suggest that 1 is non-selective in labeling the protein. In contrast, only a single new peak corresponding to the protein with one modification was found in the βLGb labelled with 8. To further pinpoint modification sites on βLGb, modified proteins were digested and subjected to MS/MS analysis (Fig. S23–S30†). Only the free cysteine site was modified with 8. However, modifications of other nucleophilic residues including lysine, tyrosine and threonine were found with 1. Interestingly, the primary amine of lysine was not double-labeled, as seen with small molecule nucleophiles. This is possibly due to the competition from other nucleophilic residues.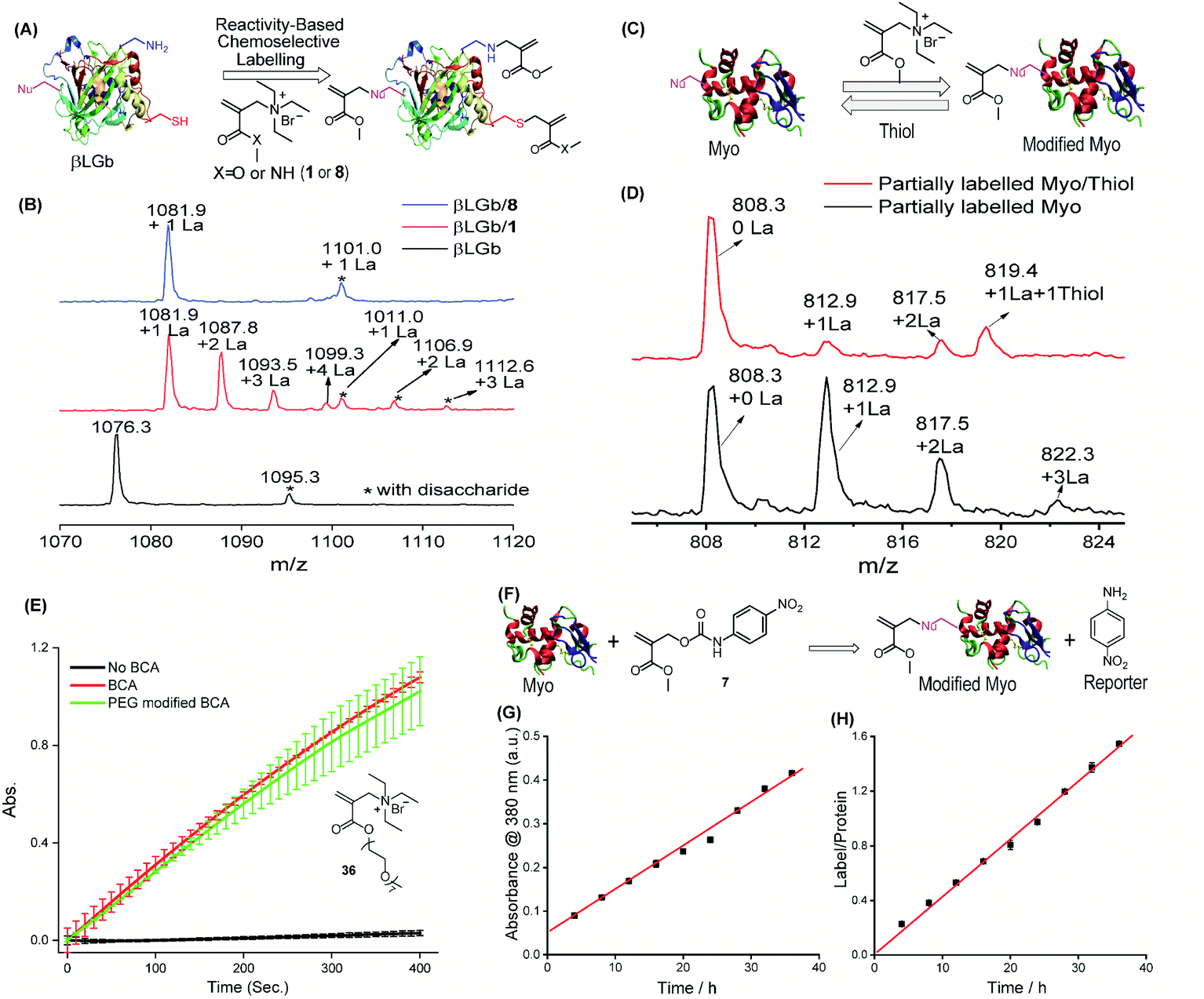 | ||
| Fig. 4 Protein modification with the TMAc chemical switch. (A) Schematic illustration of cysteine-selective labeling of βLGb, driven by the reactivity difference between 1 and 8. (B) Mass spectra of unlabeled βLGb (bottom), and βLGb labeled with 1 (middle) and 8 (top). Note: the spectra show only one charge state (+17) and the full spectra are shown in Fig. S22.† 1La stands for 1 modification. The peak highlighted by * corresponds to βLGb with disaccharide PTM. (C) Schematic illustration of reversible modification of myoglobin using compound 1. (D) Mass spectra of myoglobin modified with 1 and further incubated with and without thiol. Note: one charge state is shown and spectra with all charge states are shown in Fig. S31.† 1La stands for 1 modification. (E) Influence of PEG modification using 36 on BCA activity evaluated by using a chromogenic assay. (F) Use of the conjugate-to-release strategy to spectroscopically monitor modification of myoglobin by following the absorbance of the reporter, 4-nitroaniline. (G) 4-Nitroaniline absorbance evolution during myoglobin modification with compound 7. (H) The number of modifications on myoglobin corresponding to (G). | ||
As with the small molecules above, the reversibility of the covalent modification of the proteins was also tested, as this possibility has potential implications as a “catch and release” strategy for protein purification or protein function manipulation. To test this possibility, myoglobin (Myo) was modified with 1, followed by incubation with 10 equivalents of a thiol for 2 hours (Fig. 4C). Multiple modifications were observed on myoglobin, when treated with 1 (Fig. 4D and S31†). Modified proteins were subsequently reversed to unmodified myoglobin after incubating with a thiol, as shown by the intensity reduction of modified protein peaks accompanied by an increase in that of the unmodified protein peak. The extent of recovery is consistent with that observed in the small molecule study above. To assess the influence of the covalent modification on protein structures, Myo with ∼3 modifications was investigated for changes by CD spectroscopy, using the melting temperature and absorption spectrum of the heme region (Fig. S32 and S33†). The lack of any significant change in Myo's physical properties shows that the modification has a negligible effect on protein structures. Similarly, PEG modified bovine carbonic anhydrase (BCA) (with ∼3 modifications using 36) does not exhibit any effect on its enzymatic activity (Fig. 4E and S34†).
Additionally, note that our method also offers the ability to simultaneously monitor protein labeling within this reaction format itself when the released leaving group is a reporter (Fig. 4F). To test this idea, we tested the modification of Myo with 7, where a nucleophile induced conjugate-to-release reaction would form 4-nitroaniline, the absorbance of which can report on the progress of protein modification. The correlation between the molecular release and the extent of protein modification is indeed clear (Fig. 4G and H).
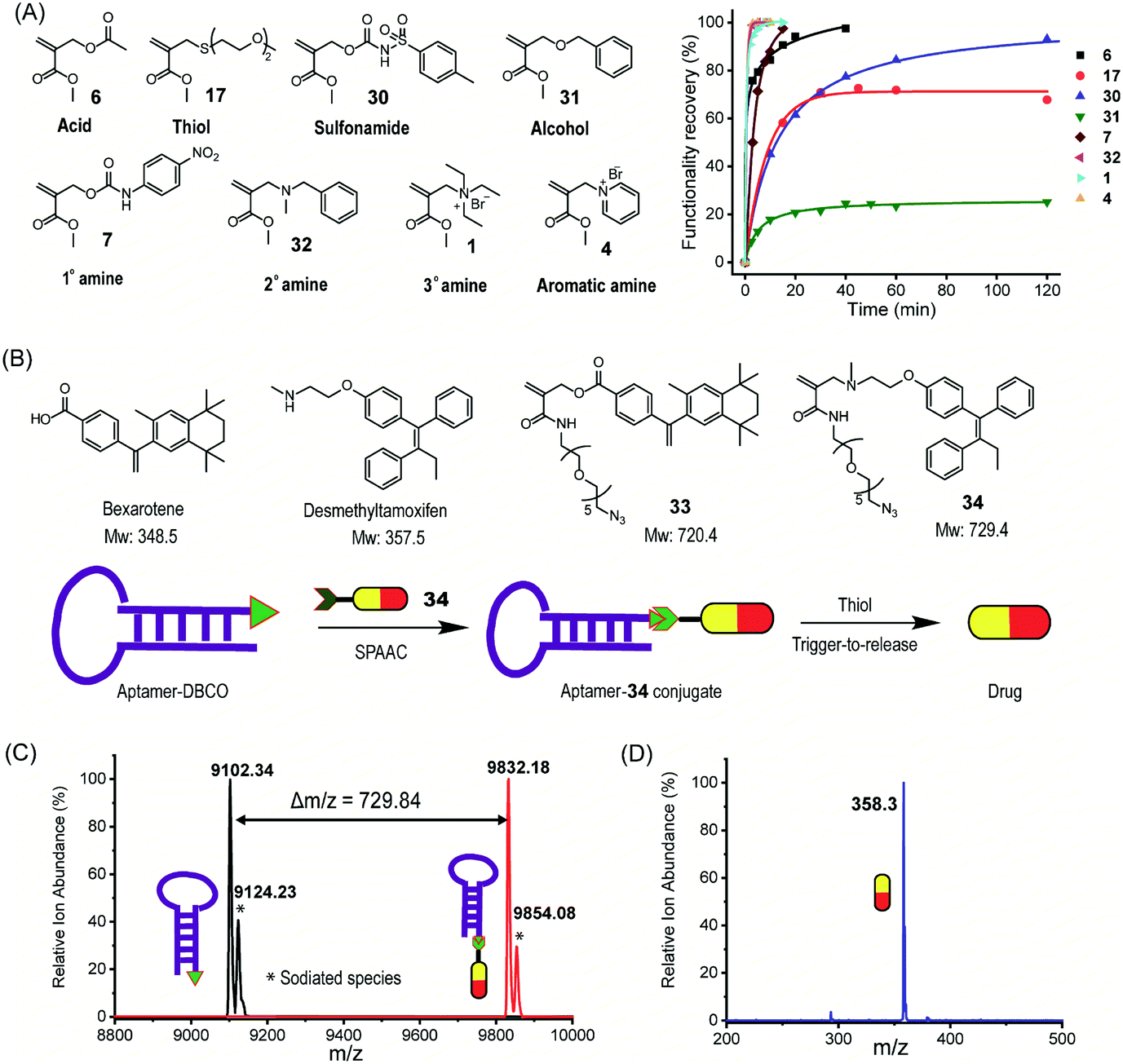 | ||
| Fig. 5 Trigger-to-release as a universal strategy for uncaging functionalities. (A) Functionalities can be masked and uncaged from the chemical switch (left), and the efficiency of thiol-triggered recovery of these functionalities. (B) Caging bexarotene and desmethyltamoxifen with an amide-based clickable TMAc (top) and preparation of the aptamer–34 conjugate and thiol-triggered release of desmethyltamoxifen from the conjugate (bottom). (C) Mass spectrometry analysis of the aptamer–DBCO (black) and aptamer–34 conjugate (red). (D) Mass spectrometry analysis of the released small molecule from the aptamer–34 conjugate. | ||
Next, an azide-functionalized prodrug 34 was conjugated to an AS1411 aptamer which is modified with a complementary reactive DBCO group through strain-promoted alkyne azide cycloaddition (SPAAC). The conjugation was characterized by mass spectrometry (Fig. 5C). Successful conjugation of 34 to an aptamer is supported by the observation of the molecular weight shift of 729.84 which is identical to the molecular weight of 34. Release of the parent drug, desmethyltamoxifen, from the aptamer–34 conjugate in the presence of thiol was followed by LC-MS. Mass spectrometry analysis (Fig. 5D) shows that the molecular weight of released small molecules is identical to that of desmethyltamoxifen suggesting the thiol-induced drug release from the conjugate. Using the same strategy, we also conjugated 33 to DBCO modified polyethylene glycol to form a polymer–drug conjugate, where the release of bexarotene molecules from the conjugate was also triggered by thiol (Fig. S49 and S50†). As a proof of concept study, we show here that the chemical switch can serve as a general platform to uncage drugs with different functionalities without linker chemistry redesign. Note that to make the system suitable for delivery applications, structural engineering on the linker is still required to achieve optimal release kinetics and on-target thiol-triggered release. The primary objective of this work is to simply illustrate the potential utility of the methodology developed here.
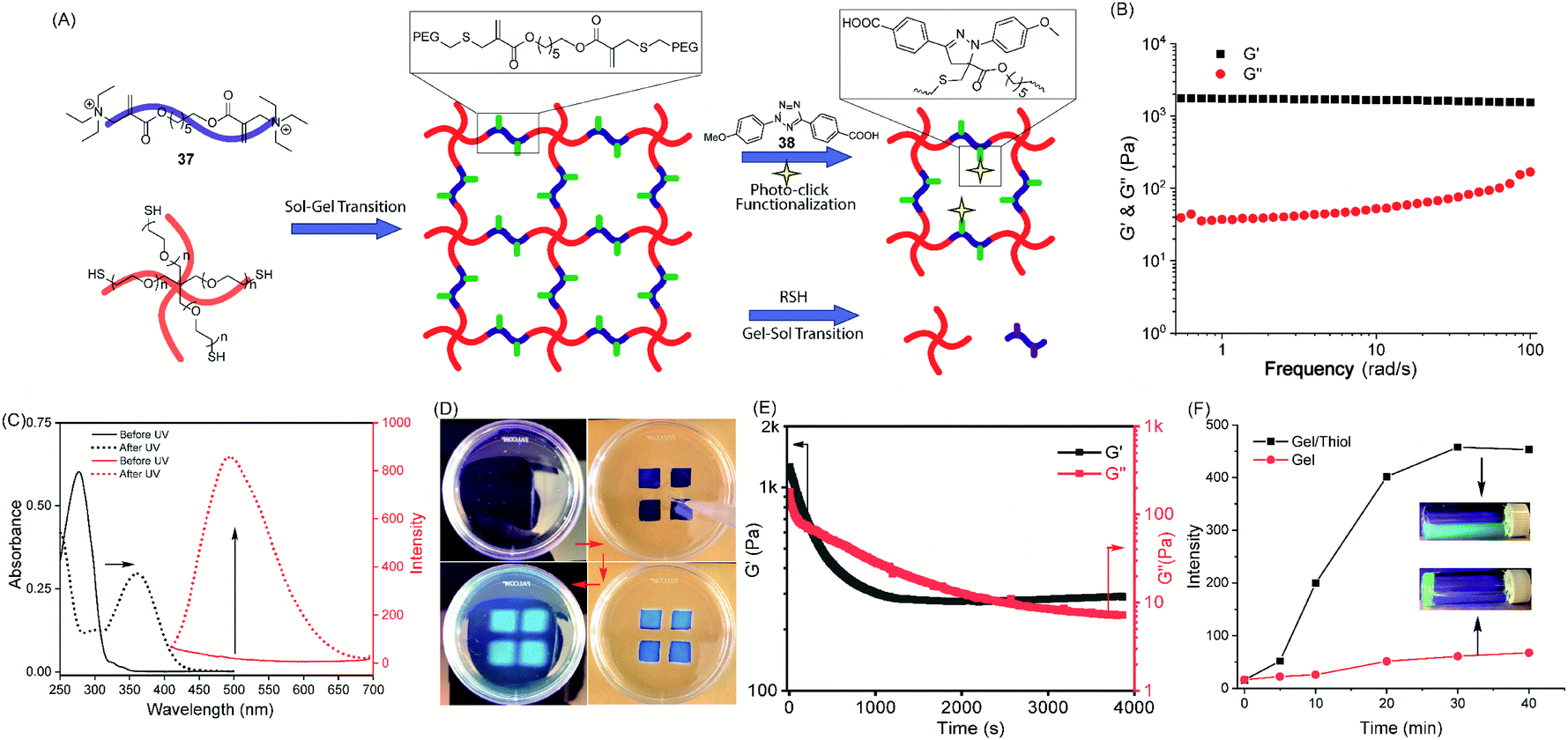 | ||
| Fig. 6 TMAc chemical switch for hydrogel preparation and orthogonal manipulation. (A) Illustration of the hydrogel network formation and orthogonal manipulation of post-gelation properties. (B) Frequency sweep of the storage modulus (G′) and loss modulus (G′′) of the pre-cured hydrogel. (C) Photo-click of a tetrazole (38) with the TMAc (18) followed by UV-vis and fluorescence spectroscopy. (D) Images of the photo-patterning hydrogel. Top left: hydrogel before tetrazole addition; top right: addition of a tetrazole to the hydrogel above a pattern-masked UV lamp. Bottom right: the hydrogel with the tetrazole exposed to UV irradiation for 2 minutes. Bottom left: the patterned hydrogel after removal of unreacted tetrazole. (E) In situ rheological measurement of thiol-triggered hydrogel dissolution. (F) Hydrogel for cargo encapsulation and thiol-triggered release measured by the evolution of fluorescence in the supernatant. Inset: corresponding photographs of the hydrogel incubated with and without the thiol. | ||
Conclusions
We have developed a simple and versatile Triggerable Michael Acceptor (TMAc) based molecular switch that concurrently possesses the key attributes of trigger-to-release and trigger-to-reverse reactions. Five different locations in a relatively small molecular volume have been utilized to achieve fine control over the reaction kinetics of both processes. Identification of the structural features that underlie the reaction kinetics, with respect to different nucleophiles, offers the opportunity to use it in a variety of applications that require efficient bonding and/or debonding features. We demonstrated this versatility in three different ways. By judiciously choosing the reactivity of the molecule, we have shown that this reaction can be used to either chemoselectively modify cysteines over other nucleophilic residues or indiscriminately label nucleophilic residues on protein surfaces. While the selectivity of the former has implications in site-specific labeling of proteins, the reactivity and reversibility of the latter has implications in proteomics and protein delivery applications. As a second demonstration of the fact that these can be incorporated onto a variety of scaffolds, we have introduced these functionalities as part of macromolecule–drug conjugates, using either a polymer or a DNA aptamer as the macromolecule. These conjugates are then triggered to release the drug molecules with high efficiency at biologically relevant thiol concentrations. Finally, we have utilized the chemistry to achieve reversible hydrogels that can be spatiotemporally addressed using the TMAc functionalities as the reactive handles. Overall, the versatility of the molecular switch, introduced here, is illustrated with the ability to bond and debond with a variety of functional groups with tunable kinetics, selectivity and reversibility. Because of their tunable features, these reactions hold the promise of impacting on a variety of areas ranging from biology to materials science.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the National Science Foundation (CHE-1740597) for supporting this work.Notes and references
- N. Haugaard, Ann. N. Y. Acad. Sci., 2000, 899, 148 CrossRef CAS PubMed.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486 CrossRef CAS PubMed.
- R. N. Zuckermann, E. J. Martin, D. C. Spellmeyer, G. B. Stauber, K. R. Shoemaker, J. M. Kerr, G. M. Figliozzi, D. A. Goff, M. A. Siani, R. J. Simon, S. C. Banville, E. G. Brown, L. Wang, L. S. Richter and W. H. Moos, J. Med. Chem., 1994, 37, 2678 CrossRef CAS PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, CA, 2nd edn, 2008 Search PubMed.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724 CrossRef CAS.
- M. R. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518 CrossRef CAS PubMed.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14 CrossRef CAS.
- J. H. Ko and H. D. Maynard, Chem. Soc. Rev., 2018, 47, 8998 RSC.
- G. Liu, G. Shi, H. Sheng, Y. Jiang, H. Liang and S. Liu, Angew. Chem., Int. Ed., 2016, 56, 8686 CrossRef PubMed.
- C. E. Holy, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- A. Abbas, B. Xing and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491 CrossRef CAS PubMed.
- V. X. Truong and A. P. Dove, Angew. Chem., Int. Ed., 2013, 52, 4132 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day, S. J. Paluck and H. D. Maynard, J. Am. Chem. Soc., 2017, 139, 1145 CrossRef PubMed.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS PubMed.
- B. Y. Li, I. Akiba, S. Harrisson and K. L. Wooley, Adv. Funct. Mater., 2008, 18, 551 CrossRef.
- C. Ott, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 3516 RSC.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS PubMed.
- A. Saha, S. De, M. C. Stuparu and A. Khan, J. Am. Chem. Soc., 2012, 134, 17291 CrossRef CAS PubMed.
- M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66 RSC.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034 CrossRef CAS PubMed.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146 CrossRef CAS PubMed.
- K. L. Diehl, I. V. Kolesnichenko, S. A. Robotham, J. L. Bachman, Y. Zhong, J. S. Brodbelt and E. V. Anslyn, Nat. Chem., 2016, 8, 968 CrossRef CAS.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960 CrossRef CAS PubMed.
- G. Joshi and E. V. Anslyn, Org. Lett., 2012, 14, 4714 CrossRef CAS PubMed.
- V. Hong, A. A. Kislukhin and M. G. Finn, J. Am. Chem. Soc., 2009, 131, 9986 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- W. A. Ogden and Z. Guan, J. Am. Chem. Soc., 2018, 140, 6217 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492 CrossRef CAS PubMed.
- B. S. Sumerlin, Science, 2018, 362, 150 CrossRef CAS PubMed.
- L. R. Staben, S. G. Koenig, S. M. Lehar, R. Vandlen, D. Zhang, J. Chuh, S. F. Yu, C. Ng, J. Guo, Y. Liu, A. Fourie-O'Donohue, M. Go, L. Xin, N. L. Segraves, T. Wang, J. Chen, B. Wei, G. D. Phillips, K. Xu, K. R. Kozak, S. Mariathasan, J. A. Flygare and T. H. Pillow, Nat. Chem., 2016, 8, 1112 CrossRef CAS PubMed.
- S. J. Moon, S. V. Govindan, T. M. Cardillo, C. A. D'Souza, H. J. Hansen and D. M. Goldenberg, J. Med. Chem., 2008, 51, 6916 CrossRef CAS PubMed.
- T. H. Pillow, M. Schutten, S.-F. Yu, R. Ohri, J. Sadowsky, K. A. Poon, W. Solis, F. Zhong, G. Del Rosario, M. A. T. Go, J. Lau, S. Yee, J. He, L. Liu, C. Ng, K. Xu, D. D. Leipold, A. V. Kamath, D. Zhang, L. Masterson, S. J. Gregson, P. W. Howard, F. Fang, J. Chen, J. Gunzner-Toste, K. K. Kozak, S. Spencer, P. Polakis, A. G. Polson, J. A. Flygare and J. R. Junutula, Mol. Cancer Ther., 2017, 16, 871 CrossRef CAS PubMed.
- S. Goggins, B. J. Marsh, A. T. Lubben and C. G. Frost, Chem. Sci., 2015, 6, 4978 RSC.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638 CrossRef CAS PubMed.
- N. Hananya and D. Shabat, ACS Cent. Sci., 2019, 56, 949 CrossRef PubMed.
- T. Wang, A. Riegger, M. Lamla, S. Wiese, P. Oeckl, M. Otto, Y. Wu, S. Fischer, H. Barth, S. L. Kuan and T. Weil, Chem. Sci., 2016, 7, 3234 RSC.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. W. Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133 RSC.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946 CrossRef CAS PubMed.
- S. Sakamoto and I. Hamachi, Anal. Sci., 2019, 35, 5 CrossRef CAS PubMed.
- J. Liu, W. Liu, I. Weitzhandler, J. Bhattacharyya, X. Li, J. Wang, Y. Qi, S. Bhattacharjee and A. Chilkoti, Angew. Chem., Int. Ed., 2015, 54, 1002 CrossRef CAS PubMed.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617 CrossRef CAS PubMed.
- X. Hu, G. Liu, Y. Li, X. Wang and S. Liu, J. Am. Chem. Soc., 2015, 137, 362 CrossRef CAS PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 33, 1700141 CrossRef PubMed.
- K. Dutta, D. Hu, B. Zhao, A. E. Ribbe, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2017, 39, 5676 CrossRef PubMed.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed.
- J. C. Grim, T. E. Brown, B. A. Aguado, D. A. Chapnick, A. L. Viert, X. Liu and K. S. Anseth, ACS Cent. Sci., 2018, 4, 909 CrossRef CAS PubMed.
- M. D. Konieczynska and M. W. Grinstaff, Acc. Chem. Res., 2017, 50, 151 CrossRef CAS PubMed.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05841a |
| This journal is © The Royal Society of Chemistry 2020 |