
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Hitchhiker's guide to biocatalysis: recent advances in the use of enzymes in organic synthesis
Roger A.
Sheldon
 *ab,
Dean
Brady
a and
Moira L.
Bode
a
*ab,
Dean
Brady
a and
Moira L.
Bode
a
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg, South Africa. E-mail: Roger.Sheldon@wits.ac.za
bDepartment of Biotechnology, Delft University of Technology, Delft, The Netherlands
First published on 13th February 2020
Abstract
Enzymes are excellent catalysts that are increasingly being used in industry and academia. This perspective is primarily aimed at synthetic organic chemists with limited experience using enzymes and provides a general and practical guide to enzymes and their synthetic potential, with particular focus on recent applications.
 Roger A. Sheldon | Roger Sheldon, a recognised authority on Green Chemistry, is widely known for developing the E factor for assessing environmental impacts of chemical processes. He received the RSC Green Chemistry Award and the Biocat Lifetime Achievement Award for his contributions to biocatalysis, was made a Fellow of the Royal Society in 2015 and Honorary Fellow of the Royal Society of Chemistry in 2019. He has a PhD from Leicester University (UK) and previously worked at Shell Research Amsterdam (1969–1980), DSMAndeno (1980–1990), was Professor at Delft University of Technology (NL) (1991–2007), and CEO of CLEA Technologies (2006–2015). He is currently Distinguished Professor of Biocatalysis Engineering at the University of the Witwatersrand (RSA). |
 Dean Brady | Dean Brady obtained his PhD from Rhodes University, under the supervision of Professor John Duncan. He then joined AECI (African Explosives and Chemical Industries) in 1994, working on R&D in industrial biotechnology. In 2001 he joined Professor Roger Sheldon at Delft University of Technology (NL) as a Visiting Research Fellow and then at CSIR (RSA) as a Research Group Leader and Chief Research Scientist, specialising in Biocatalysis. He joined the University of the Witwatersrand in 2013 as a Professor and the Head of the School of Chemistry. |
 Moira L. Bode | Moira Bode obtained her PhD from Rhodes University under the supervision of Professor Perry Kaye. Her main research interests are the design and synthesis of compounds with activity against HIV enzymatic targets (reverse transcriptase and protease) and the use of enzymes in organic synthesis. She worked at the Agricultural Research Council, at the CSIR (RSA), and in the laboratories of Prof Roger Sheldon at Delft University of Technology (NL). She is currently an Associate Professor of organic chemistry at the University of the Witwatersrand. |
1. Introduction
Faced with having to perform hundreds of reactions in what is essentially one pot (the prokaryotic cell), life has had to provide catalysts that are highly efficient and very selective. There is an enzyme suited to every natural compound. These enzymes can also catalyse non-natural synthetic reactions, and enzymes are even evolving in vivo to specifically handle non-natural industrial chemicals.1 Moreover, biocatalysts can all function in the same readily available green solvent, water, a capability that the chemical industry needs to attain if it is to be sustainable.2In this article we look at how the organic chemist can obtain these nifty catalysts, what they do and how they work, and how they can be adapted to industrial use, integrated into chemical processes, and modified to meet specific process requirements.
2. Sources and commercial availability of enzymes in the metagenome era
Although the chemist James Sumner had already isolated the enzyme urease in 1926 and proved its protein structure, it would take some time before this knowledge was translated into new chemical processes. However, by the beginning of the millennium quite a few companies were using biocatalysis3 and Straathof et al. could already list 134 industrial biocatalytic processes or biotransformations.4 Biocatalysis research accelerated from about 2000 onwards, largely as a result of the spectacular advances in (meta)genome sequencing and protein engineering5 and by 2019 Woodley observed that several hundred processes are now in operation,6 especially in the pharmaceutical industry.7The extent of their impact on chemicals manufacture is immense. The largest use of isolated enzymes in a single process is the liquefaction and saccharification of starch to produce glucose. Another process uses glucose isomerase to produce 107 tons per annum of high fructose corn syrup from glucose.8 The enzymatic conversion of waste cellulosic biomass to provide sugars for bioethanol will no doubt eventually surpass this in scale. In the chemical industry, nitrile hydratases are used to catalyse the conversion of acrylonitrile to the industrial monomer, acrylamide, on a multi hundred thousand ton per annum scale (see Section 5). In the pharmaceutical industry, penicillin G amidase is used in the industrial synthesis of thousands of tons of a variety of beta lactam antibiotics, such as amoxicillin.9 Enzymes are also used on a large scale in the more traditional applications: food and beverage processing, detergents, pulp and paper manufacture, and in the textiles and leather industries.
Enzymes can be sourced from plants, e.g. carrots10 and soy beans,11 fungi12,13 (such as baker's yeast, filamentous fungi and mushrooms), bacteria14 and archaea.15 The early use of readily available enzymes from animal organs, e.g. pig liver, obtained from abattoirs, has been superseded by advances in biotechnology, which enabled the cheaper production of enzymes through over-expression using recombinant DNA technology in microbial expression hosts.16 Future developments are likely to involve the production of enzymes in plant hosts that can be grown, harvested and easily processed to give ultra-cheap enzymes.17
The globally traded enzyme market is worth about $7 bn (this excludes production for in-house use). The majority of commercially traded enzymes are produced and sold by Novozymes, followed by DuPont (incorporating Danisco and Genencor). Other large scale producers include DSM, Amano and BASF (incorporating Verenium), Advanced Enzyme Technologies Limited and AB Enzymes. Other companies such as Thermo Fischer Scientific, Hoffmann La Roche, Procter and Gamble, BBI Enzymes, Puratos, Novus International and Chr. Hansen are also producers of significant quantities of commercial enzymes. These companies tend to produce enzyme formulations for large scale applications, such as food and beverage production, household care (e.g. detergents) and bioenergy or feed, but increasingly also for pharmaceutical production and biotechnology. Carbohydrases, proteases, and to a lesser extent, lipases are the major enzyme classes sold and used on a large scale. The advantage of using these enzymes is that they are readily available, consistent from batch to batch, and are produced in sufficient quantities to support commercial biocatalytic processes. The downside is the limited range of enzymes available. Commercial enzyme producers often kindly provide enzyme samples to researchers in quantities that are sufficient for student projects. However, industry limits the available information on the enzymes for reasons of intellectual property and hence the species origin and protein sequence is becoming increasingly difficult to identify. This can be a problem if the product is discontinued, or if the enzyme is modified during formulation improvement. However, if an enzyme is already being used at scale in a chemical process the company can usually accommodate continued production.
Smaller companies, such as Codexis (USA), Biocatalysts (Wales, UK) and BRAIN (Germany), are focused on enzyme development. Interestingly, Johnson Matthey, the speciality chemicals company that produces catalysts, now has a wide range of biocatalysts among their products.
The smallest companies tend to be university spin-outs and include Enzymicals AG (Germany) and Prozomix (UK). They have the advantage of offering wide novel enzyme selections, catering to previously unexplored enzyme classes. They are also often willing to develop enzyme reactions according to client requirements. Enzymes may be purchased as single enzymes (often as kits containing co-factors, test substrates and other required reagents) and as enzyme selections (usually 10–50 enzymes of the same activity type). Research Chemical companies, such as Sigma-Aldrich, also sell enzymes, sourced from companies of all sizes.
Caution should be taken as many enzyme preparations are only partially purified from the cell, and may therefore contain multiple enzyme types and exhibit more than one activity.18 Moreover, some enzymes have the capacity for more than one enzyme activity (e.g. fatty acid synthase).
New enzymes with different substrate profiles or even catalytic mechanisms can be obtained in various ways. Microbial collections are often cross-referenced for the catalytic activities each microbial isolate has shown. Increasingly the genomes of all species are being sequenced and annotated for the genes and the proteins (including enzymes) which they encode and the data deposited in publicly accessible data bases that can be “mined” for biocatalysts. Examples of such databases are the National Centre for Biological Information (NCBI)19 and Uniprot.20 If the gene sequence is known it can be ordered on-line and the plasmid ready for cloning in a suitable host will be delivered to your doorstep in a couple of weeks.
If an ideal enzyme cannot be found, new enzymes can be obtained directly from the environment using metagenomics, whereby DNA is extracted from an environmental sample of e.g. soil or water and fragments are filed into a “library” in individual host bacteria (e.g. E. coli). The library can be screened against target molecules, often using colorimetric assays in large arrays or automated chromatography systems. This enables the discovery of novel sequences and, with a suitable chemical screen, novel activities.21–23 For example, the recent surge of interest in transamination motivated the discovery of novel, robust transaminases derived from the metagenome of the microbial population of the human mouth,24 and domestic drain.25 If a suitable enzyme has not been found it is now usual to modify the enzyme at the genetic level using advanced directed evolution techniques.26 These use in vitro mutation techniques, often through recombination of variants, to create permutations of the enzyme's amino acid sequence. Although the theoretical number of potential variants is astronomical, using enzyme structural data to plan mutations allows this to be reduced to a more tractable number.27,28
3. Enzyme structure, functions, mechanisms and scope in organic synthesis
Enzymes consist of linear polypeptide chains (proteins) of about 300 alpha L-amino acids (hence a mass of about 33 kDa).29 This varies considerably as 4-oxalocrotonate tautomerase is only 62 amino acids long30 while fatty acid synthase is 2461 amino acids long. They can also form associations of multiple proteins, and will therefore often be larger in practice. Proteins are typically comprised of the 20 proteinogenic amino acids that are encoded for on the DNA, but there is an additional pair, seleneocysteine and pyrrolysine, that are formed during translation and may be located in the enzyme's active site.Typically only a handful of these amino acids perform catalytic functions in any one protein, while the other 99% are often structural or regulatory. Some amino acids, such as histidine, are frequent contributors to the catalytic chemistry of enzymes, while others, such as glycine, do not contribute.31,32 Structural amino acids align the catalytic amino acids in order to enable the catalytic mechanism. Their physical proximity and spatial arrangement also limits the range of acceptable substrates, thereby creating catalytic specificity.
The catalytic active sites provide an exclusive reaction environment and the active site is therefore usually buried at the end of a channel lined with bulky and aromatic amino acids that controls access of substrates and solvents. Consequently the amino acid composition in the channel varies with the enzyme type (e.g. more hydrophilic for ligases but hydrophobic for oxidoreductases).33 Mechanical components, e.g. lipid activated lids to the active site of lipases, also help define substrate type.34 These structures endow enzymes with exquisite chemo-, regio- and most importantly stereo-selectivity.35 Engineering of these channels is a developing area of research.36
The non-catalytic amino acids also provide the structural backbone of the enzyme in that the hydrophobic regions fold inwards, away from the hydrophilic aqueous environment, to provide the core; while the hydrophilic regions form the surface and enhance enzyme solubility in the aqueous environment.
Enzyme catalysis can also include non-protein elements, referred to as cofactors, which greatly expand the catalytic repertoire of that enzyme class. There are 27 small molecule organic enzyme cofactors37 and a number of commonly used metals38 including Mg, V, Mn, Fe, Ni, Co, Cu, Mo, Zn, Se and W,39 many of which are tightly bound to the enzyme. Exploration of the repertoire of biocatalysts is far from reaching its limits and is not constrained by existing biochemistry. Further expansion can be realised through cofactor engineering,40–42 whereby existing metal or organic cofactors can be replaced by artificial analogs. Another possibility is to reprogram the amino acids that constitute the enzyme backbone with non-canonical amino acids (ncAAs).43
In short, largely as a result of in silico metagenome mining and directed evolution, biocatalysis has evolved into a cost-effective and broadly applicable technology that has been successfully integrated into mainstream organic synthesis.7,44 Indeed, the rapidly expanding biocatalysis toolbox45 has created a situation where it is eminently feasible to apply a biocatalytic retrosynthetic approach to identify enzymatic routes by ‘deconstructing’ target molecules.46–49
4. Advantages and limitations of enzymes: improving performance
Organic chemists who are unfamiliar with enzymes are often discouraged by real or perceived limitations with regard to their application in organic synthesis. Indeed, it is still widely believed that enzymatic processes are slow and occur at very low substrate concentrations in water, resulting in long reaction times and poor catalyst and volumetric productivities (kg product per kg enzyme and kg product per L per h), respectively. This is certainly not observed with enzymes catalysing their natural substrates under physiological conditions. It is, however, often true of wild-type enzymes transforming molecules which are structurally very different to their natural counterparts under the demanding conditions of industrial processes with elevated temperatures and in the presence of organic solvents.Nonetheless, advances in protein engineering and directed evolution techniques over the last two decades have dramatically changed this situation. Enzymes can be evolved, in a relatively short time and at relatively limited expense, to perform well with non-natural substrates under non-natural conditions. i.e. at substrate concentrations of >100 g L−1, volumetric productivities of >10 g L−1 and catalyst productivities of >100 kg product per kg enzyme.50
Another perceived drawback is that enzymes have very limited stability under the conditions encountered in organic synthesis. It is true that many, particularly wild-type enzymes, may be deactivated by unfolding (denaturing) at elevated temperatures, in the presence of organic solvents or under high shear caused by mixing or gas bubble surfaces. However, a number of extracellular enzyme classes, such as proteases, amylases, lipases, laccases and cellulases, are generally more robust and have been widely applied in detergents and industrial processes.
A good source of more stable enzymes is from organisms that live in extreme environments (extremophiles), such as thermophiles that thrive around geological vents.51 For example, enzymes were discovered that function at temperatures up to 120 °C, at extremes of pH between 0 and 12 and at pressures of 1000 bar, as well as in pure solvents and in slurries. Some have already been harnessed for industrial use, such as the glucoamylases and amylases which perform admirably in the hydrolysis of starch at temperatures above 100 °C. Some enzymes, such as lipases, are stable in highly hydrophobic solvents.52 Alternatively, enzymes can be evolved to increase their stability under various conditions of temperature, pH, in the presence of organic solvents, etc.
Yet another practical issue with enzymes is that it is challenging to recover them from aqueous media and they are, therefore, used on a single-use throw away basis. This is neither conducive to a waste-free circular economy nor a cost-effective process. One possible solution to the problem is to immobilise the enzyme by transforming it into a heterogeneous solid catalyst which can be readily recovered from an aqueous medium and reused.53–59 This substantially reduces the enzyme costs per kg product and the environmental footprint, thereby driving competitiveness and sustainability. Immobilisation also results in a decrease in the flexibility of the enzyme molecule, affording an increase in operational stability by suppressing its propensity to unfold at elevated temperatures or in the presence of organic solvents. In short, immobilisation can enable applications that would not be economically viable with the free enzyme and it can facilitate their use in cost-effective continuous flow processing.60,61
Immobilisation typically involves binding the enzyme to a prefabricated carrier (support) by physical adsorption, ionic interactions or the formation of covalent bonding via the amino acids in the hydrophilic surface shell (usually lysine). Alternatively, the enzyme can be encapsulated in a polymeric matrix formed in the presence of the enzyme. However, the use of a support inevitably leads to ‘dilution of activity’, owing to the introduction of a large portion of non-catalytic ballast, giving rise to lower catalyst productivities.
Immobilisation by cross linking of enzyme molecules to each other, in contrast, affords carrier-free immobilised enzymes with high productivities and avoids the extra costs of a carrier. For example, cross-linked enzyme aggregates (CLEAs) are formed by precipitation of the enzyme from aqueous buffer, without perturbation of their tertiary structure, and cross-linking with a bifunctional reagent, such as glutaraldehyde.62 Immobilised enzymes are more robust and can be recycled using filtration or centrifugation. Magnetic recovery is also possible if a magnetisable feature is introduced into the support or the cross-linking procedure.63 Whole cell biocatalysts have the advantage that enzymes are immobilised within the cell wall and the cells can be recovered by centrifugation. Enzymes can also be made more robust through protein engineering techniques,64 such as directed evolution, sometimes with dramatic improvements. For example, carbonic anhydrase was evolved to tolerate temperatures up to 107 °C at pH > 10, with a 4 × 106 improvement in stability over the wild-type enzyme.65
5. Stoichiometric and chemocatalytic vs. biocatalytic processes in organic synthesis
In order to be green and sustainable, organic syntheses should generate minimum waste and avoid the use of toxic and/or hazardous materials.66 Major causes of waste in organic syntheses are the use of stoichiometric inorganic or organic reagents and solvents in multi-step syntheses. Hence, in order to minimise waste it is necessary to use step-67 and atom-economic,68 catalytic methods, preferably under solvent-free conditions. Where use of a solvent cannot be avoided it should involve limited amounts of green, environmentally acceptable solvents.69Enzymatic processes are green and sustainable50 conforming to 10 of the 12 principles of green chemistry.70 The catalyst is produced from renewable biomass and is biocompatible, biodegradable and essentially non-hazardous and non-toxic. It provides an attractive alternative to the use of scarce precious metals as catalysts and avoids the costs of removing traces of noble metals from end products. Enzymatic reactions generally avoid the need for functional-group activation, protection, and deprotection steps, affording routes that are more step-economic and generate less waste than conventional organic syntheses. Moreover, they are conducted in standard reactors at close to ambient temperatures and pressures in water, rendering them both environmentally attractive and cost-effective. Furthermore, enzymatic processes are generally highly selective: distinguishing between regio- and stereo-isomers in addition to various functional groups (chemoselective). In particular enzymatic processes have become the method of choice in the synthesis of enantiomerically pure pharma intermediates based on the magnificent, close to absolute enantioselectivities that are obtainable with highly engineered enzymes.71–74
5.1 Hydrolytic processes
Traditionally, enzymatic processes were used industrially in the hydrolysis and formation of, inter alia, ester, amide and glycoside bonds, especially where very high selectivities were required (chemo-, regio- or stereo-selectivity). Typical examples include the processing of fats and oils and the synthesis of beta-lactam antibiotics.Enzymatic processes have the advantage of producing substantially less waste compared with the use of mineral acids or metal compounds as catalysts. A prime example, involving the production of a commodity chemical, is the Mitsubishi process for the hydrolysis of acrylonitrile to acrylamide catalysed by a nitrile hydratase (NHase) from the bacterium Rhodococcus rhodochrous J1 (Scheme 1a).
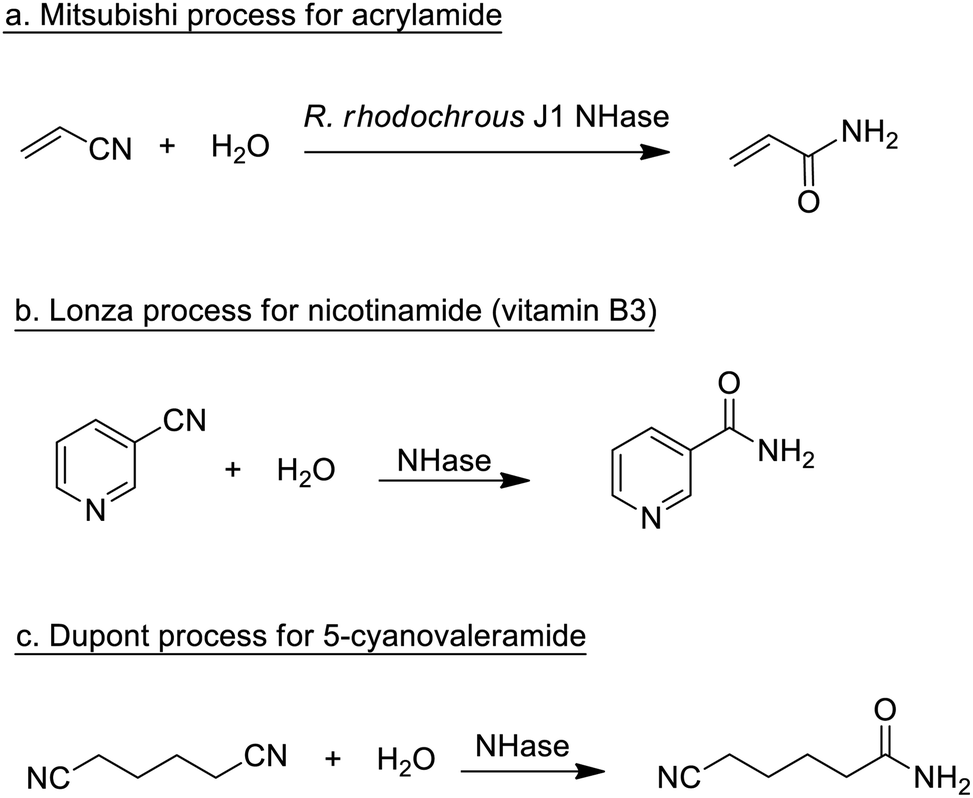 | ||
| Scheme 1 Nitrile hydratase catalysed hydrations of nitriles. | ||
The process, originally developed in the 1980s,75 currently accounts for the global production of several hundred thousand tons per annum of acrylamide. The latter is mainly applied in the production of polyacrylamide, which requires very pure monomer as starting material. Hence, an important advantage of the NHase process, compared to the copper-catalysed hydration that it replaced, is its close to perfect chemoselectivity (>99.99%). The NHase is used as whole (dead) cells because of the limited stability of the active enzyme which is a tetramer that readily dissociates outside the cell.
NHase-catalysed hydration of the corresponding nitrile is also used in the Lonza process for the industrial production of nicotinamide (vitamin B3)76 where the enzymatic process (Scheme 1b) is more selective, with respect to competing formation of the corresponding acid, than chemocatalytic alternatives. Similarly, 5-cyanovaleramide, a herbicide intermediate is produced by DuPont via regioselective NHase catalysed hydration of adiponitrile (Scheme 1c),77 generating fewer byproducts and less waste than processes using metal catalysts such as RANEY®Cu or MnO2.
Other well-known commercial applications of hydrolases include the use of the protease thermolysin for synthesis of the artificial sweetener aspartame,78 penicillin G amidase/acylase (E.C. 3.5.1.11) for synthesis of semi-synthetic β-lactam antibiotics,79 and amylase for the production of glucose via hydrolysis of starch.80
Pharmaceuticals tend to be lipophilic and therefore easy to handle in organic solvents81 and lipases are enzymes that are particularly tolerant of hydrophobic environments.82 Therefore, it is not surprising that pharmaceutical syntheses using biocatalysis have been dominated by lipase catalysed reactions.83–85 Lipases perform hydrolysis and esterification reactions, generating alcohols, carboxylic acids and esters, often with high stereo-selectivity when required. Moreover, they are catalytically versatile and can catalyse reactions such as aminolysis (the conversion of esters to amides, using amines as the nucleophile) and direct amidation of carboxylic acids.86
An interesting recent development in reverse hydrolysis reactions, such as the formation of carboxylic esters, is the use of the acyltransferase from Mycobacterium smegmatis (MsAcT) to catalyse the synthesis of flavour esters by transesterification in water.87 MsAcT is characterised by a hydrophobic tunnel leading to the active site, where access of water is disfavoured. Hence, transesterifications are favoured over hydrolysis, even in water.88,89
In the case of chiral substrates, enzymes can be used to synthesise the pure enantiomers. In hydrolytic processes, such as ester and amide hydrolyses this generally involves a kinetic resolution that affords a maximum yield of 50%. The maximum yield of 50% of the unwanted enantiomer (the distomer) constitutes waste that has to be disposed of or recycled back to the racemate via racemisation. In some cases the racemisation step can be performed in situ, affording a so-called dynamic kinetic resolution.90,91
The application of lipases for enzymatic kinetic resolution (EKR) of chiral intermediates in commercial syntheses includes pregabalin92 (Scheme 2a) and (R)-1-phenylethylamine93 (Scheme 2b) both of which involved the use of commercially available lipases. Hence, hydrolases such as lipases can now be considered mainstream organic synthetic tools, and their popularity continues unabated.94–96 Research is being directed increasingly to adapting the enzymes for industrial applications.53,97
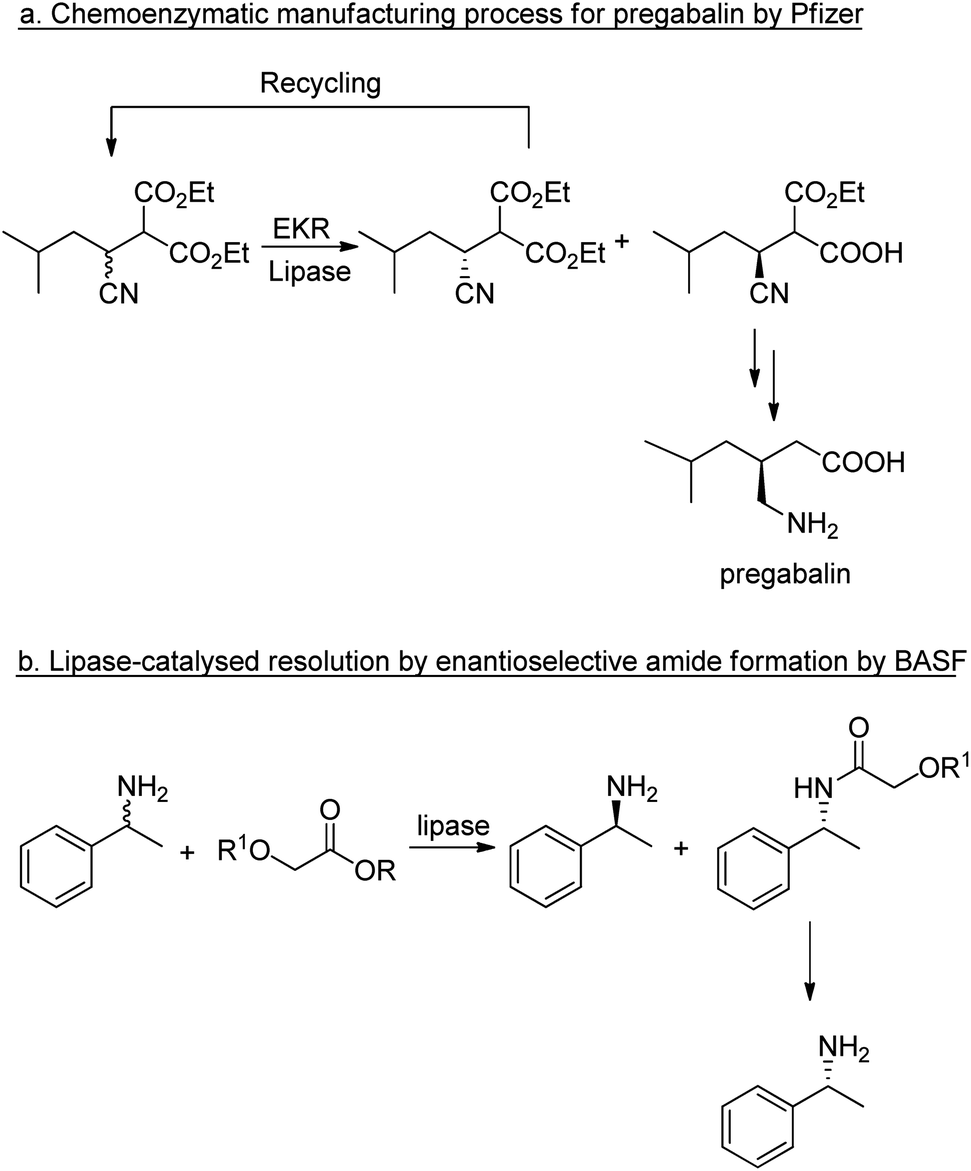 | ||
| Scheme 2 Enzymatic kinetic resolutions using lipases. | ||
Up until fairly recently it was generally thought that kinetic resolutions would be dominated by biocatalytic processes and asymmetric syntheses would be largely the domain of precious metal complexes with chiral ligands as catalysts in reactions such as asymmetric hydrogenations and hydroformylations. This situation has changed in the last two decades, resulting in a decrease in the use of lipases for kinetic resolutions98 and the broadening application of enzymatic redox processes in industrial organic synthesis as reflected in patent applications by industry.99–102 Indeed, in one study of 547 biocatalysis based patents filed between 2000 and 2015, only 10% involved hydrolases, while 68% were based on oxidoreductases.103 Hence, in the next section we will focus on recent developments in oxidoreductases.
5.2. Redox processes
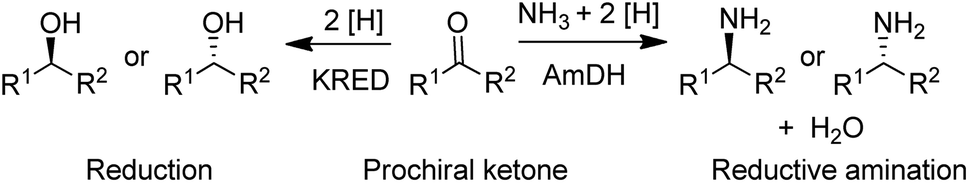 | ||
| Scheme 3 Reduction and reductive amination of ketones. | ||
In the 1990s chiral alcohols were typically produced either by stoichiometric reduction of ketones with chiral boron reagents or asymmetric hydrogenation catalysed by homogeneous chiral complexes of ruthenium and other precious metals.104 Ketoreductases (KREDs), also known as alcohol dehydrogenases (ADHs) catalyse oxidative dehydrogenation of (secondary) alcohols and the reverse reaction: the enantioselective reduction of ketones. The reducing equivalents are provided by stoichiometric amounts of nicotinamide cofactors, NADH or NADPH and for industrial viability these need to be recycled in situ. This is achieved by adding a large excess of an alcohol co-substrate, such as isopropanol, or a second enzyme in combination with an inexpensive co-substrate (Scheme 4). The most used combinations are formate/formate dehydrogenase (FDH) and glucose/glucose dehydrogenase (GDH).
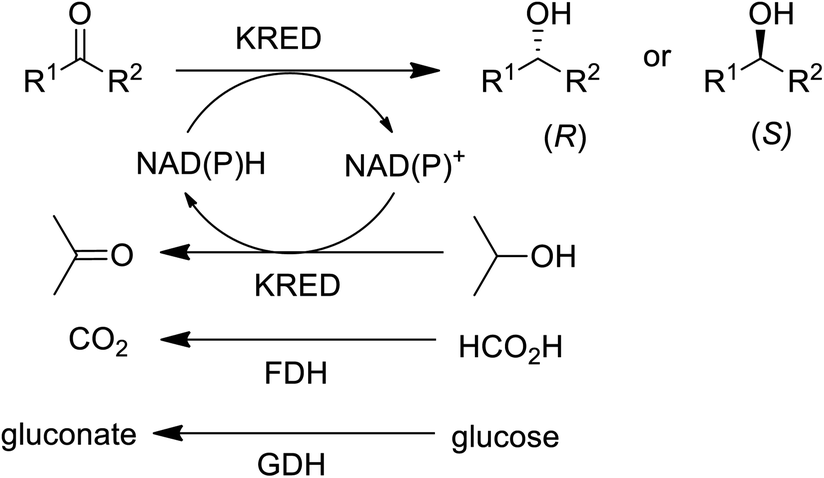 | ||
| Scheme 4 KRED catalysed reduction of prochiral ketones. | ||
Thanks to key advances in protein engineering by directed evolution, together with gene sequencing and DNA synthesis, KREDs have been successfully applied to the synthesis of a variety of enantiomerically pure secondary alcohols as key intermediates for a variety of chiral drugs,105,106 including atorvastatin,107 montelukast,108 ezetimibe, sulopenem and licarbazepine (Scheme 5).
 | ||
| Scheme 5 Examples of enantiomerically pure secondary alcohols produced with engineered KREDs. | ||
A widely used method for producing amines involves chemocatalytic reductive amination of ketones with a mixture of hydrogen and ammonia. This suggests that what was missing from the enzyme toolbox was an asymmetric reductive amination of prochiral ketones with ammonia catalysed by NAD(P)H-dependent amine dehydrogenases (AmDHs). Such enzymes are rare in vivo where amine catabolism involves aerobic oxidation catalysed by amine oxidases. Amino acid dehydrogenases, in contrast, are well-known and form an obvious starting point for the development of AmDHs by directed evolution.
Bommarius and co-workers109 used leucine dehydrogenase (LeuDH) as a protein scaffold for developing an enzymatic reductive amination of structurally related methyl isobutyl ketone. Similarly, phenylalanine dehydrogenase (PheDH) was used as a starting point for engineering a highly active and highly selective (>99.8% ee) AmDH for both aliphatic and benzylic ketones110 in combination with glucose/GDH for cofactor regeneration (Scheme 6). The same group subsequently used domain shuffling of the two parent AmDHs to produce a novel chimeric AmDH, with altered substrate scope. Further practical improvements were realised using a combination of ammonium formate and formate dehydrogenase (FDH) as both a source of ammonia and cofactor regeneration in a biphasic organic solvent/water system.111 Mutti and co-workers112 similarly used three different AmDHs, in combination with formate and FDH, for the reductive amination of a wide variety of prochiral ketones, including aromatic, benzylic, aliphatic and ketones with bulky substituents. They further suggested that practical utility could be improved by co-immobilisation of the two enzymes or co-expression in a single microorganism. Likewise, Li and co-workers113 developed an AmDH for the reductive amination of phenylacetone and 4-phenyl-2-butanone by protein engineering of PheDH (Scheme 6).
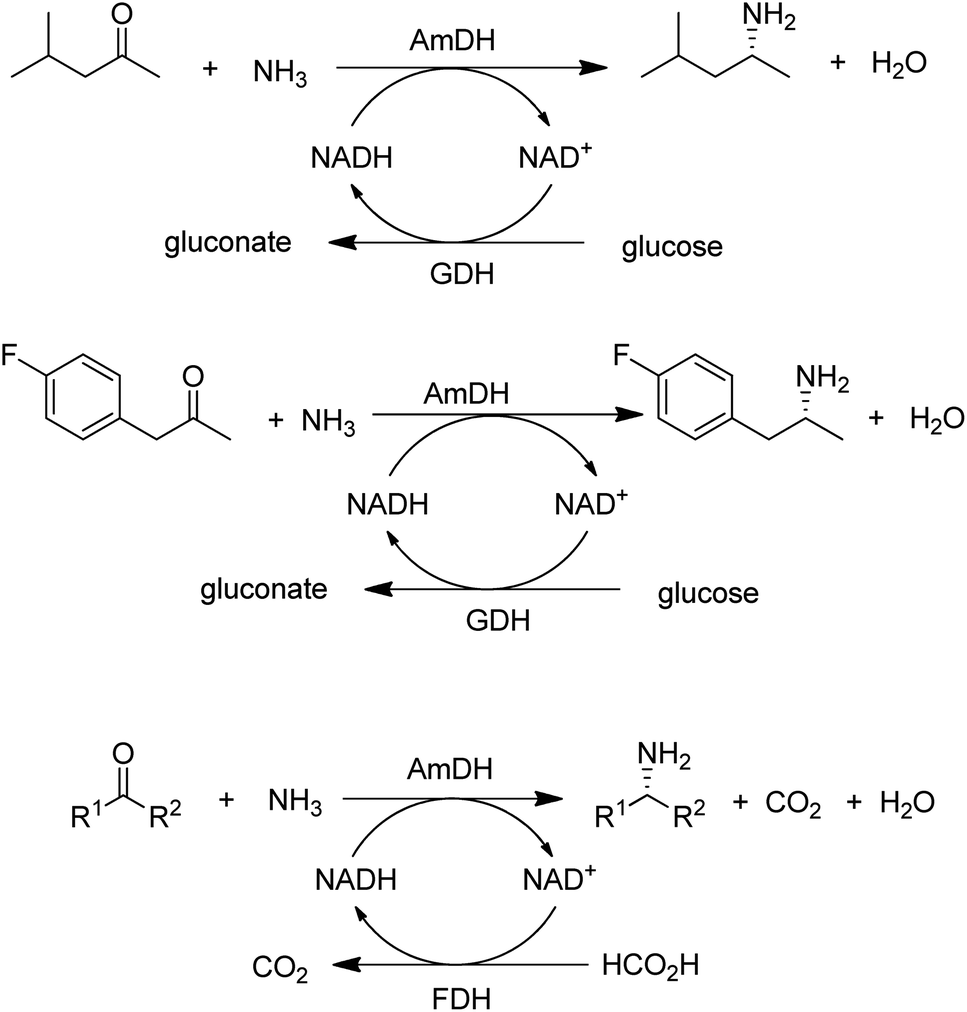 | ||
| Scheme 6 Enzymatic primary amine synthesis with amine dehydrogenases. | ||
Another fairly recent development is the emergence of imine reductases (IREDs)114–119 for the enantioselective reduction of cyclic imines derived from secondary amines and the asymmetric reductive amination of ketones with primary amines (Scheme 7).120–123
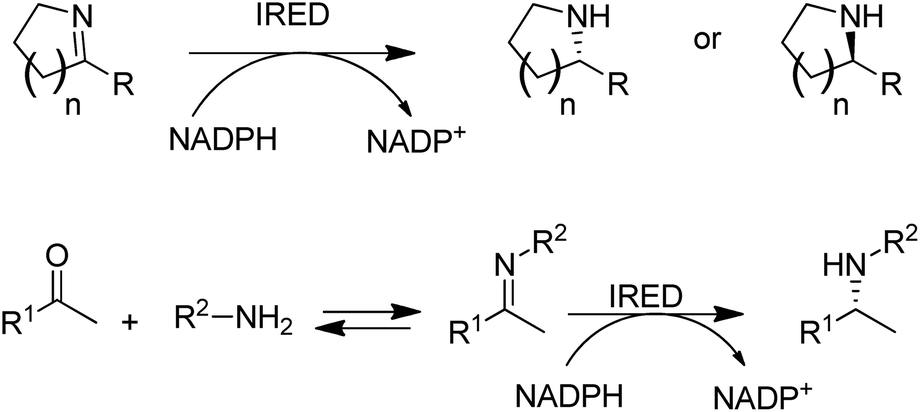 | ||
| Scheme 7 Imine reductases and amine dehydrogenase for reductive aminations with amines. | ||
The concept of chiral amine synthesis via enzymatic reductive amination was taken to a new level of sophistication by applying the concept of hydrogen borrowing (itself having been borrowed from the chemocatalytic literature).124 For example, the combination of an ADH and an AmDH catalysed a redox-neutral conversion of a racemic alcohol to a single enantiomer of the corresponding amine (Scheme 8).125 Ironically, the success of this method depends on the ADH being aselective126 as it needs to catalyse the oxidation of both enantiomers of the alcohol, which is not a simple task as most ADHs are highly enantioselective. The overall efficiency of the process could be improved by co-immobilisation of the ADH and AmDH.127 Overall the reaction constitutes a nucleophilic substitution of a hydroxyl group in a secondary alcohol, in this case by an amino group, with an equivalent of water as the sole co-product. Such a reaction is one of the nine reactions in a recently published list of 10 key research areas for a greening of the pharmaceutical industry.128
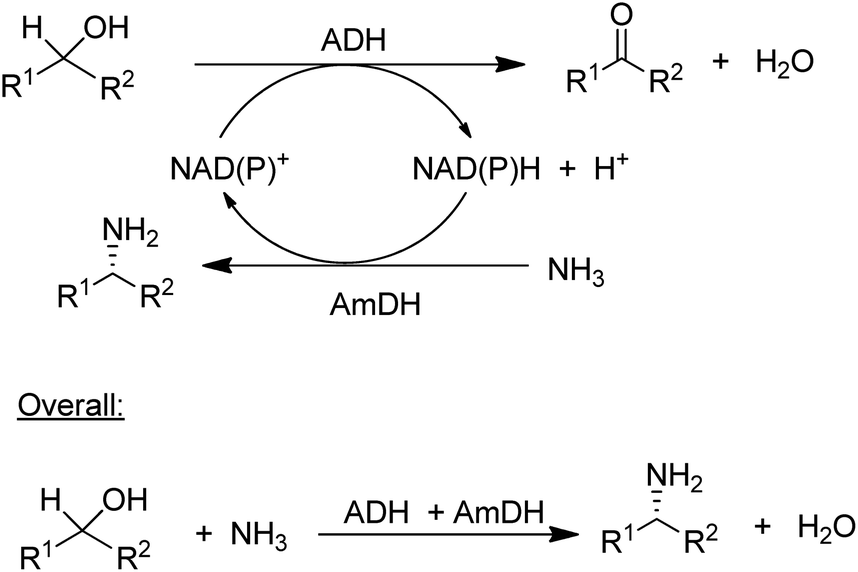 | ||
| Scheme 8 Enantioselective conversion of prochiral alcohols to chiral amines via hydrogen borrowing. | ||
The most commonly used enzymes for the oxidation of alcohols are alcohol oxidases (AOx) that use dioxygen as the oxidant and form H2O2 as the co-product. In practice, catalase is added in order to rapidly remove the H2O2 to avoid degradation of the enzyme. They are involved in carbohydrate oxidations where they tend to be very substrate specific. Examples include galactose oxidase (GOase) and glucose oxidase (GOX) which are copper and flavin-dependent, respectively.
In the last decade many new flavin- and Cu-dependent AOxs, with altered substrate specificities and stabilities have been developed with the aid of genome mining and directed evolution. Examples include GOase variants able to catalyse the oxidation of secondary alcohols,136 amino alcohols137 and 5-hydroxymethyl-furfural138 and a choline oxidase which was evolved to become a broad spectrum primary alcohol oxidase (Scheme 9).
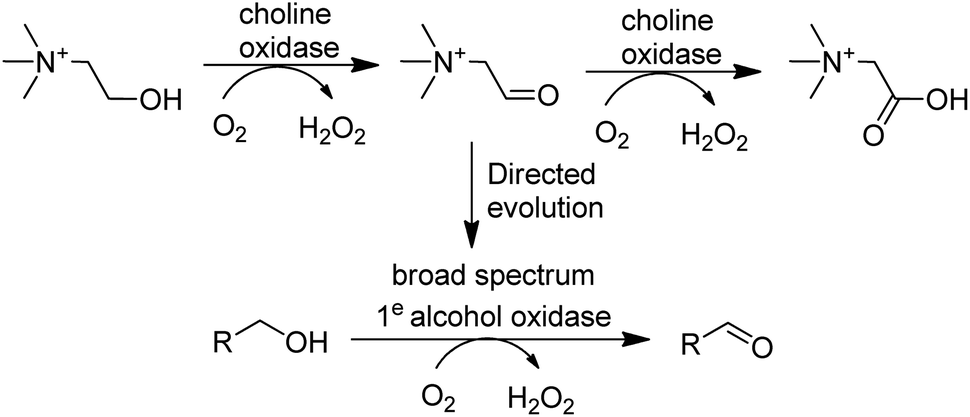 | ||
| Scheme 9 An evolved choline oxidase as a broad spectrum primary alcohol oxidase. | ||
A variant of a flavin-dependent oxidase catalysed the oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid (FDCA), involving two alcohol oxidation steps and one aldehyde oxidation (Scheme 10).139 Since many aldehydes are present in water mainly as the hydrate (gem-diol), it is not so surprising that an alcohol oxidase can also catalyse the aerobic oxidation of aldehydes.
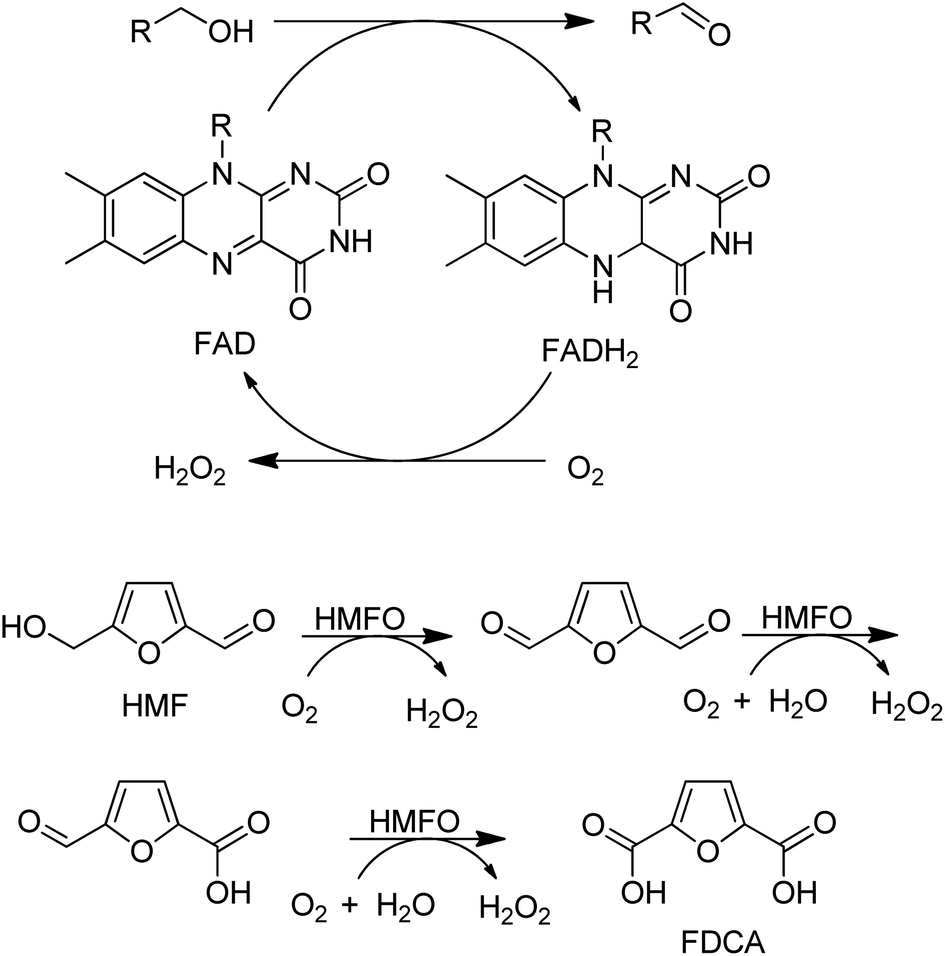 | ||
| Scheme 10 HMFA oxidation with flavin dependent AOx. | ||
The above-mentioned variant of GOase also catalysed the ammoxidation of primary alcohols140 to nitriles (Scheme 11).
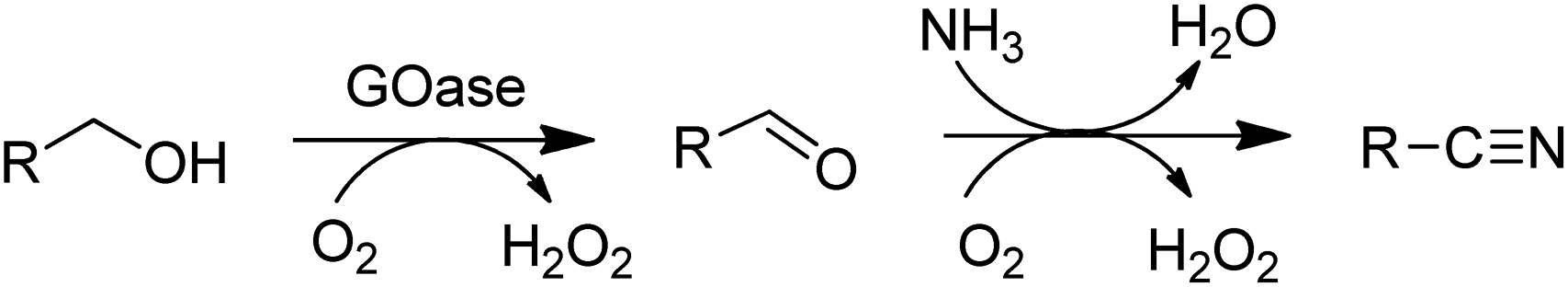 | ||
| Scheme 11 AOx catalysed ammoxidation of an alcohol. | ||
Alternatively, a combination of the Cu-dependent oxidase, laccase, with stable nitroxyl radicals, such as TEMPO, catalyses the aerobic oxidation of primary alcohols to the corresponding aldehydes.141 It was also effective in the oxidation of the secondary alcohol isosorbide to the corresponding diketone (Scheme 12).142
 | ||
| Scheme 12 Laccase/TEMPO catalysed oxidations of alcohols. | ||
Alcohol dehydrogenases (ADHs) catalyse the oxidation of alcohols and require regeneration of the nicotinamide cofactor using an excess of a co-substrate.143–145 Alternatively, NAD(P)H oxidase (NOx) can be employed to catalyse reoxidation of the cofactor by dioxygen.146,147
NAD-dependent aldehyde dehydrogenase (ALDH) catalyses the aerobic oxidation of aldehydes to the corresponding carboxylic acids and three recombinant ALDHs have been used, in combination with NOx for cofactor recycle, for the highly selective aerobic oxidation of a wide variety of aldehydes.148 Combination of an ALDH with an ene-reductase (ERED) afforded a hydrogen-borrowing strategy for the conversion of α,β-unsaturated aldehydes to the corresponding α-substituted carboxylic acids in high chemo- and stereoselectivities (Scheme 13).149
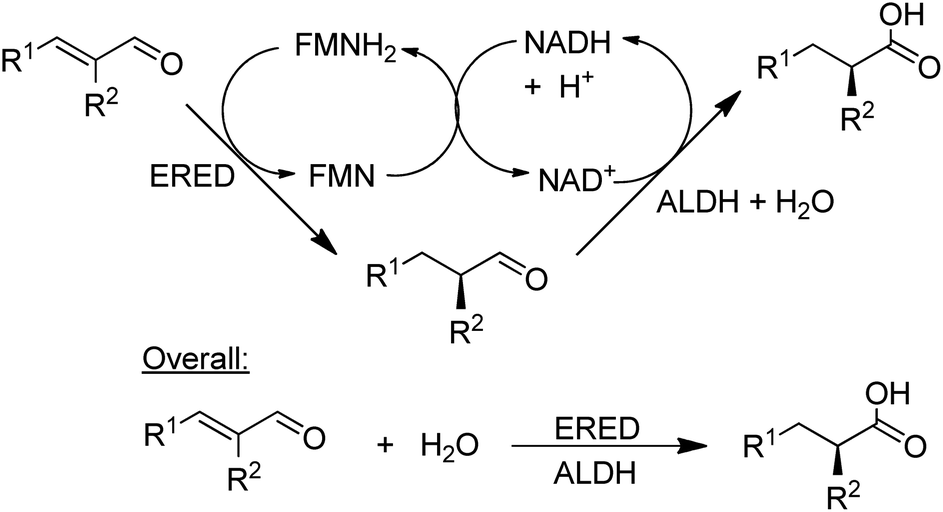 | ||
| Scheme 13 Combination of an ALDH with an ERED. | ||
Baeyer–Villiger Monooxygenases (BVMOs) are synthetically interesting flavin-dependent enzymes that catalyse the aerobic oxidation of ketones and cyclic ketones to esters,150,151 and lactones, respectively. They can be combined with ADHs, for example as a fused ADH–BVMO,152,153 to afford hydrogen-borrowing cascades for conversion of an alcohol to an ester or a smart synthesis of caprolactone from 1,6-hexanediol.154 Similarly, an elegant redox neutral process155 for highly enantioselective sulfoxidations with dioxygen was designed by combining an evolved cyclohexanone monooxygenase (CHMO) with a KRED (Scheme 14). It was used in the production of, for example, the single enantiomer drug esomeprazole (S-omeprazole).156
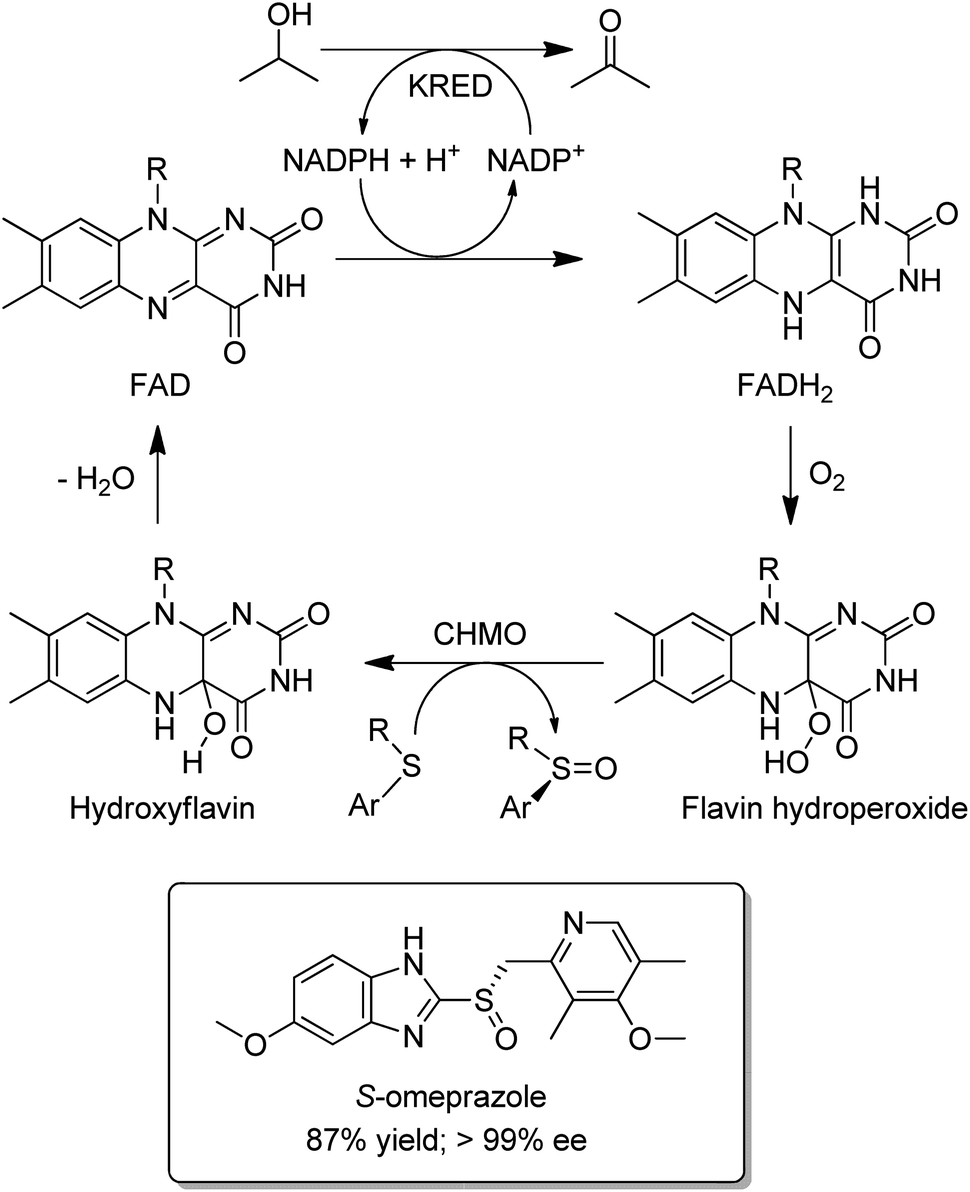 | ||
| Scheme 14 Enantioselective sulfoxidation using an ADH/BVMO cascade. | ||
Flavin-dependent monoamine oxidases (MAOs) catalyse the aerobic oxidation of amines to the corresponding imines.157. Turner and coworkers158 used directed evolution to develop an MAO for the deracemisation of structurally diverse primary, secondary, and tertiary amines. The process is a kinetic resolution but can afford high yields of one enantiomer by combining it with repeated aselective chemical reduction of the imine. MAO variants were also used for the enantioselective desymmetrisation of prochiral amines, e.g. in the synthesis of the hepatitis C drug, Boceprevir159 (Scheme 15).
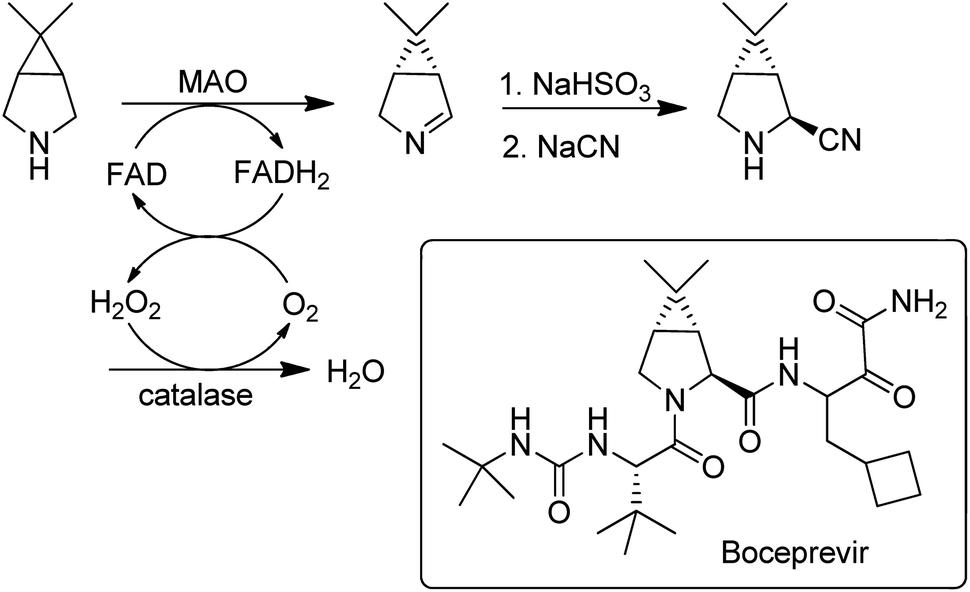 | ||
| Scheme 15 Desymmetrisation of a prochiral amine with an MAO. | ||
5.3. C–O, C–N and C–C bond forming reactions
The reactions discussed in the preceding two sections involved functional group transformations via hydrolytic or redox processes. In this section we shall discuss the formation of C–O and C–N containing functional groups by enzymatic conversion of hydrocarbons and the formation of C–C bonds.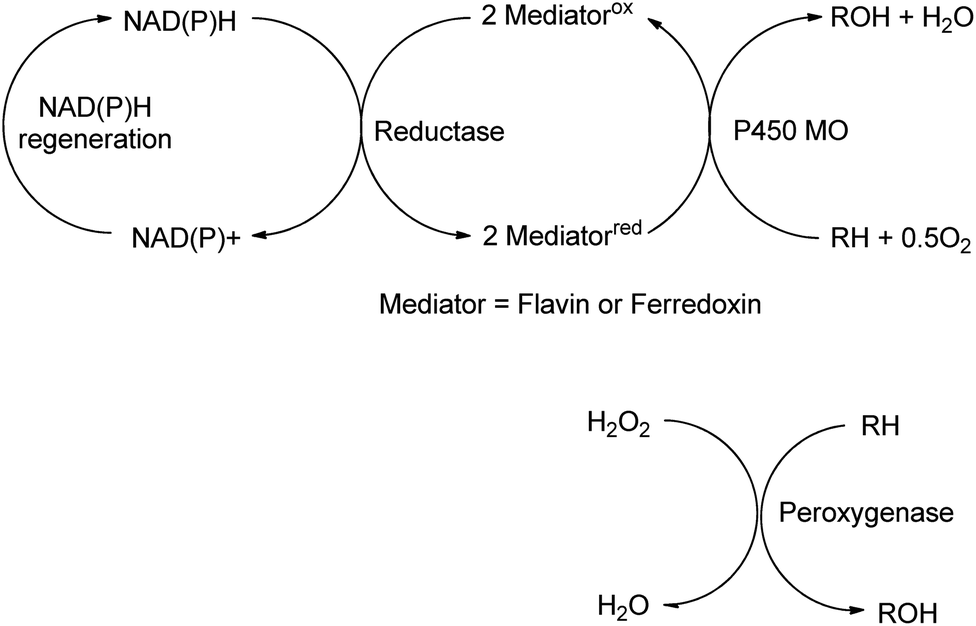 | ||
| Scheme 16 Monooxygenase and peroxygenase pathways. | ||
Mono-and dioxygenases catalyse the formation of new C–O bonds by the oxidation of hydrocarbons by dioxygen. Cyt-P450-dependent monooxygenases, for example, are multicomponent biocatalysts in which reducing equivalents are supplied by NAD(P)H indirectly through a flavin or ferredoxin mediator (see Scheme 16). They catalyse a broad range of regio- and stereoselective hydroxylations of alkanes, alkenes and aromatic hydrocarbons employing dioxygen as the terminal oxidant, accounting for about 95% of all enzyme catalysed oxidations.162
Catalytically self-sufficient P450s containing the P450 and reductase domains in the same polypeptide chain, such as P450BM3 from Bacillus megaterium,163 are of particular interest in this context and a panel of new thermostable, self-sufficient P450s with broad substrate scope from thermophilic organisms has been reported.164 However, the best result – 1.6 g L−1 product concentration after 10 h in the hydroxylation of diclofenac – is still 1–2 orders of magnitude lower than what is needed for industrial viability but can be seen as a starting point for further engineering by directed evolution.165
In addition to catalysing oxygen insertion into C–H bonds of alkanes and alkylaromatics and allylic C–H bonds in olefins, Cyt P450s166 and peroxygenases167 catalyse asymmetric epoxidations and an engineered non-haem iron dependent Rieske dioxygenase (RO) catalysed cis-dihydroxylations of olefins (Scheme 17).168 The introduction of a single point mutation in the dioxygenase improved conversions (>99%) and selectivities (>95%). The method constitutes a greener, more sustainable alternative to the use of toxic osmium catalysts in the Sharpless cis-dihydroxylation.169 Despite the synthetic potential of these transformations their reduction to commercial practice remains a significant challenge170 as they are generally characterised by low product concentrations and space time yields. Although replacing dioxygen and oxygenases by hydrogen peroxide with peroxygenases171 circumvents the need for cofactors it is also fraught with difficulties, such as low stabilities of the enzymes towards the hydrogen peroxide oxidant. The methods seem to be en route but have not yet reached their destination.
 | ||
| Scheme 17 Olefin cis-dihydroxylation catalysed by a dioxygenase. | ||
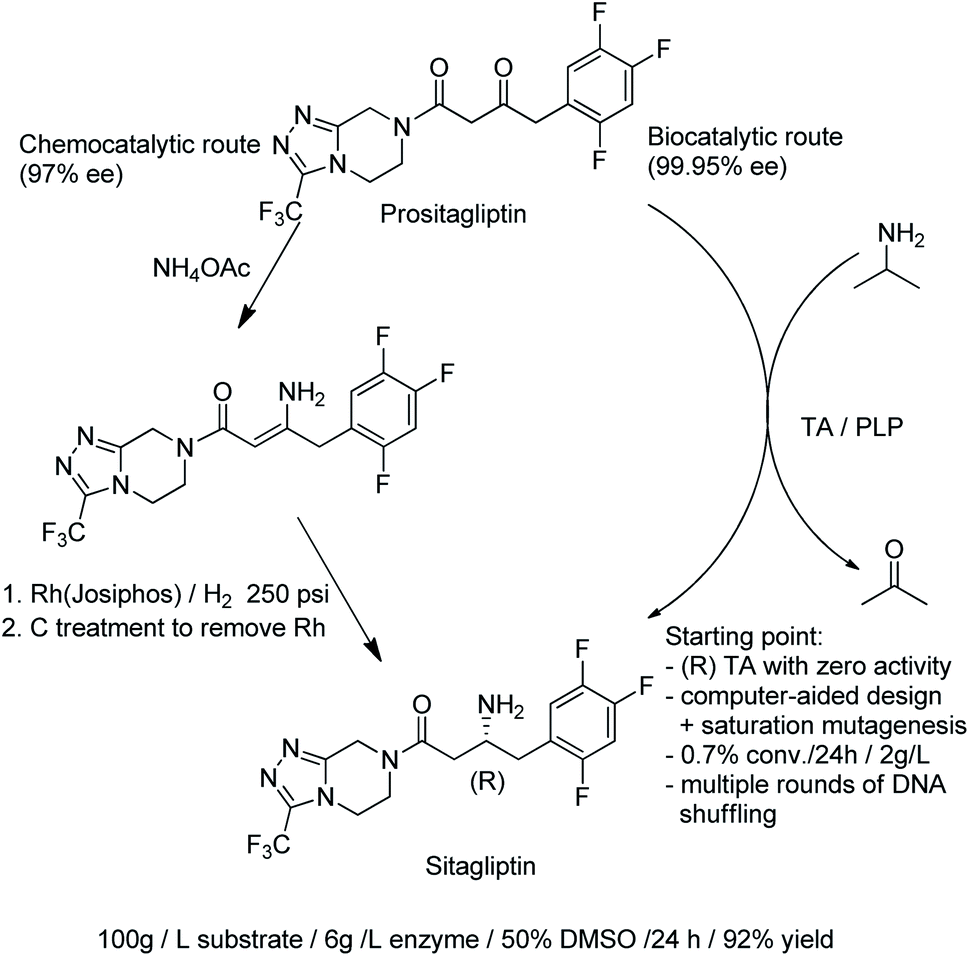 | ||
| Scheme 18 Synthesis of sitagliptin. | ||
Protein engineering was also used to develop highly active, (S)-selective TAms for the asymmetric synthesis of bulky chiral amines in up to >99.9% ee.177 A disadvantage of TAms is that several equivalents of the sacrificial amine are generally required to drive the unfavourable equilibrium. Interestingly, robust TAms, requiring less equivalents of isopropylamine, were recently developed from a domestic drain metagenome (see Section 2).
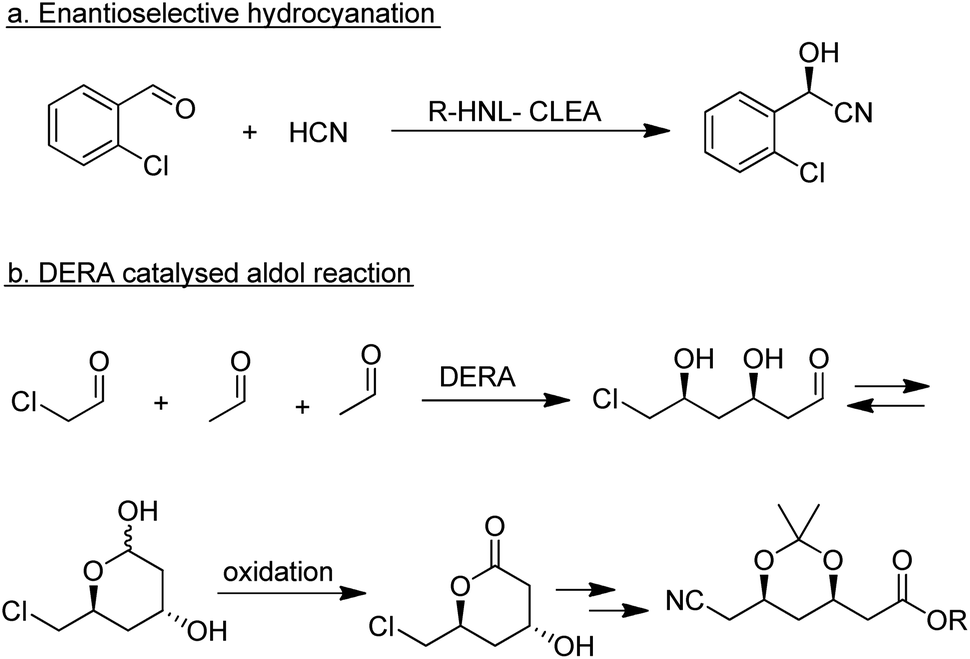 | ||
| Scheme 19 Enzymatic C–C bond forming reactions. | ||
Among the many types of aldolases, 2-deoxy-D-ribose-5-phosphate aldolase (DERA) is of particular interest. In vivo it catalyses the synthesis of 2-deoxy-D-ribose-5-phosphate from acetaldehyde and glyceraldehyde-3-phosphate but in vitro it also catalyses the aldol reaction of acetaldehyde with numerous other simple aldehydes.184 Its synthetic potential is limited by its low tolerance to industrially relevant aldehyde concentrations but this hurdle has been overcome by protein engineering. DSM, for example, developed a process for the synthesis of an advanced atorvastatin intermediate by aldol reaction of two equivalents of acetaldehyde with chloroacetaldehyde (Scheme 19b) that operates at 100 g L−1 substrate concentration.185
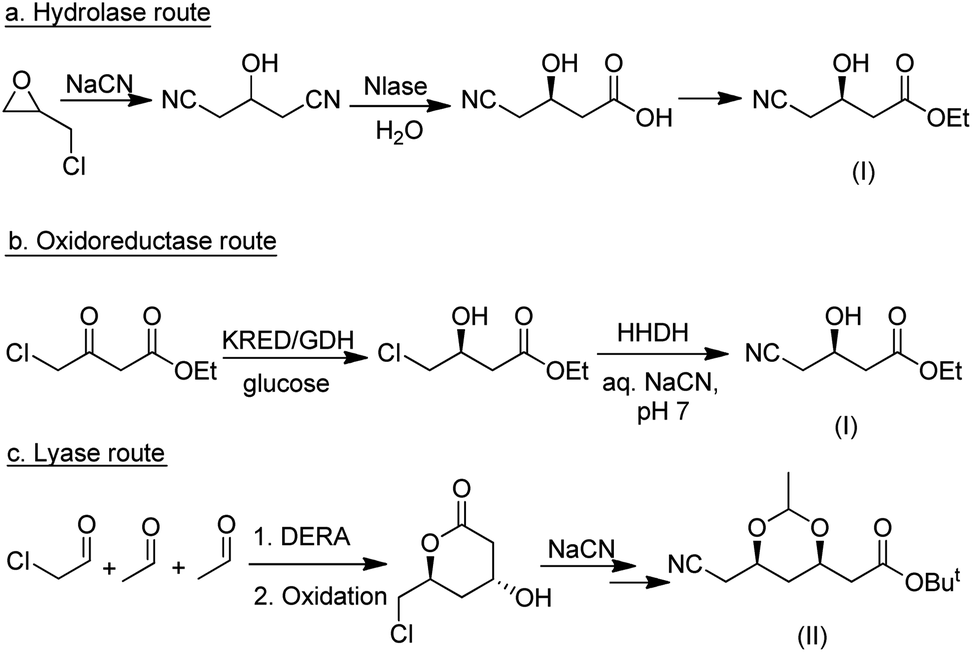 | ||
| Scheme 20 Competing biocatalytic routes to atorvastatin intermediates. | ||
6. Integration: biocatalytic and chemoenzymatic cascade processes
The 8th principle of Green Chemistry is reduction in the number of steps through reduced derivatisation. The high specificity of enzymes obviates the need for protection and deprotection steps.62 Examples of the successful integration of biocatalysis steps into a traditional synthetic process are many, including diltiazem,186 sitagliptin,173 pregabalin,89 atorvastatin104,187 and other statins,188 to name but a few.In traditional multistep organic syntheses a simple step-by-step approach is used whereby intermediates are isolated and often purified before proceeding to the next step. The result is: low volumetric productivities, protracted recycle loops and considerable waste generation. Hence, integration of several catalytic steps into step economic, one-pot procedures, with no isolation of intermediates, will automatically lead to less waste formation. This telescoping of multistep syntheses has various practical advantages: fewer unit operations, smaller reactor volume, shorter cycle times, higher productivities and less waste.
This is nothing special in nature where cellular metabolism requires a vast array of enzymes working together in multi-step reactions in a single “pot” to make a multitude of products with high specificity and negligible accumulation of intermediates.
With a rapidly expanding toolbox of biocatalytic conversions, which proceed under approximately the same conditions – in water at roughly ambient temperature and pressure – the construction of enzymatic cascade reactions has become increasingly popular.44,189–194 It provides tremendous advantages to chemists as the effort and yield losses associated with work-up after each step are eliminated, increasing the overall yield. Moreover, it can be used to drive equilibria towards product formation. For example, in a 3 step reaction involving enzymatic hydrocyanation of a ketone followed by two sequential enzymatic hydrolytic steps the equilibrium of the reversible hydrocyanation step was shifted towards product by its removal in the hydrolysis step.195
The outstanding benefits of biocatalytic cascades are illustrated in a synthesis of islatravir (an HIV reverse transcriptase translocation inhibitor), recently developed in a collaboration between Codexis and Merck (Scheme 21). The three-step biocatalytic cascade involves nine enzymes in a single aqueous solution without the need for isolation of intermediates. Four of the enzymes were ancillary, including sucrose phosphorylase, which catalysed a reaction incorporated into the cascade to shift the entire equilibrium forward. This route halved the total number of reaction steps required, greatly improved atom economy, and permitted an excellent overall islatravir yield of 51% from simple building blocks.196
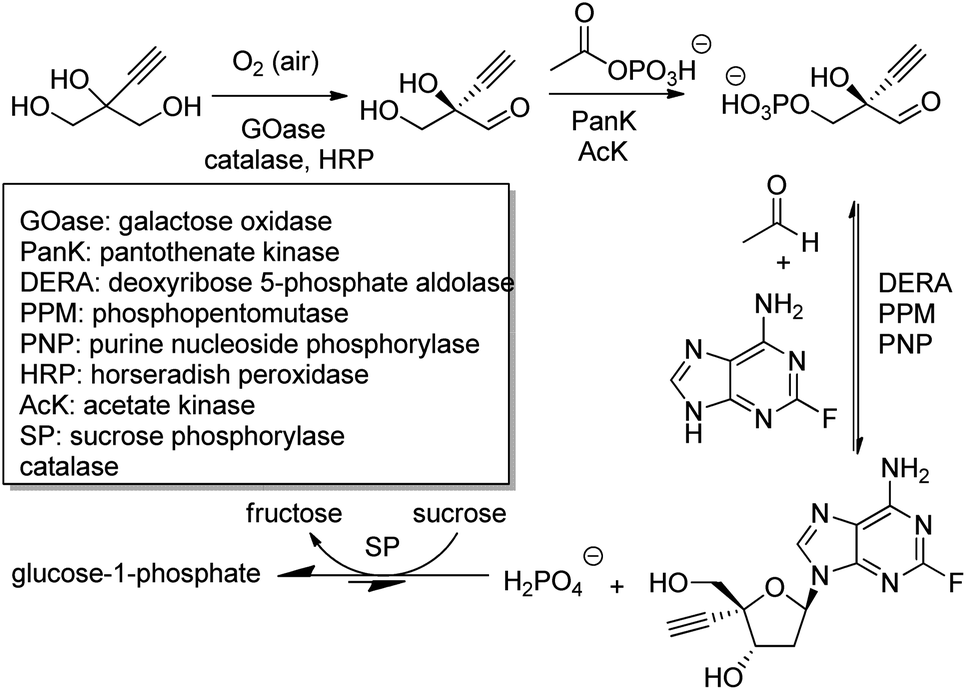 | ||
| Scheme 21 Biocatalytic cascade synthetic route to islatravir. | ||
The majority of biocatalytic cascade processes involve redox reactions, where reactions are coupled to integrate the use and regeneration of nicotinamide cofactors (NADH and NADPH) thereby making the overall process redox neutral. We have already discussed striking examples of such hydrogen borrowing in the preceding section.
Catalytic cascade processes can also involve the integration of biocatalytic and chemocatalytic steps to afford chemoenzymatic cascade processes.197–199 Chemoenzymatic approaches have also proved useful in the synthesis of natural products.200–203 A recent important publication by Lipshutz and coworkers has shown that the formation of micelles in the reaction medium can facilitate the combination of chemo- and bio-catalysis in a single reactor as the substrates, products and catalysts partition into the different compartments.204 For an extensive review of the recent literature on biocatalytic cascade reactions see Kroutil and coworkers.205
7. New to nature reactions
Continued expansion of the biocatalysis toolbox for synthetic chemists is being made possible by the discovery of new enzymes with new functions,7,206 as well as by repurposing of existing enzymes to catalyse reactions not observed in nature. These so-called “new-to-nature” reactions can arise naturally207,208 but have been driven, in particular, by directed evolution.26,209 This approach relies on enzyme catalytic promiscuity, the ability of an enzyme to catalyse more than one distinct type of chemical reaction within the same active site.210 This alternative chemistry that is available to the enzyme is often present naturally at a very low level, but can be selected for and enhanced in the process of directed evolution.211 An area also receiving considerable attention is the use of artificial enzymes for conducting new-to-nature reactions.212,2137.1 Haem metalloenzyme-mediated reactions
Cytochrome P450 monooxygenases catalyse a variety of oxidations of hydrocarbons with dioxygen whereby an oxygen atom is inserted into a C–H bond or a C–C double bond of an olefin. The key step in the latter reactions involves oxygen transfer from a high-valent oxoiron porphyrin complex to the double bond of the olefin, affording an epoxide. By analogy with this oxene transfer, it was shown that P450BM3, under anaerobic conditions, could catalyse carbene214 and nitrene transfer reactions, to afford cyclopropanes and aziridines, respectively. For example, cyclopropane derivatives were formed by reaction of styrene with ethyl diazoacetate.215 Depending on the mutation(s) introduced, either the cis or trans product could be obtained in high enantioselectivity (Scheme 22). In a similar manner, modified myoglobin, another haem protein, was also found to be an effective cyclopropanation catalyst.216 The synthesis of a number of drug precursors containing the cyclopropane moiety was demonstrated at preparative scale using these engineered haem proteins.217 In a variation on this theme, Hartwig and co-workers used an artificial iridium porphyrin-containing P450 enzyme optimised through directed evolution for alkene cyclopropanation.218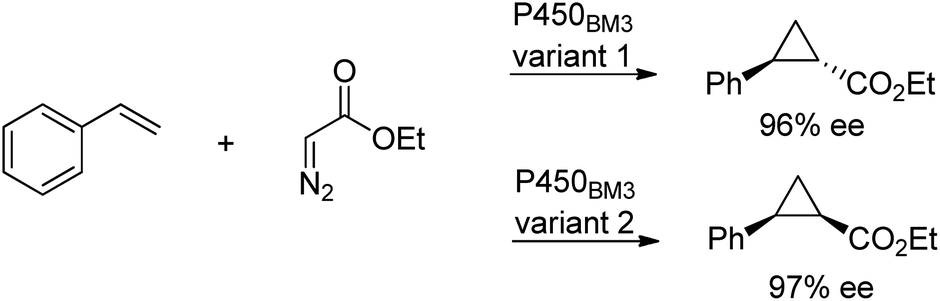 | ||
| Scheme 22 Stereoselective cyclopropanation reaction of P450BM3. | ||
Similarly, enantioselective aziridination of styrene derivatives by reaction with tosyl azide was achieved using a P411 variant of cytochrome P450BM3.219 Arnold and co-workers also exploited the nitrene-transfer capacity of a P450 enzyme variant to catalyse intramolecular benzylic C–H amination,220 a reaction first reported with wild type Cyt P450 by Breslow and co-workers in 1985.221 Arnold et al. followed up this work with a demonstration of regioselective C–H amination either at the benzylic position or the homo-benzylic position (Scheme 23a).222
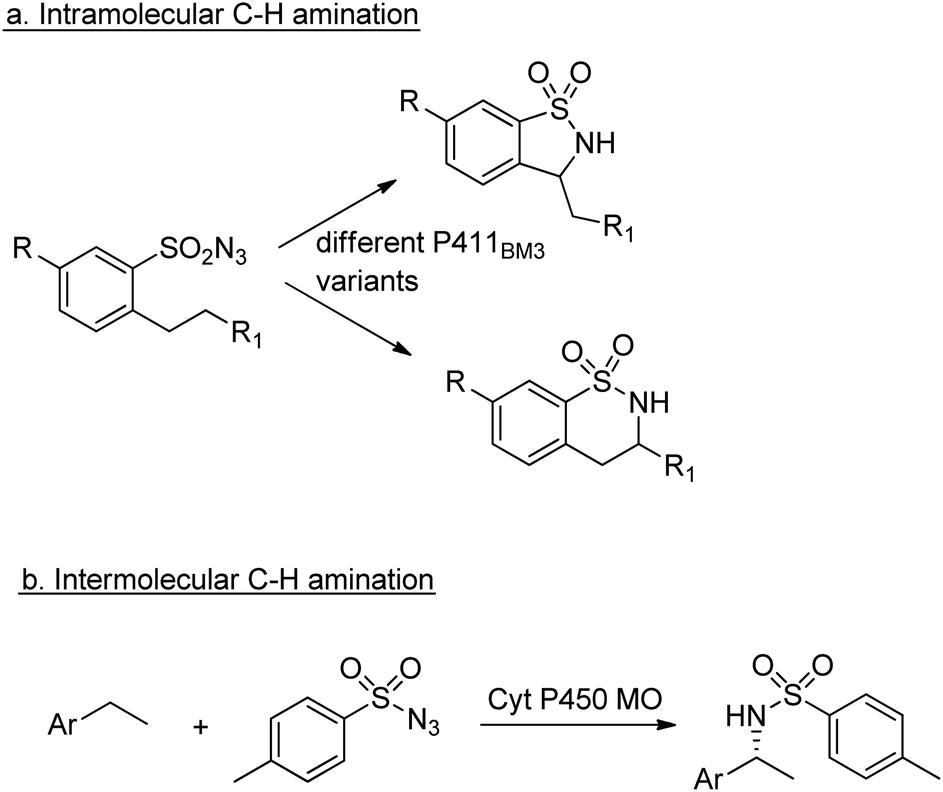 | ||
| Scheme 23 C–H aminations catalysed by Cyt P450s. | ||
Similarly, engineered Cyt P450s catalysed intermolecular benzylic C–H amination via the insertion of nitrenoid species, derived from tosyl azide, into unactivated C–H bonds223 to afford amines (Scheme 23b). This reaction has been extended to allow introduction of the NHTs group to position C-2 of N-methylindole.224
The utility of modified myoglobins was also expanded to include catalysis of olefination reactions of aromatic aldehydes using diazo acetate reagents (Scheme 24).225
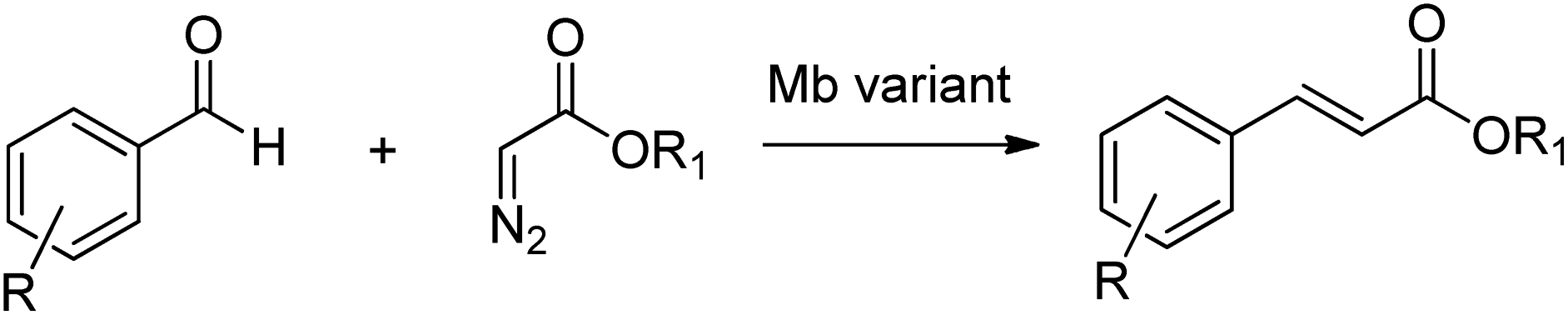 | ||
| Scheme 24 Olefination of aromatic aldehydes. | ||
Enzymatic carbene transfer reactions mediated by CytP450 mutants were also recently demonstrated with heterocyclic substrates such as indoles and pyrroles226 and in the enantioselective C–H fluoroalkylation at the α-C–H bond of amines (Scheme 25).227
 | ||
| Scheme 25 C–H fluoroalkylation of amines. | ||
Notwithstanding the elegance of such new-to-nature carbene and nitrene insertion reactions, it is worth noting that the reactions shown here are not possible in nature because of the absence of diazo esters and tosyl azide. However, it is interesting to note that Ohnishi et al. have recently proposed an in vivo nitrene transfer mechanism, involving the formation of an iron nitrenoid intermediate by cytochrome P450 BezE catalysed elimination of acetic acid from an O-acetyl hydroxylamine functionality, to explain an intramolecular amination reaction.228 The authors suggested the name P450 nitrene transferase for BezE.
Other examples of non-natural reactions catalysed by engineered haem proteins can be found in recent reviews.26,229
7.2 Reactions catalysed by promiscuous hydrolases
A number of hydrolases, such as lipases, have been demonstrated to catalyse a wide range of reactions that do not occur in vivo.230,231The lipase-catalysed addition of thiols to vinyl esters was achieved by Lin et al. in organic solvent, and depending on the choice of solvent either Markovnikov or anti-Markovnikov addition was observed (Scheme 26).235
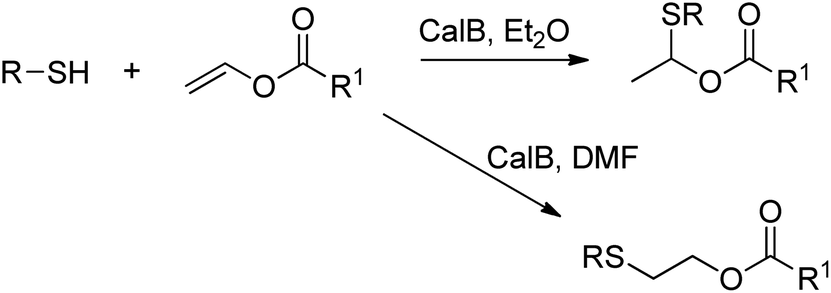 | ||
| Scheme 26 Addition of thiols to vinyl esters. | ||
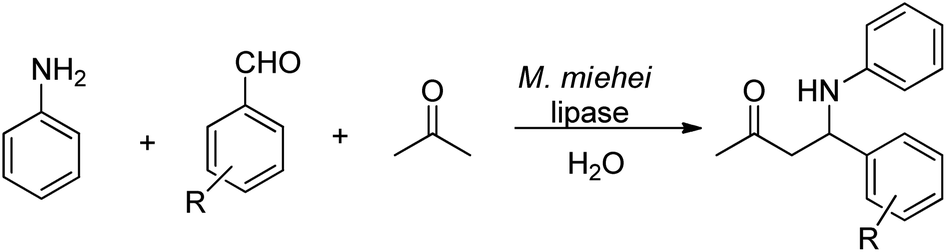 | ||
| Scheme 27 Lipase-catalysed Mannich reaction. | ||
Further examples of non-natural conversions catalysed by promiscuous hydrolases can be found in a number of reviews.240–242
7.3 Other reactions
An artificial “metathase” was prepared by Ward and co-workers by the combination of streptavidin with a biotinylated Grubbs–Hoveyda metathesis catalyst as the cofactor, and ring-closing metathesis was demonstrated in cellulo.243 Siegel et al. designed artificial enzymes able to catalyse an intermolecular Diels–Alder reaction stereoselectively.244 Photoexcitation has been demonstrated by Hyster et al. as a successful approach to accessing new non-natural oxidoreductase activity.245,246 It is evident that the range of biocatalytic reactions useful to organic chemists is increasing by leaps and bounds.8. Conclusions and future prospects
The phenomenal advances in new enzyme discovery, through the fruitful combination of metagenomics and bioinformatics for collection and analysis of gene sequences and directed evolution for enzyme improvement have completely revolutionised biocatalysis in the last two decades. Biocatalysis has been promoted to a method of choice in mainstream organic synthesis. Methodology development continues to play a pivotal role in the directed evolution of chemo-, regio- and stereo-selective enzymes247 and, since directed evolution produces huge amounts of data, it is ideally suited to benefit from machine learning.248–250 Similarly, computer-aided retrosynthetic planning of biocatalytic routes can be used effectively to design new enzymatic processes.251Another important technological development is the use of flow chemistry and microfluidics.252–255 This will facilitate the design of biocatalytic cascade reactions as these can easily be performed in series in a flow reactor. Moreover, the protective and low shear environment of micro-reactors will mean that less effort will be required for increasing the robustness of the enzymes.
Other emerging technologies will help to drive the future of biocatalysis. Synthetic biology, for example, has advanced to the point where defined microorganisms can be composed from scratch. This permits the production of better defined biocatalysts with reduced contamination by other enzymes.256 It has also allowed the redundancy in the gene code to be rationalised to provide dedicated codes for non-canonical amino acids,257 the potential contribution of which to new biocatalysis was mentioned above.
Good primers on the subject of biocatalysis include books by Grogan258 and Faber,259 while a more comprehensive contemporary review has been compiled by Faber, Fessner and Turner260 and Whittall, Sutton and Kroutil.261 The excellent paper by Lin et al.262 provides a convenient correlation of how enzyme catalysed reactions correspond with named organic chemical reactions that organic chemists are familiar with.
In conclusion, enzymes, long used in diverse large scale industries, are increasingly being applied in all areas of synthetic organic chemistry. If you are a synthetic chemist who is new to the application of enzymes in your research, Don't Panic, once you become familiar with biocatalysts you will find them indispensable.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
We would like to acknowledge Douglas Adams, the late acclaimed author, for inspiration, 40 years after the publication of the Hitchhiker's Guide to the Galaxy.References
- L. Esquirol, T. S. Peat, M. Wilding, J.-W. Liu, N. G. French, C. J. Hartley, H. Onagi, T. Nebl, C. J. Easton, J. Newman and C. Scott, J. Biol. Chem., 2018, 293, 7880–7891 CrossRef CAS PubMed.
- M. A. F. Delgove, A. B. Laurent, J. M. Woodley, K. V. Bernaerts and Y. van der Meer, ChemSusChem, 2019, 12, 1349–1360 CrossRef CAS PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- A. J. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13, 548–556 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, Chem. Commun., 2018, 54, 6088–6104 RSC.
- J. M. Woodley, Appl. Microbiol. Biotechnol., 2019, 103, 4733–4739 CrossRef CAS PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- S. H. Bhosale, M. B. Rao and V. V. Deshpandi, Microbiol. Rev., 1996, 60, 280–300 CrossRef CAS.
- R. A. Sheldon and F. van Rantwijk, Aust. J. Chem., 2004, 57, 281 CrossRef CAS.
- A. T. Omori, F. G. Lobo, A. Carolina, G. do Amaral and C. de Souza de Oliveira, J. Mol. Catal. B: Enzym., 2016, 127, 93–97 CrossRef CAS.
- C. Sanfilippo, A. Paterna, D. M. Biondi and A. Patti, Bioorg. Chem., 2019, 93, 103325 CrossRef CAS PubMed.
- F. Garzón-Posse, L. Becerra-Figueroa, J. Hernández-Arias and D. Gamba-Sánchez, Molecules, 2018, 23, 1241 CrossRef PubMed.
- S. G. Burton, Catal. Today, 1994, 22, 459–487 CrossRef CAS.
- C. C. C. R de Carvalho, Microb. Biotechnol., 2017, 10, 250–263 CrossRef PubMed.
- J. A. Littlechild, Archaea, 2015, 147671 Search PubMed.
- L. Liu, H. Yang, H. D. Shin, R. R. Chen, J. Li, G. Du and J. Chen, Bioengineered, 2013, 4, 212–223 CrossRef PubMed.
- D. Tusé, T. Tu and K. A. McDonald, BioMed Res. Int., 2014, 256135 Search PubMed.
- M. L. Bode, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2003, 84, 710–713 CrossRef CAS PubMed.
- https://blast.ncbi.nlm.nih.gov/Blast.cgilast, accessed, 5 November 2019.
- https://www.uniprot.org/last, accessed, 5 November 2019.
- M. Tuffin, D. Anderson, C. Heath and D. A. Cowan, Biotechnol. J., 2009, 4, 1671–1683 CrossRef CAS PubMed.
- F. Berini, C. Casciello, G. L. Marcone and F. Marinelli, FEMS Microbiol. Lett., 2017, 364, fnx211 CrossRef PubMed.
- K. Rashamuse, V. Magomani, T. Ronneburg and D. Brady, Appl. Microbiol. Biotechnol., 2009, 83, 491–501 CrossRef CAS PubMed.
- D. Baud, J. W. E. Jeffries, T. S. Moody, J. M. Ward and H. C. Hailes, Green Chem., 2017, 19, 1134–1143 RSC.
- L. Leipold, D. Dobrijevic, J. W. E. Jeffries, M. Bawn, T. S. Moody, J. M. Ward and H. C. Hailes, Green Chem., 2019, 21, 75–86 RSC.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- U. T. Bornscheuer, B. Hauer, K. E. Jaeger and U. Schwaneberg, Angew. Chem., Int. Ed., 2019, 58, 36–40 CrossRef CAS PubMed.
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131–157 CrossRef CAS PubMed.
- A. Tiessen, P. Pérez-Rodríguez and L. J. Delaye-Arredondo, BMC Res. Notes, 2012, 5, 85 CrossRef CAS PubMed.
- L. H. Chen, G. I. Kenyon, F. Curtin, S. Harayama, M. E. G. Bembenek, C. P. Hajipour and C. P. Whitman, J. Biol. Chem., 1992, 267, 17716–17721 CAS.
- A. J. M. Ribeiro, G. I. Holliday, N. Furnham, J. D. Tyzack, K. Ferris and J. M. Thornton, Nucleic Acids Res., 2018, 46(D1), D618–D623 CrossRef CAS PubMed.
- G. J. Bartlett, C. T. Porter, N. Borkakoti and J. M. Thornton, J. Mol. Biol., 2002, 24, 105–121 CrossRef.
- L. Pravda, K. Berka, R. Svobodová Vařeková, D. Sehnal, P. Banáš, R. A. Laskowski, J. Koča and M. Otyepka, BMC Bioinf., 2014, 15, 379 CrossRef PubMed.
- C. Cheng, T. Jiang, Y. Wu, L. Cui, S. Qin and B. He, Bioengineered, 2018, 119, 1211–1217 CAS.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- S. Kaushik, Z. Prokop, J. Damborsky and R. Chaloupkova, FEBS J., 2017, 284, 134–148 CrossRef CAS PubMed.
- J. D. Fischer, G. L. Holliday and J. M. Thornton, Bioinformatics, 2010, 26, 2496–2497 CrossRef CAS.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, Bioinformatics, 2009, 25, 2088–2089 CrossRef CAS.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, J. Biol. Inorg Chem., 2008, 13, 1205–1218 CrossRef CAS PubMed.
- C. E. Paul and F. Hollmann, Appl. Microbiol. Biotechnol., 2016, 100, 4773–4778 CrossRef CAS PubMed.
- C. K. Prier and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13992–14006 CrossRef CAS PubMed.
- C. Martinoli, H. M. Dudek, R. Orru, D. E. Edmondson, M. W. Fraaije and A. Mattevi, ACS Catal., 2013, 3, 3058–3062 CrossRef CAS PubMed.
- F. Agostini, J.-S. Völler, B. Koksch, C. G. Acevedo-Rocha, V. Kubyshkin and N. Budisa, Angew. Chem., Int. Ed., 2017, 56, 9680–9709 CrossRef CAS PubMed.
- A. M. Foley and A. R. Maguire, Eur. J. Org. Chem., 2019, 3713–3734 CrossRef CAS.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 17, 2–25 Search PubMed.
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- N. J. Turner and L. Humphreys, Biocatalysis in organic synthesis: the retrosynthetic approach, Royal Society of Chemistry, Cambridge, 2018 Search PubMed.
- A. P. Green and N. J. Turner, Perspectives in Science, 2016, 9, 42–48 CrossRef.
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem.–Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- A. Krüger, C. Schäfers, C. Schröder and G. Antranikian, New Biotechnol., 2018, 40, 144–153 CrossRef PubMed.
- D. Brady, A. Rousseau, L. H. Steenkamp, J. Chaplin, S. Reddy, R. Mitra, B. Mboniswa, C. J. Parkinson, T. Moutlana, S. F. Marais and N. S. Gardener, J. Mol. Catal. B: Enzym., 2012, 75, 1–10 CrossRef CAS.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 2885–2904 CrossRef CAS.
- S. Prasad and I. Roy, Recent Pat. Biotechnol., 2018, 12, 33–56 CAS.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 74–88 CrossRef PubMed.
- R. A. Sheldon, in Biocatalysis: An Industrial Perspective, ed. G. de Gonzalo and P. Dominguez de Maria, Royal Society of Chemistry, 2018, ch. 14 Search PubMed.
- R. A. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- M. J. Liszka, M. E. Clark, E. Schneider and D. S. Clark, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 77–102 CrossRef CAS PubMed.
- O. Alvizo, L. J. Nguyen, C. K. Savile, J. A. Bresson, S. L. Lakhapatri, E. O. Solis, R. J. Fox, J. M. Broering, M. R. Benoit, S. A. Zimmerman, S. J. Novick, J. Liang and J. J. Lalonde, Proc. Natl. Acad. Sci., 2014, 111, 16436–16441 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- P. A. Wender, S. T. Handy and D. L. Wright, Chem. Ind., 1997, 765–769 CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS.
- R. A. Sheldon, Curr. Opin. Green Sust. Chem., 2019, 18, 13–19 Search PubMed.
- P. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, UK, 1998 Search PubMed.
- J. Lalonde, Curr. Opin. Biotechnol., 2016, 42, 152–158 CrossRef CAS.
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS PubMed.
- K. Rosenthal and S. Lütz, Curr. Opin. Green Sust. Chem., 2018, 11, 58–64 Search PubMed.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G. -D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS.
- T. Nagasawa, H. Shimizu and H. Yamada, Appl. Microbiol. Biotechnol., 1993, 40, 189–195 CrossRef CAS.
- B. Li, J. Su and J. Tao, Org. Process Res. Dev., 2011, 15, 291–293 CrossRef CAS.
- E. C. Hann, A. Eisenberg, S. K. Fager, N. E. Perkins, F. G. Gallagher, S. M. Cooper, J. E. Gavagan, B. Stieglitz, S. M. Hennessey and R. DiCosimo, Bioorg. Med. Chem., 1999, 10, 2239–2245 CrossRef.
- Y. Isowa, M. Ohmori, T. Ichikawa, K. Mori, Y. Nonaka, K. Kihara, K. Oyama, H. Satoh and S. Nishimura, Tetrahedron Lett., 1979, 20, 2611–2612 CrossRef.
- A. Bruggink, E. C. Roos and E. de Vroom, Org. Process Res. Dev., 1998, 2, 128–133 CrossRef CAS.
- C. Bertoldo and G. Antranikian, Curr. Opin. Chem. Biol., 2002, 6, 151–160 CrossRef CAS.
- C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS PubMed.
- A. Salihu and M. Z. Alam, Process Biochem., 2015, 50, 86–96 CrossRef CAS.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS.
- R. N. Patel, Bioorg. Med. Chem., 2018, 26, 1252–1274 CrossRef CAS PubMed.
- A. C. Carvalho, T. de Fonseca, M. C. de Mattos, M. de Oliveira, T. L. de Lemos, F. Molinari, D. Romano and I. Serra, Int. J. Mol. Sci., 2015, 16, 29682–29716 CrossRef PubMed.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- I. C. Perdomo, S. Gianolio, A. Pinto, D. Romano, M. L. Contente, F. Paradisi and F. Molinari, J. Agric. Food Chem., 2019, 67, 6517–6522 CrossRef CAS PubMed.
- N. de Leeuw, G. Torrelo, C. Bisterfeld, V. Resch, L. Mestrom, E. Straulino, L. van der Weel and U. Hanefeld, Adv. Synth. Catal., 2018, 360, 242–249 CrossRef CAS.
- L. Mestrom, J. G. R. Claessen and U. Hanefeld, ChemCatChem, 2019, 11, 2004–2010 CrossRef CAS.
- O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- Y. Kim, J. Park and M.-J. Kim, ChemCatChem, 2011, 3, 271–277 CrossRef CAS.
- C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kelleher and L. Tully, Org. Process Res. Dev., 2008, 12, 392–398 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed.
- N. Kishi and H. Kojima, ChemistrySelect, 2019, 4, 9570–9572 CrossRef CAS.
- W. P. Juma, V. Chhiba, D. Brady and M. L. Bode, Tetrahedron: Asymmetry, 2017, 28, 1169–1174 CrossRef CAS.
- S. Dasetty, M. A. Blenner and S. Sarupria, Mater. Res. Express, 2017, 4, 114008 CrossRef.
- K. de Godoy Daiha, R. Angeli, S. D. de Oliveira and R. V. Almeida, PLoS One, 2015, 10, e0131624 CrossRef PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2016, 20, 700–706 CrossRef CAS.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 1063–1080 CrossRef CAS.
- P. Domínguez de María, G. de Gonzalo and A. R. Alcántara, Catalysts, 2019, 9, 802 CrossRef.
- G. W. Huisman and S. J. Collier, Curr. Opin. Chem. Biol., 2013, 17, 284–292 CrossRef CAS PubMed.
- R. Buller, K. Hechta, M. A. Mirata and H.-P. Meyer, in Biocatalysis: An Industrial Perspective, ed. G. de Gonzalo and P. Domínguez de María, Royal Society of Chemistry, 2017, pp. 3–43, ISBN 978-1-78262-619-0 Search PubMed.
- M. Yoshimura, S. Tanaka and M. Kitamura, Tetrahedron Lett., 2014, 55, 3635–3640 CrossRef CAS.
- R. Zhang, Y. Xu and R. Xiao, Biotechnol. Adv., 2015, 33, 1671–1684 CrossRef CAS PubMed.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS.
- M. J. Abrahamson, E. Vazquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 CrossRef CAS PubMed.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS.
- S. K. Au, B. R. Bommarius and A. S. Bommarius, ACS Catal., 2014, 4, 4021–4026 CrossRef CAS.
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119 CrossRef CAS.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleis, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, ChemBioChem, 2014, 15, 2201–2204 CrossRef CAS PubMed.
- Z. Maugeri and D. Rother, Adv. Synth. Catal., 2016, 358, 2745–2750 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- G. Grogan and N. J. Turner, Chem.–Eur. J., 2016, 22, 1900–1907 CrossRef CAS PubMed.
- S. P. France, R. M. Howard, J. Steflik, J. M. Weise, J. Mangas-Sanchez, S. L. Montgomery, R. Crook, R. Kumar and N. J. Turner, ChemCatChem, 2018, 10, 510–514 CrossRef CAS.
- P. N. Scheller, M. Lenz, S. C. Hammer, B. Hauer and B. M. Nestl, ChemCatChem, 2015, 7, 3239 CrossRef CAS.
- M. Lenz, J. Meisner, L. Quertinmont, S. Lutz, J. Kastner and B. M. Nestl, ChemBioChem, 2017, 18, 253–256 CrossRef CAS PubMed.
- D. Wetzl, M. Gand, A. Ross, H. Meller, P. Matzel, S. P. Hanlon, M. Meller, B. Wirz, M. Hçhne and H. Iding, ChemCatChem, 2016, 8, 2023–2026 CrossRef CAS.
- P. Matzel, M. Gand and M. Hçhne, Green Chem., 2017, 19, 385–389 RSC.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- M. P. Thompson and N. J. Turner, ChemCatChem, 2017, 23, 3833–3836 CrossRef.
- W. Böhmer, T. Kraus and F. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberthm, Green Chem., 2018, 20, 5082–5103 RSC.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Organomet. Chem., 2015, 63, 91–174 CrossRef CAS.
- R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef CAS PubMed.
- R. A. Sheldon, I. W. C. E. Arends, G. J. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, in Liquid Phase Aerobic Oxidation Catalysis, ed. S. S.Stahl and P. J. Alsters, 2016, pp. 85–96 Search PubMed.
- J. E. Steves, Y. Preger, J. R. Martinelli, C. J. Welch, T. W. Root, J. M. Hawkins and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1548–1553 CrossRef CAS.
- J. Liu, S. Wu and Z. Li, Curr. Opin. Chem. Biol., 2018, 43, 77–86 CrossRef CAS PubMed.
- F. Escaletters and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef PubMed.
- S. Herter, S. M. McKenna, A. R. Frazer, S. Lehmkαhler, A. J. Carnell and N. J. Turner, ChemCatChem, 2015, 7, 2313–2317 CrossRef CAS.
- S. M. McKenna, S. Leimkühler, S. Herter, N. J. Turner and A. Carnell, Green Chem., 2015, 17, 3271–3275 RSC.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS.
- J. Vilim, T. Kraus and F. Mutti, Angew. Chem., Int. Ed., 2018, 57, 14240–14244 CrossRef CAS PubMed.
- A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 RSC.
- J. Gross, K. Tauber, M. Fuchs, N. G. Schmidt, A. Rajagopalan, K. Faber, W. M. F. Fabian, J. Pfeiffer, T. Haas and W. Kroutil, Green Chem., 2014, 16, 2117–2121 RSC.
- A. Weckbecker, H. Gröger and W. Hummel, Adv. Biochem. Eng./Biotechnol., 2010, 120, 195–242 CAS.
- J. K. B. Cahn, C. A. Werlang, A. Baumschlager, S. Brinkman-Chen, S. I. Mayo and F. Arnold, ACS Synth. Biol., 2017, 6, 326–333 CrossRef CAS PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Adv. Synth. Catal., 2004, 346, 125–142 CrossRef CAS.
- J. D. Zhang, Z. M. Cui, X. J. Fan, H. L. Wu and H. H. Chang, Bioprocess Biosyst. Eng., 2016, 39, 603–611 CrossRef CAS PubMed.
- T. Knaus, V. Tseliou, L. D. Humphreys, N. S. Scrutton and F. G. Mutti, Green Chem., 2018, 20, 3931–3943 RSC.
- T. Knaus, F. G. Mutti, L. D. Humphreys, N. J. Turner and N. S. Scrutton, Org. Biomol. Chem., 2015, 13, 223–233 RSC.
- K. Bulke, M. Kadow, H. Mallin, S. Sass and U. T. Bornscheuer, Org. Biomol. Chem., 2012, 10, 6249–6265 RSC.
- M. Bucko, P. Gemeiner, A. Scenkmayerova, T. Kajovic, F. Rudroff and M. D. Mihovilovic, Appl. Microbiol. Biotechnol., 2016, 100, 6585–6599 CrossRef CAS PubMed.
- H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165 CrossRef CAS PubMed.
- E. Y. Jeon, A. H. Beek, U. T. Bornscheuer and J. B. Park, Appl. Microbiol. Biotechnol., 2015, 99, 6267–6275 CrossRef CAS PubMed.
- A. Bornadel, R. Hatti-Kaul, F. Hollmann and S. Kara, ChemCatChem, 2015, 7, 2442–2445 CrossRef CAS.
- R. Wilson, Chim Oggi, 2015, 33, 50–52 Search PubMed ; see also G. de Gonzalo and A. Franconetti, Enzyme Microb. Technol., 2018, 113, 24–28 CrossRef CAS PubMed.
- Y. Zang, Y. Q. Wu, N. Xu, Q. Zhao, H. L. Yu and J. H. Xu, ACS Sustainable Chem. Eng., 2019, 7, 7218–7226 CrossRef.
- G. Grogan, Curr. Opin. Chem. Biol., 2018, 43, 15–52 CrossRef CAS PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed.
- B. Mijts, S. Muley, J. Liang, L. M. Newman, X. Zhang, J. Lalonde, M. D. Clay, J. Zhu, J. Gruber, J. Colbeck, J. D. Munger Jr, J. Mavinahalli and R. Sheldon, US Pat., 8574876 B2, 2013.
- J. Dong, E. Fernandez-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- S. Bormann, A. G. Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 RSC.
- F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed.
- A. W. Munro, D. G. Leys, K. J. McLean, K. R. Marshall, T. W. B. Ost, S. Daff, C. S. Miles, S. K. Chapman, D. A. Lysek, C. C. Moser, C. C. Page and P. L. Dutton, Trends Biochem. Sci., 2002, 27, 250–257 CrossRef CAS.
- M. Tavanti, J. L. Porter, S. Sabatini, N. J. Turner and S. L. Flitsch, ChemCatChem, 2018, 10, 1042–1051 CrossRef CAS.
- J. N. Kolev, J. M. Zaengle, R. Ravikumar and R. Fasan, ChemBioChem, 2014, 15, 1001–1010 CrossRef CAS PubMed.
- T. Kubo, P. Meinhold, M. W. Peters and F. H. Arnold, Chem.–Eur. J., 2006, 12, 1216–1220 CrossRef CAS PubMed.
- S. Peter, M. Kinne, R. Ulrich, G. Kayser and M. Hofrichter, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef CAS PubMed.
- C. Gally, B. Nestl and B. Hauer, Angew. Chem., Int. Ed., 2015, 54, 12952–12956 CrossRef CAS PubMed.
- H. C. Kolb, M. S. Van Nieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- D. Holtmann and F. Hollmann, ChemBioChem, 2016, 17, 1391–1398 CrossRef CAS PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- J. S. Shin and B. G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS PubMed.
- G. W. Matcham and A. R. S. Bowen, Chem. Today, 1996, 14, 20–24 CAS.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS PubMed.
- J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 Search PubMed.
- C. K. Saville, J. M. Maney, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef PubMed.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed.
- K. Fesko and M. Gruber-Khadjawi, ChemCatChem, 2013, 5, 1248–1272 CrossRef CAS.
- S. K. Panday, Tetrahedron: Asymmetry, 2011, 22, 1817–1847 CrossRef CAS.
- S. Bhowmick, A. Mondal, A. Ghosh and K. C. Bhowmick, Tetrahedron: Asymmetry, 2015, 26, 1215–1244 CrossRef CAS.
- P. Bracco, H. Busch, J. von Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- E. Lanfranchia, K. Steiner, A. Glieder, I. Hajnal, R. A. Sheldon, S. van Pelt and M. Winkler, Recent Pat. Biotechnol., 2013, 7, 197–206 CrossRef PubMed.
- L. M. van Langen, R. P. Selassa, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2005, 7, 327–329 CrossRef CAS PubMed.
- M. Haridas, E. M. M. Abdelraheem and U. Hanefeld, Appl. Microbiol. Biotechnol., 2018, 102, 9959–9971 CrossRef CAS PubMed.
- S. Jennewein, M. Schurmann, M. Wolberg, I. Hilker, R. Luiten, M. Wubbolts and D. Mink, Biotechnol. J., 2006, 1, 537–548 CrossRef CAS PubMed.
- R. A. Sheldon, Chimia, 1996, 50, 418EP0343714A1, 1989 Search PubMed.
- R. J. Fox, S. C. Davis, E. Mundorff, L. Newman, V. Gavrilovic, S. Ma, L. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. Whitman, R. A. Sheldon and G. W. Huisman, Nat. Biotechnol., 2007, 25, 338–344 CrossRef CAS PubMed.
- P. Hoyos, V. Pace and A. R. Alcantara, Catalysts, 2019, 9(260), 1–32 Search PubMed.
- R. A. Sheldon, in Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis, ed. E. Garcia Junceda, Wiley-VCH, Weinheim, 2008, pp. 109–135 Search PubMed.
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC.
- R. Xue and J. M. Woodley, Bioresour. Technol., 2012, 115, 183–195 CrossRef CAS PubMed.
- V. Köhler and N. J. Turner, Chem. Commun., 2015, 13, 223–233 Search PubMed.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- G. E. R. Gordon, M. L. Bode, D. F. Visser, M. J. Lepuru, J. G. Zeevaart, N. Ragubeer, M. Ratsaka, D. R. Walwyn and D. Brady, Org. Process Res. Dev., 2011, 15, 258–265 CrossRef CAS.
- D. Brady, A. Beeton, C. Kgaje, J. Zeevaart, F. van Rantwijk and R. A. Sheldon, Appl. Microbiol. Biotechnol., 2004, 64, 76–85 CrossRef CAS PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez- Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yan, Science, 2019, 1255–1259 CrossRef CAS PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- A. V. Ruales-Salcedo, J. C. Higuita, J. Fontalvo and J. M. Woodley, Z. Naturforsch., 2019, 74, 77–84 CAS.
- E. Garcia-Junceda, I. Lavandera, D. Rother and J. H. Schrittwieser, J. Mol. Catal. B: Enzym., 2015, 114, 1–6 CrossRef CAS.
- J. B. Pyser, S. A. Baker Dockrey, A. Rodríguez Benítez, L. A. Joyce, R. A. Wiscons, J. L. Smith and A. R. H. Narayan, J. Am. Chem. Soc., 2019, 141, 18551–18559 CrossRef CAS PubMed.
- T. J. Doyon, J. C. Perkins, S. A. Baker Dockrey, E. O. Romero, K. C. Skinner, P. M. Zimmerman and A. R. H. Narayan, J. Am. Chem. Soc., 2019, 141, 20269–20277 CrossRef CAS PubMed.
- J. Li, X. Zhang and H. Renata, Angew. Chem., Int. Ed., 2019, 58, 11657–11660 CrossRef CAS PubMed.
- C. R. Zwick III and H. Renata, J. Org. Chem., 2018, 83, 7407–7415 CrossRef PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J.-Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 2169 CrossRef PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- U. T. Bornscheuer, Philos. Trans. R. Soc., A, 2017, 376, 20170063 CrossRef PubMed.
- H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed.
- S. Noor, M. C. Taylor, R. J. Russell, L. S. Jermiin, C. J. Jackson, J. G. Oakeshott and C. Scott, PLoS One, 2012, 7, e39822 CrossRef CAS PubMed.
- S. C. Hammer, A. M. Knight and F. H. Arnold, Curr. Opin. Green Sus. Chem., 2017, 7, 23–30 Search PubMed.
- U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032–6040 CrossRef CAS PubMed.
- F. H. Arnold, Q. Rev. Biophys., 2015, 48, 404–410 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- T. K. Hyster and T. R. Ward, Angew. Chem., Int. Ed., 2016, 55, 7344–7357 CrossRef CAS PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 127, 1764–1768 CrossRef.
- O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, Z. Liu, J. Y.-E. Rha, A. Nazarenko, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 302–308 CrossRef CAS PubMed.
- C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed.
- J. A. McIntosh, P. S. Coelho, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed.
- E. W. Svastits, J. H. Dawson, R. Breslow and S. Gellman, J. Am. Chem. Soc., 1985, 107, 6427–6428 CrossRef CAS.
- T. K. Hyster, C. C. Farwell, A. R. Buller, J. A. McIntosh and F. H. Arnold, J. Am. Chem. Soc., 2014, 136, 15505–15508 CrossRef CAS PubMed.
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkman-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed.
- O. F. Brandenberg, D. C. Miller, U. Markel, A. O. Chaib and F. H. Arnold, ACS Catal., 2019, 9, 8271–8275 CrossRef CAS PubMed.
- V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 2512–2516 CrossRef CAS PubMed.
- O. F. Brandenberg, K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 8989–8995 CrossRef CAS PubMed.
- J. Zhang, X. Huang, R. K. Zhang and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 9798–9802 CrossRef CAS PubMed.
- H. Tsutsumi, Y. Katsuyama, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-ya and Y. Ohnishi, J. Am. Chem. Soc., 2018, 140, 6631–6639 CrossRef CAS PubMed.
- J. G. Gober and E. M. Brustad, Curr. Opin. Chem. Biol., 2016, 35, 124–132 CrossRef CAS PubMed.
- B. P. Dwivedee, S. Soni, M. Sharma, J. Bhaumik, J. K. Laha and U. C. Banerjee, ChemistrySelect, 2018, 3, 2441–2466 CrossRef CAS.
- D. Koszelewski and R. Ostaszewski, ChemCatChem, 2019, 11, 2554–2558 CrossRef CAS.
- Q. Wu, B.-K. Liu and X.-F. Lin, Curr. Org. Chem., 2010, 14, 1966–1988 CrossRef CAS.
- M. Svedendahl, K. Hult and P. Berglund, J. Am. Chem. Soc., 2005, 127, 17988–17989 CrossRef CAS PubMed.
- M. Svedendahl, B. Jovanović, L. Fransson and P. Berglund, ChemCatChem, 2009, 1, 252–258 CrossRef CAS.
- F.-W. Lou, B.-K. Liu, Q. Wu, D.-S. Lv and X.-F. Lin, Adv. Synth. Catal., 2008, 350, 1959–1962 CrossRef CAS.
- R. Kazlauskas, Curr. Opin. Chem. Biol., 2005, 9, 195–201 CrossRef CAS PubMed.
- C. Li, X.-W. Feng, N. Wang, Y.-J. Zhou and X.-Q. Yu, Green Chem., 2008, 10, 616–618 RSC.
- R. C. Tang, Z. Guan, Y.-H. He and W. Zhu, J. Mol. Catal. B: Enzym., 2010, 63, 62–67 CrossRef CAS.
- K. Li, T. He, C. Li, X.-W. Feng, N. Wang and X.-Q. Yu, Green Chem., 2009, 11, 777–779 RSC.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
- M. López-Iglesias and V. Gotor-Fernández, Chem. Rec., 2015, 15, 743–759 CrossRef PubMed.
- X. Ge, Y. Lai and X. Chen, Chin. J. Org. Chem., 2013, 33, 1686–1696 CrossRef CAS.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS PubMed.
- T. K. Hyster, Synlett, 2019, 30, A–G Search PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201901491.
- G. Li, Y. Dong and M. T. Reetz, Adv. Synth. Catal., 2019, 361, 2377–2386 CAS.
- Y. Saito, M. Oikawa, H. Nakazawa, T. Niide, T. Kameda, K. Tsuda and M. Umetsu, ACS Synth. Biol., 2018, 7, 2014–2022 CrossRef CAS PubMed.
- F. Cadet, N. Fontaine, G. Li, J. Sanchis, M. N. F. Chong, R. Pandjaitan, I. Vetrivel, B. Offmann and M. T. Reetz, Sci. Rep., 2018, 8, 16757 CrossRef PubMed.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- X. W. Diefenbach, I. Farasat, E. D. Guetschow, C. J. Welch, R. T. Kennedy, S. Sun and J. C. Moore, ACS Omega, 2018, 3, 1498–1508 CrossRef CAS.
- P. Žnidaršič-Plazl, Biotechnol. J., 2019, 14, 1800580 CrossRef PubMed.
- T. Peschke, P. Bitterwolf, K. S. Rabe and C. M. Niemeyer, Chem. Eng. Technol., 2019, 42(10), 2009–2017 CrossRef CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamiso and K. F. Jensen, Science, 2019, 365(6453), eaax1566 CrossRef CAS.
- A. Currin, N. Swainston, P. J. Dayace and D. B. Kell, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC.
- J. Fredens, K. Wang, D. de la Torre, L. F. H. Funke, W. E. Robertson, Y. Christova, T. Chia, W. H. Schmied, D. L. Dunkelmann, V. Beránek, C. Uttamapinant, A. Gonzalez Llamazares, T. S. Elliott and J. W. Chin, Nature, 2019, 569, 514–518 CrossRef CAS PubMed.
- G. Grogan, Practical Biotransformations: A Beginner's Guide, Wiley-Blackwell, 2009, ISBN: 978-1-405-17125-0 Search PubMed.
- K. Faber, Biotransformations in Organic Chemistry, 2011, Springer, ISBN 978-3-319-61590-5 Search PubMed.
- Biocatalysis in Organic Synthesis. Science of Synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, 2015, vol. 1–3, ISBN 978-3132028715 Search PubMed.
- Practical Methods for Biocatalysis and Biotransformations, ed. J. Whittall, P. W. Sutton and W Kroutil, Wiley, 2009, 2012 and 2016, vol. 1–3, ISBN: 9780470519271, 9781119991397 and ISBN: 9781118605257 Search PubMed.
- C.-I. Lin, R. M. McCarty and H.-W. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446–3489 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |