
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible reduction drives anion ejection and C60 binding within an FeII4L6 cage†
Zhenpin
Lu
 ,
Tanya K.
Ronson
and
Jonathan R.
Nitschke
*
,
Tanya K.
Ronson
and
Jonathan R.
Nitschke
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: jrn34@cam.ac.uk
First published on 5th December 2019
Abstract
FeII4L6 tetrahedral cage 1 was prepared from a redox-active dicationic naphthalenediimide (NDI) ligand. The +20 charge of the cage makes it a good host for anionic guests, with no binding observed for neutral aromatic molecules. Following reduction by Cp2Co, the cage released anionic guests; subsequent oxidation by AgNTf2 led to re-uptake of anions. In its reduced form, however, 1 was observed to bind neutral C60. The fullerene guest was subsequently ejected following cage re-oxidation. The guest release process was found to be facilitated by anion-mediated transport from organic to aqueous solution. Cage 1 thus employs electron transfer as a stimulus to control the uptake and release of both neutral and charged guests, through distinct pathways.
Introduction
Self-assembled metal–organic cages1 have found uses across various fields, ranging from chemical separations,2 catalyzing organic reactions,3 sensing specific analytes4 and acting as photoreactors,5 among others. These applications are often based on encapsulation of guests within the well-defined inner cavities of cages. Guest uptake and release by a host molecule can be controlled using stimuli such as heat,6 light,7 pH8 and competing guests,9 as understanding has increased as to how to design stimuli-responsive behaviour.10 The use of redox stimuli is particularly attractive11 because electrons are ‘clean’ stimuli, producing no chemical by-products. Thus far, several redox-active metal–organic cages have been successfully synthesized.11c Recently Sallé, Goeb and co-workers reported several tetrathiafulvalene based coordination cages, which can reversibly uptake and release perfluorocarborate11d or corronene11a guests under redox control. Inspired by these achievements, we sought to develop new redox-active metal–organic cage systems capable of reversible guest uptake. In the present system, electron transfer was used to stimulate the uptake and release of both anionic and neutral guests, via distinct pathways.Naphthalenediimides (NDIs) and their derivatives are redox-active electron-deficient compounds, and can be readily substituted with a wide variety of functional groups,12 making them ideal building blocks for metal–organic cages.13 We have reported several NDI-diamine based tetrahedral metal–organic cages, with the redox behaviour of one catalyzing the oxidative coupling of arylborates to give biphenyls.14 However, reduction of that cage14 resulted in precipitation due to charge neutralization. This behaviour prevented further study of its redox dependent host–guest chemistry in solution. In order to solve this issue, we designed a new dicationic subcomponent, A (Fig. 1). We hypothesised that the permanent charges of A would improve the solubility of the corresponding cage 1, following reduction of the NDI moieties. This FeII4L6 cage, 1, did indeed remain in solution upon NDI reduction, enabling different guests to be taken up and released upon reduction and oxidation of the cage.
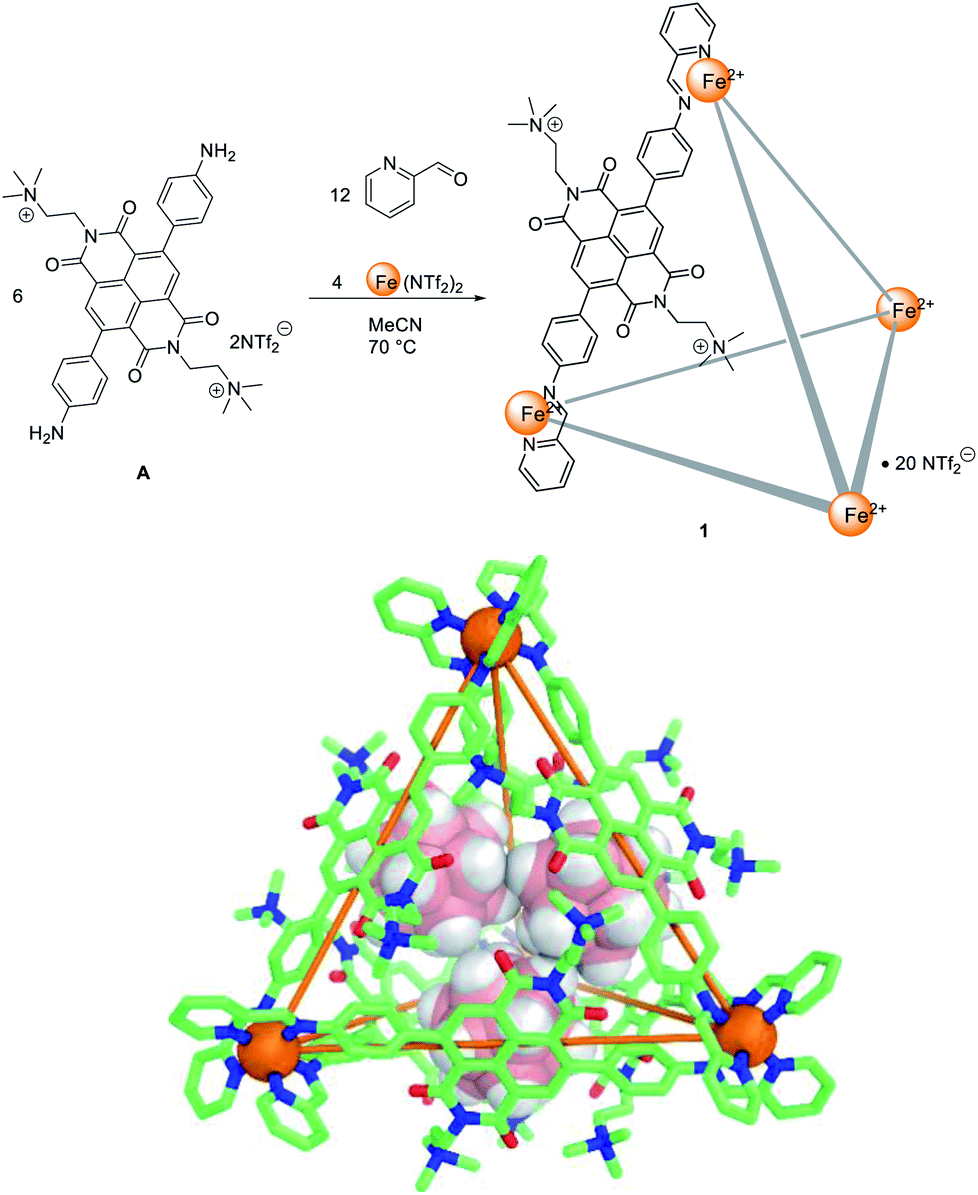 | ||
| Fig. 1 The synthesis of FeII4L6 cage 1 (top) and crystal structure of 1 with three carborate anions encapsulated (bottom). Cage hydrogen atoms, counterions, solvents and disorder are omitted for clarity. | ||
Results and discussion
Tetrahedral FeII4L6 cage 1 was synthesized from quaternary-ammonium-functionalized NDI subcomponent A (6 equiv.), 2-formylpyridine (12 equiv.) and Fe(NTf2)2 (4 equiv.) in acetonitrile (Fig. 1). The 1H NMR spectrum of 1 was consistent with a single symmetric species (Fig. S2–S7†). ESI-MS of 1 confirmed the formation of an assembly with FeII4L6 stoichiometry (Fig. S8–S11†). Subcomponent A was not observed to form the Zn4L6 analogue of 1 when Zn(NTf2)2 was used in place of the iron salt. We infer this lack of reactivity to be due to the weaker metal–ligand bonds involving zinc not being able to compensate for coulombic repulsion among the cationic ligands.Single crystals of 1 were obtained from vapour diffusion of diisopropyl ether into an acetonitrile solution of 1 containing cesium carborane (10 equiv.). The crystal structure of 1 revealed a T-symmetric framework (Fig. 1), with six ligands bridging four octahedral iron(II) centres of the same handedness. The solid state structure is consistent with NMR data, in which all ligands are magnetically equivalent. The metal–metal distances are in the range 18.771(3)–19.345(2) Å (average 19.1 Å). The NDI moieties lie tangent to the edges of the tetrahedron, affording an enclosed cavity which is further blocked by the –(CH2)2N+(Me)3 substituents of the ligands. A cavity volume of 1100 Å3 was determined using VOIDOO15 (Fig. S26†). Three carborate anions were found in the cavity in the solid state.
The electrochemical properties of cage 1 and subcomponent A were investigated by cyclic voltammetry, carried out in 0.1 M nBu4N+Tf2N− in MeCN at a scan rate of 500 mV s−1. Similar to other NDI derivatives,14,16 cage 1 exhibited a quasi-reversible process upon reduction (Fig. 2), in which the first reduction wave appeared at −0.81 V vs. Fc/Fc* and the second occurred at −1.19 V. The related oxidation waves were found at −1.29 and −0.78 V. Reduction was reversible over several cycles. In comparison, two similar reduction waves (at −0.96 and −1.36 V) were also observed for subcomponent A (Fig. S25†). However, the intensity of the CV signals decreased with each cycle in the case of A, consistent with irreversible reactions following redox events for A, but not for 1. We thus infer that self-assembly rendered the NDI panels more robust to redox processes.
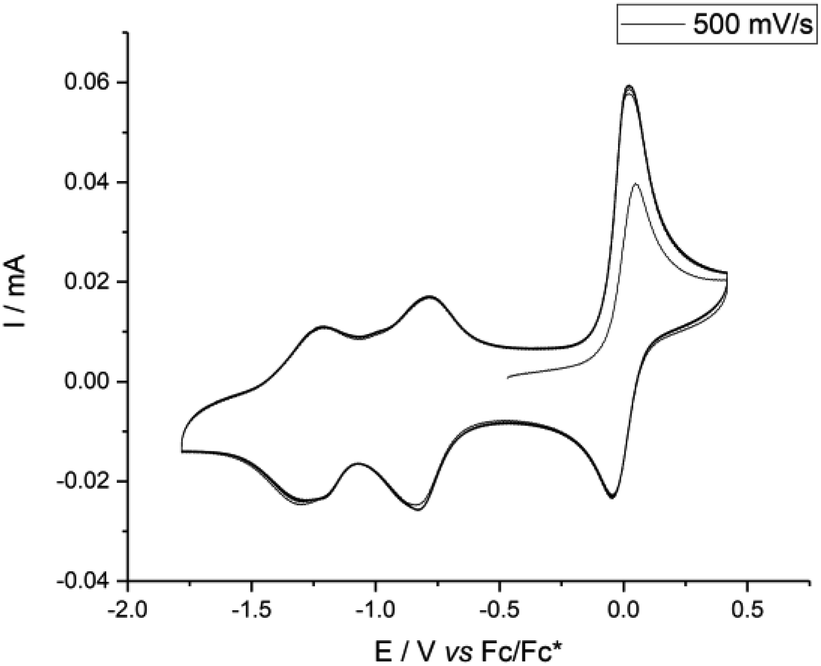 | ||
| Fig. 2 Cyclic voltammetry (5 scans, 500 mV s−1) of 1 in MeCN (0.1 M nBu4N+Tf2N−) at 25 °C. | ||
In light of our electrochemical studies, we investigated the reactions of 1 with chemical reductants and oxidants. Cp2Co and AgNTf2 were selected as an appropriate one-electron reductant and oxidant, respectively, for 1. Following the addition of Cp2Co (10 equiv.), a sharp signal attributed to Cp2Co+ appeared at 5.67 ppm in the 1H NMR spectrum, indicating that cage reduction had occurred. Following reduction, the cage signals became NMR silent due to the formation of radical species, as was observed previously in the case of related systems.14,17 When AgNTf2 (12 equiv.) was added to the mixture, the cage signals reappeared cleanly (Fig. S41†), demonstrating the reversibility of the process.
Next we investigated the binding behaviour of 1 with various prospective guests using 1H NMR spectroscopy in CD3CN. Cage 1 did not show measurable affinity towards the neutral species investigated (Fig. 3 and S27–S32†) despite the enclosed cavity observed in the crystal structure (Fig. 1).
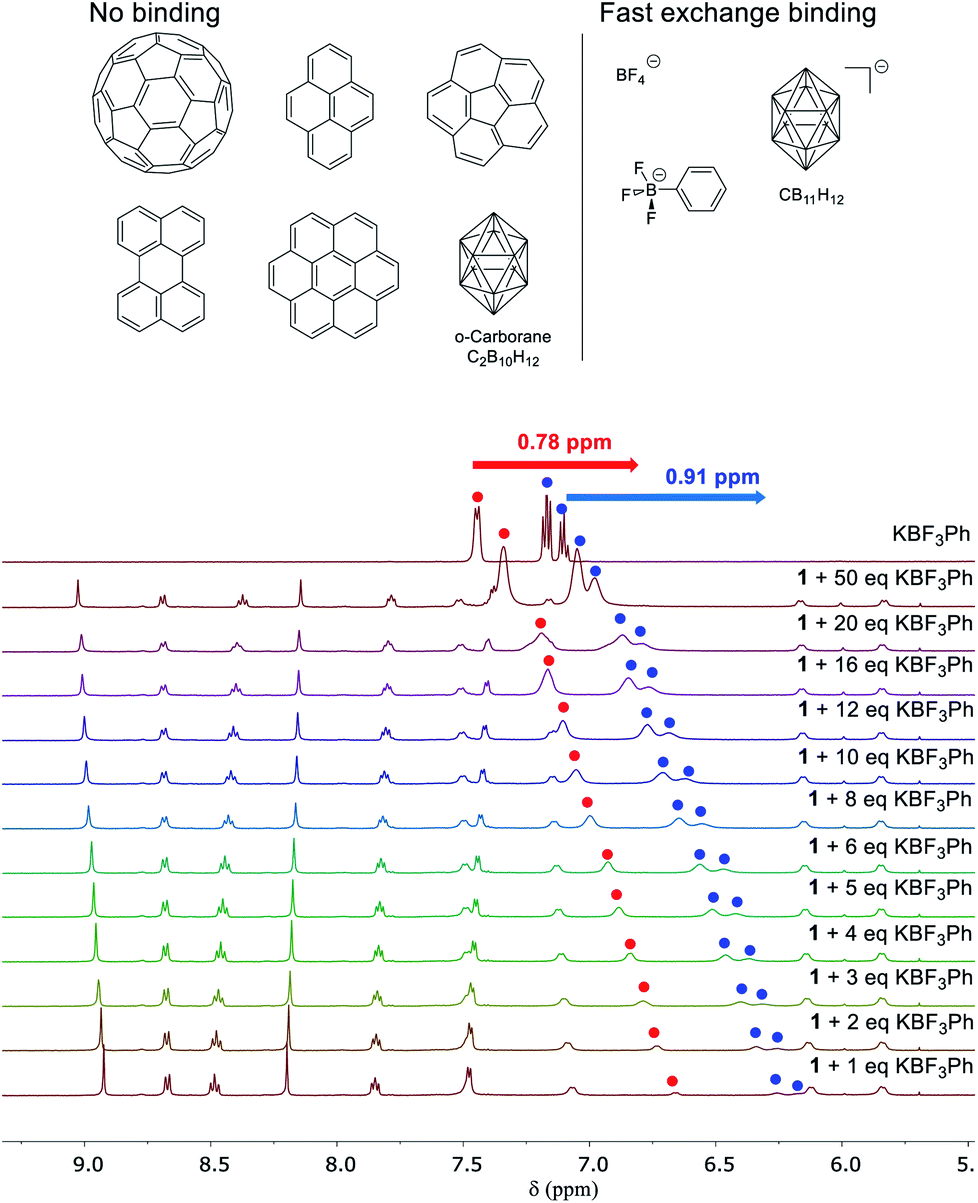 | ||
| Fig. 3 Top, neutral aromatic non-guests and anionic guests for cage 1; bottom, a stacked plot of the 1H NMR titration (500 MHz, 298 K) of KBF3Ph into a solution of 1 (0.17 mM) in CD3CN. The signals from the guest (KBF3Ph) have been labelled with red and blue dots, and the others belong to cage 1. | ||
Only the anions BF4−, PhBF3− and CB11H12− were observed to bind in fast exchange on the NMR chemical-shift timescale (Fig. 3 and S33–S40†). For instance, the 1H NMR signals of PhBF3− were shifted upfield by up to 0.91 ppm in the presence of 1. Similar chemical shift changes were also observed in the 19F NMR spectrum of this anion (Fig. S37†); host signals were also observed to shift in the presence of guests. However, the binding stoichiometries of these anions in cage 1 could not be established due to their fast exchange binding, and attempts to crystallize these host–guest adducts were also not successful. The 19F NMR signal of triflimide shifted upon the addition of KBF3Ph, suggesting that the encapsulated Tf2N− anions were released in the presence of the competing guest PhBF3− (Fig. S36†). We infer the cationic nature of subcomponent A to impart the cage with a higher binding affinity for anionic guests relative to neutral guests.
PhBF3− was chosen as a model guest to probe the binding behaviour of 1 under redox control. During the stepwise addition of Cp2Co, the 1H NMR signals of PhBF3− gradually shifted downfield, towards the values for the free guest (Fig. 4).
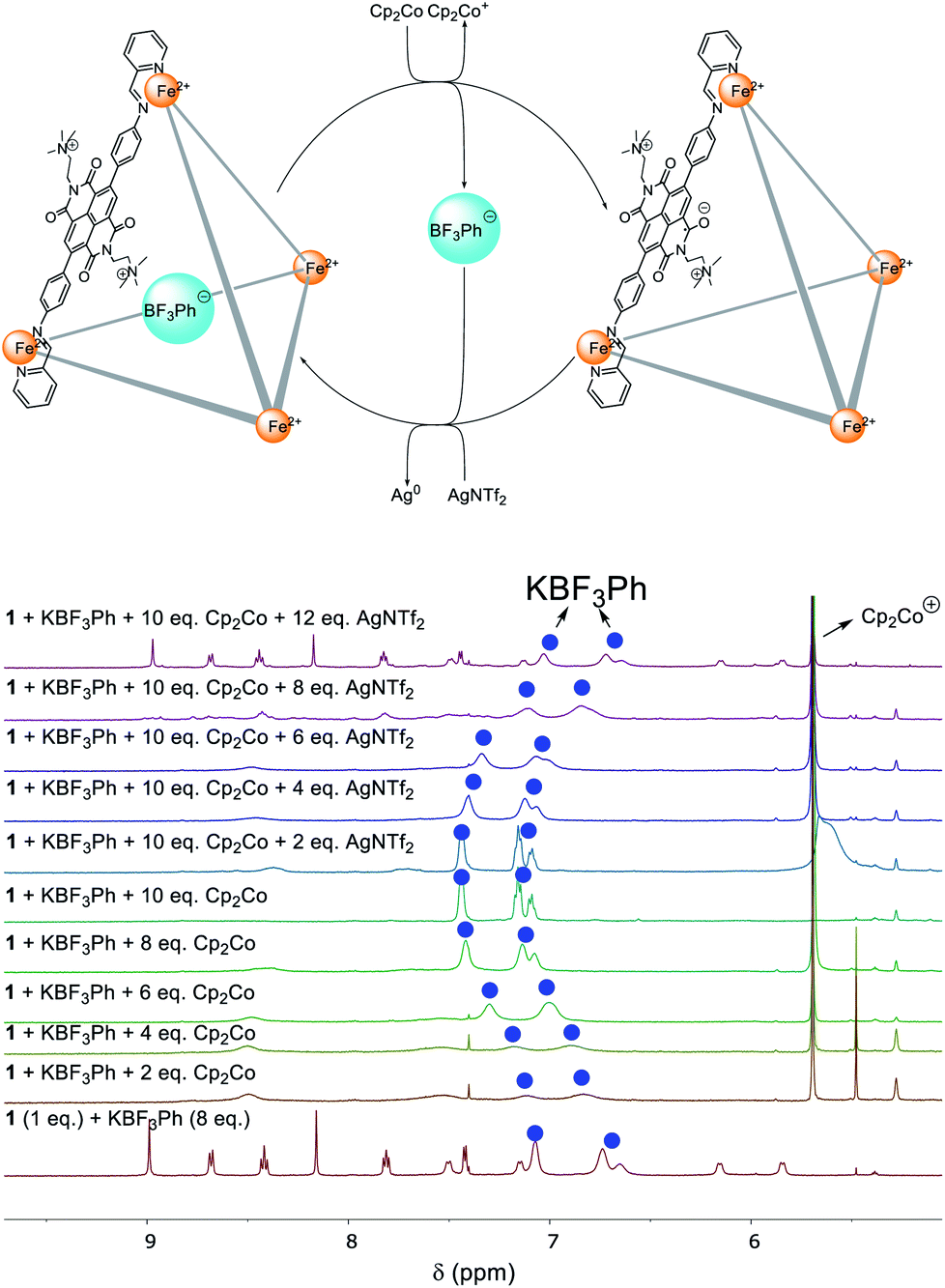 | ||
| Fig. 4 Redox control of KBF3Ph binding within 1. | ||
We infer the reduction of the NDI panels of 1 to result in repulsion between the anionic guest and the reduced cage panels, leading to release of the bound guests. The signals of 1 broadened into the baseline during the reduction process due to the formation of radical anion species. After the addition of 10 equivalents of Cp2Co, the 1H NMR signals of PhBF3− were found at the same chemical shift values as the free anion, suggesting complete ejection of the guest from the cage cavity. Full recovery of the 1H NMR spectrum of PhBF3− ⊂ 1 was observed after the addition of AgNTf2 (12 equiv.).
We next investigated the use of electron transfer to control the uptake and release and of neutral molecules. Although cage 1 possesses a +20 charge, which favours the binding of anions, we reasoned that reduction of the NDI panels with Cp2Co would partially neutralize the charge and potentially modify the guest preference.
The X-ray structure of 1 (Fig. 1) suggested that cage 1 would have a suitable volume (Fig. S26†) to accommodate C60, and analogous cages have been shown to bind fullerenes well.14,18 C60 thus appeared to be an ideal guest molecule to test the catch-and-release cycle shown in Fig. 5. This cycle is inferred to have three distinct stages. First, after the addition of Cp2Co, reduced cage 1 released the anionic guest in favour of neutral C60. Second, treatment with AgNTf2 oxidized the cage back to its initial state, giving C60 ⊂ 1 as a kinetically-trapped species. Third, the thermodynamically-unfavourable C60 ⊂ 1 released neutral C60, generating the more stable triflimide adduct.
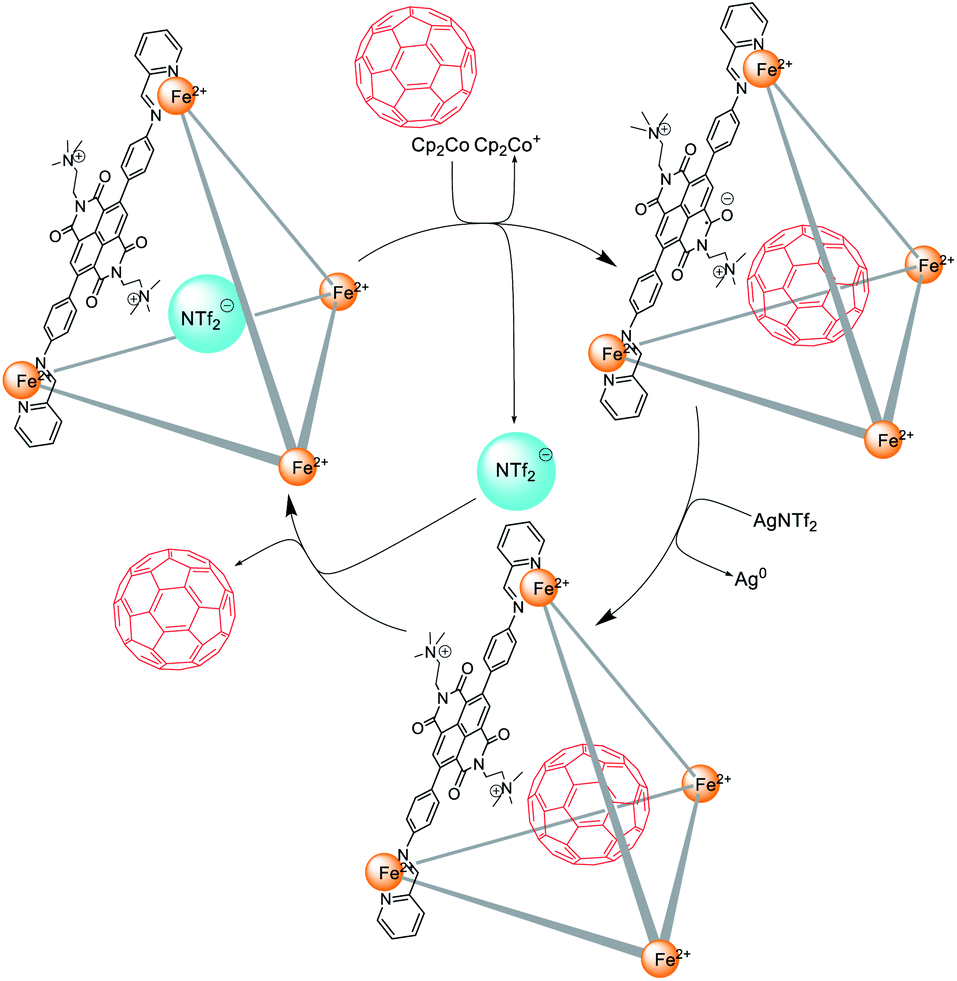 | ||
| Fig. 5 The encapsulation and release of C60 within cage 1 following reduction and subsequent re-oxidation. | ||
To test the cycle of Fig. 5, a solution of reduced cage 1, prepared through addition of Cp2Co (10 equiv.) to 1 in CD3CN, was mixed with C60 (4 equiv.), and the mixture was kept at room temperature overnight. AgNTf2 (12 equiv.) was added to oxidize the cage back to its initial state. The presence of C60 was confirmed by 13C NMR (Fig. S15 and S21†) in CD3CN, a solvent in which free C60 displays negligible solubility.19
Cage 1 exhibited T point symmetry in solution, as reflected in its 1H NMR spectra,20 however, the encapsulation of C60 within 1 resulted in the formation of diastereomers having all possible combinations of Δ and Λ metal stereochemistries with T, S4, and C3 point symmetries (Fig. S13†).13d,20 Interestingly, the C60 ⊂ 1 complex was found to be a kinetically trapped species. After 5 days the S4 and C3 diastereomers20 (Fig. S19†) of C60 ⊂ 1 released their encapsulated C60 with concomitant formation of free 1 and a reduction in the intensity of 13C NMR signal for C60 (Fig. S21†). Only the T diastereomer20 of C60 ⊂ 1 (14% by 1H NMR integration) remained in solution after 30 days, indicating its greater kinetic stability relative to the other diastereomers of C60 ⊂ 1 (Fig. 6).
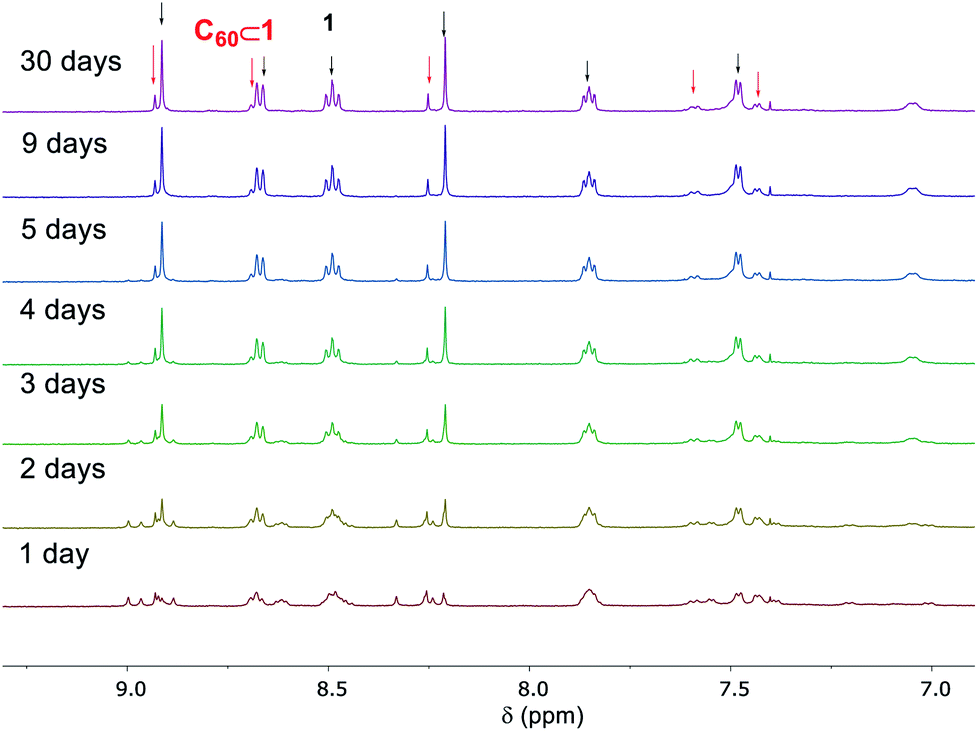 | ||
| Fig. 6 The release of C60 from C60 ⊂ 1 monitored by 1H NMR (500 MHz, CD3CN) over the course of a month. | ||
Counteranions have been shown to drive the phase transfer of cationic coordination cages, permitting guests to be conveyed across phase boundaries.1a,1c The treatment of a solution of cage 1 in MeCN/EtOAc (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with aqueous Na2SO4 resulted in transfer of 1 from the organic phase into water (Fig. S42 and S43†) as the sulfate salt.1a Treatment with aqueous Na2SO4 likewise stimulated phase transfer of the C60 ⊂ 1 host–guest complex. This complex, however, was observed to release C60 upon phase transfer, allowing cargo recovery by filtration (Fig. 7, S44 and S45†). This novel use of phase transfer to effect guest ejection could enable new means of chemical purification, whereby a guest is separated from its recyclable host in a single step, rather than requiring a separate purification step.21
:1) with aqueous Na2SO4 resulted in transfer of 1 from the organic phase into water (Fig. S42 and S43†) as the sulfate salt.1a Treatment with aqueous Na2SO4 likewise stimulated phase transfer of the C60 ⊂ 1 host–guest complex. This complex, however, was observed to release C60 upon phase transfer, allowing cargo recovery by filtration (Fig. 7, S44 and S45†). This novel use of phase transfer to effect guest ejection could enable new means of chemical purification, whereby a guest is separated from its recyclable host in a single step, rather than requiring a separate purification step.21
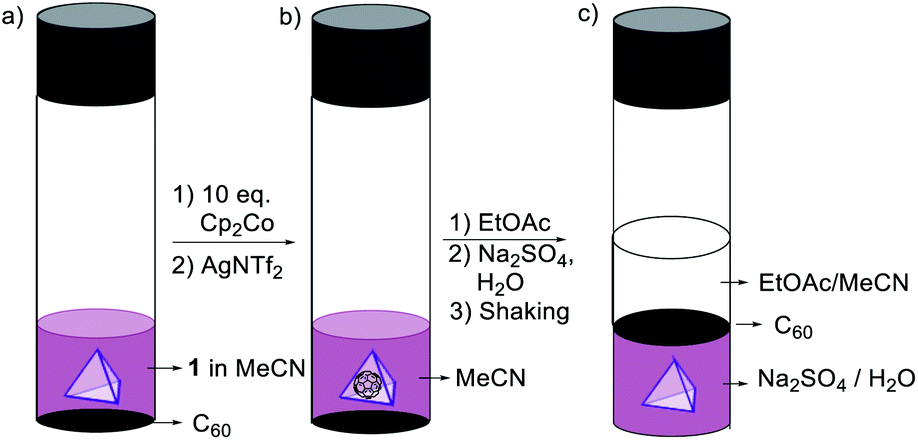 | ||
| Fig. 7 (a) Mixture of cage 1 and C60 in MeCN. (b) Formation of C60 ⊂ 1. (c) The release of C60 and transfer of cage 1 from the EtOAc/MeCN to the water layer. | ||
Conclusions
In this study, we reported a new redox-switchable FeII4L6 tetrahedral cage. Although, the cage shows binding affinity for anionic guests, following reduction the cage was observed to encapsulate neutral C60 with ejection of the anionic guests. Current efforts are focused on expanding redox-stimulated guest uptake and release to a broader set of guest species, and applying these concepts to design new systems where electrical energy may be used directly to effect chemical separations and transport.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (695009) and the UK Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1 and EP/M008258/1). Z. L. thanks the Deutsche Forschungsgemeinschaft (DFG) for a Postdoctoral fellowship. We thank Diamond Light Source (UK) for synchrotron beamtime on I19 (MT15768).Notes and references
- (a) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS; (b) J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS PubMed; (c) D. Samanta, S. Shanmugaraju, S. A. Joshi, Y. P. Patil, M. Nethajia and P. S. Mukherjee, Chem. Commun., 2012, 48, 2298–2300 RSC.
- (a) A. B. Grommet and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 2176–2179 CrossRef CAS PubMed; (b) B. S. Pilgrim, D. A. Roberts, T. G. Lohr, T. K. Ronson and J. R. Nitschke, Nat. Chem., 2017, 9, 1276–1281 CrossRef CAS; (c) A. B. Grommet, J. B. Hoffman, E. G. Percastegui, J. Mosquera, D. J. Howe, J. L. Bolliger and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 14770–14776 CrossRef CAS PubMed; (d) J. Lee, C. Y. Chuah, J. Kim, Y. Kim, N. Ko, Y. Seo, K. Kim, T. H. Bae and E. Lee, Angew. Chem., Int. Ed., 2018, 57, 7869–7873 CrossRef CAS PubMed; (e) B. Roy, A. Kumar Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916–11919 CrossRef CAS PubMed.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (b) P. L. Manna, C. Talotta, G. Floresta, M. De Rosa, A. Soriente, A. Rescifina, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2018, 57, 5423–5428 CrossRef PubMed; (c) D. Preston, J. J. Sutton, K. C. Gordon and J. D. Crowley, Angew. Chem., Int. Ed., 2018, 57, 8659–8663 CrossRef CAS PubMed; (d) W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821–2828 CrossRef CAS PubMed.
- (a) P. D. Frischmann, V. Kunz and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 7285–7289 CrossRef CAS PubMed; (b) S. Shanmugaraju and P. S. Mukherjee, Chem.–Eur. J., 2015, 21, 6656–6666 CrossRef CAS PubMed; (c) Q. Q. Wang, V. W. Day and K. Bowman-James, J. Am. Chem. Soc., 2013, 135, 392–399 CrossRef CAS PubMed.
- (a) M. Yoshizawa, Y. Takeyama, T. Kusukawa and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 1347–1349 CrossRef CAS; (b) M. Yoshizawa, S. Miyagi, M. Kawano, K. Ishiguro and M. Fujita, J. Am. Chem. Soc., 2004, 126, 9172–9173 CrossRef CAS PubMed; (c) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133–5136 CrossRef CAS PubMed; (d) N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- (a) J. H. Tang, Y. Sun, Z. L. Gong, Z. Y. Li, Z. Zhou, H. Wang, X. Li, M. L. Saha, Y. W. Zhong and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7723–7729 CrossRef CAS PubMed; (b) H. Wu, Y. Chen, L. Zhang, O. Anamimoghadam, D. Shen, Z. Liu, K. Cai, C. Pezzato, C. L. Stern, Y. Liu and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1280–1289 CrossRef CAS PubMed.
- (a) M. Han, R. Michel, B. He, Y. S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed; (b) S. E. Border, R. Z. Pavlovic, L. Zhiquan and J. D. Badjic, J. Am. Chem. Soc., 2017, 139, 18496–18499 CrossRef CAS PubMed; (c) A. J. McConnell, C. J. E. Haynes, A. B. Grommet, C. M. Aitchison, J. Guilleme, S. Mikutis and J. R. Nitschke, J. Am. Chem. Soc., 2018, 49, 16952–16956 CrossRef PubMed; (d) F. C. Parks, Y. Liu, S. R. Stutsman, S. Debnath, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed.
- (a) K. Kurihara, K. Yazaki, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2017, 56, 11360–11364 CrossRef CAS PubMed; (b) W. Cullen, K. A. Thomas, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 4025–4028 RSC; (c) X. Ji, Y. Yao, J. Li, X. Yan and F. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed; (d) I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC; (e) X. Su, S. Voskian, R. P. Hughes and I. Aprahamian, Angew. Chem., Int. Ed., 2013, 52, 10734–10739 CrossRef CAS PubMed.
- (a) A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 2342–2351 CrossRef CAS PubMed; (b) M. C. Young, L. R. Holloway, A. M. Johnson and R. J. Hooley, Angew. Chem., Int. Ed., 2014, 53, 9832–9836 CrossRef CAS PubMed.
- (a) T. Y. Kim, R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Eur. J., 2018, 24, 14878–14890 CrossRef CAS PubMed; (b) A. Goswami, S. Saha, P. K. Biswas, and M. Schmittel, Chem. Rev., DOI:10.1021/acs.chemrev.9b00159; (c) W. Wang, Y. Wang and H. B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (d) M. Schmittel, Chem. Commun., 2015, 51, 14956–14968 RSC.
- (a) G. Szalóki, V. Croué, V. Carré, F. Aubriet, O. Alévêque, E. Levillain, M. Allain, J. Aragó, E. Ortí, S. Goeb and M. Sallé, Angew. Chem., Int. Ed., 2017, 56, 16272–16276 CrossRef PubMed; (b) K. Yazaki, S. Noda, Y. Tanaka, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 15031–15034 CrossRef CAS PubMed; (c) V. Croué, S. Goeb, G. Szalóki, M. Allain and M. Sallé, Angew. Chem., Int. Ed., 2016, 55, 1746–1750 CrossRef PubMed; (d) C. Colomban, G. Szalóki, M. Allain, L. Gómez, S. Goeb, M. Sallé, M. Costas and X. Ribas, Chem.–Eur. J., 2017, 23, 3016–3022 CrossRef CAS PubMed; (e) Y. Satoh, L. Catti, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2019, 141, 12268–12273 CrossRef CAS PubMed; (f) K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS PubMed.
- M. A. Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef CAS PubMed.
- (a) Y. Wu, M. D. Krzyaniak, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 2948–2951 CrossRef CAS PubMed; (b) S. P. Black, D. M. Wood, F. B. Schwarz, T. K. Ronson, J. J. Holstein, A. R. Stefankiewicz, C. A. Schalley, J. K. M. Sanders and J. R. Nitschke, Chem. Sci., 2016, 7, 2614–2620 RSC; (c) S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed; (d) T. K. Ronson, D. A. Roberts, S. P. Black and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 14502–14512 CrossRef CAS PubMed.
- Z. Lu, R. Lavendomme, O. Burghaus and J. R. Nitschke, Angew. Chem., Int. Ed., 2019, 58, 9073–9077 CrossRef CAS PubMed.
- G. J. Kleywegt and T. A. Jones, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 178–185 CrossRef CAS PubMed.
- S. Suraru, C. Burschka and F. Würthner, J. Org. Chem., 2014, 79, 128–139 CrossRef CAS PubMed.
- G. Belanger-Chabot, A. Ali and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 9958–9961 CrossRef CAS PubMed.
- T. K. Ronson, W. Meng and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 9698–9707 CrossRef CAS PubMed.
- K. N. Semenov, N. A. Charykov, V. A. Keskinov, A. K. Piartman, A. A. Blokhin and A. A. Kopyrin, J. Chem. Eng. Data, 2010, 55, 13–36 CrossRef CAS.
- M4L6 cages are often found to present a mixture of three diastereomers, having distinct symmetries: homochiral T, with all metal centers having the same Δ or Λ configuration, heterochiral C3, with ΔΔΔΛ/ΛΔΔΔ configuration, and achiral S4, having ΔΔΛΛ metal center handednesses. See W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1935513. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05728e |
| This journal is © The Royal Society of Chemistry 2020 |