
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a high-efficiency, multivariate metal–organic framework phosphor for white LED bulbs†
William P.
Lustig
a,
Zeqing
Shen
a,
Simon J.
Teat
 b,
Nasir
Javed
a,
Ever
Velasco
a,
Deirdre M.
O'Carroll
a and
Jing
Li
*a
b,
Nasir
Javed
a,
Ever
Velasco
a,
Deirdre M.
O'Carroll
a and
Jing
Li
*a
aDepartment of Chemistry & Chemical Biology, Rutgers University, 123 Bevier Rd, Piscataway, NJ 08854, USA. E-mail: jingli@rutgers.edu
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 10th January 2020
Abstract
Developing rare-earth element (REE) free yellow phosphors that can be excited by 455 nm blue light will help to decrease the environmental impact of manufacturing energy efficient white light-emitting diodes (WLEDs), decrease their cost of production, and accelerate their adoption across the globe. Luminescent metal–organic frameworks (LMOFs) demonstrate strong potential for use as phosphor materials and have been investigated intensively in recent years. However, the majority are not suitable for the current WLED technology due to their lack of blue excitability. Therefore, designing highly efficient blue-excitable, yellow-emitting, REE free LMOFs is much needed. With an internal quantum yield of 76% at 455 nm excitation, LMOF-231 is the most efficient blue-excitable yellow-emitting LMOF phosphor reported to date. Spectroscopic studies suggest that this quantum yield could be further improved by narrowing the material's bandgap. Based on this information and guided by DFT calculations, we apply a ligand substitution strategy to produce a semi-fluorinated analogue of LMOF-231, LMOF-305. With an internal quantum yield of 88% (λem = 550 nm) under 455 nm excitation, this LMOF sets a new record for luminescent efficiency in yellow-emitting, blue-excitable, REE free LMOF phosphors. Temperature-dependent and polarized photoluminescence (PL) studies have provided insight on the mechanism of emission and origin of the significant PL enhancement.
1. Introduction
White LED (WLED) bulbs are a cost- and energy-efficient solid state lighting (SSL) technology that make up an increasing share of the global lighting market. The US Department of Energy estimates that WLEDs will comprise 84% of the US lighting market by 2030, reducing light energy consumption by 261 terawatt-hours per year, or 40% of the annual lighting energy consumption, which in turn accounts for approximately 6% of the total US energy consumption.1,2 Globally, lighting accounts for 15% of all energy used, and while government regulations will likely phase out inefficient incandescent lighting in some areas over the next several years, WLED bulbs must continue to compete with cheaper incandescent bulbs in much of the world.3 Decreasing the cost of WLED bulbs will speed their global adoption, resulting in significant energy savings. In addition, switching to phosphor materials that are more environmentally benign is equally important. One possible method to achieve these goals is through the development of low-cost and rare-earth-element (REE) free phosphors. Currently, the most cost-effective WLED design is comprised of a blue-emitting LED chip, coated with a blue-excitable REE based yellow phosphor or multicomponent phosphors; the combination of blue light from the chip and yellow light from the phosphor coating produces white light. This is known as a phosphor-converted WLED, or PC-WLED. The yellow phosphor used in commercial devices is mainly YAG:Ce—a cerium-doped yttrium aluminum garnet. However, cost and supply risks for yttrium and cerium may continue to rise due to the high and increasing demand of these REEs in many high-tech devices, and mining the REEs is environmentally very damaging.4,5 Developing new, REE free phosphor materials can both provide new cost flexibility to WLED bulbs and reduce the environmentally hazardous mining operations.Metal–organic frameworks (MOFs) are crystalline materials composed of metal ions linked into a framework by organic ligands with multiple binding sites. These framework structures are generally porous and possess highly tunable characteristics, and the properties of a given MOF depend not only on the properties of the metal and ligand molecules used to construct it, but also on the connectivity, topology, and interaction between framework elements that comprise the structure. Due to this tunability, MOFs have been explored for a wide variety of applications, including gas storage and separation, catalysis, medical imaging, drug delivery, sensing, toxin adsorption, and energy efficient lighting.6–17
Luminescent metal–organic frameworks (LMOFs) are one of the most promising candidate materials as new phosphors. Their luminescence can arise from a variety of mechanisms and interactions within the structure. Emission can be centered on ligand molecules, metal ions, occur via charge or energy transfer between different ligands or between ligands and metal ions, and involve guest molecules located within the porous framework.18 These various mechanisms provide multiple ways to influence luminescence in MOFs,19 and are part of the reason why LMOFs are such attractive phosphors. One effective strategy for designing an LMOF phosphor is through a chromophoric ligand approach, in which an organic chromophore with desirable emission properties is converted into a ligand molecule which can be built into a MOF.20 The inclusion of a ligand into a rigid framework can often improve quantum yield through immobilization/rigidification of the chromophore, which reduces or eliminates vibrational and rotational pathways leading to nonradiative exciton recombination.21,22
A good example of the effectiveness of this strategy is LMOF-231, or Zn2(tcbpe) (H4tcbpe = 1,1,2,2-tetrakis(4-(4-carboxy-phenyl)phenyl)ethene).21 The blue-excitable, yellow-emitting chromophore ligand H4tcbpe was designed by starting from the known AIE-active tetraphenylethene (tpe) core and using DFT calculations to as a screening method to identify molecules with the appropriate electronic structure for yellow emission. LMOF-231 boasts an impressive quantum yield of 76% under 455 nm blue excitation. However, despite being the highest quantum yield reported for an LMOF phosphor with properties appropriate for use in a PC-WLED, the commercial phosphor YAG:Ce has a higher quantum yield of 96% at the same excitation energy. And despite being reported in 2015, no further performance improvements have been made in LMOF phosphors for PC-WLEDs. More significant spectroscopic investigations of energy transfer and other optoelectronic processes in MOFs may provide the opportunity to deepen our understanding of luminescence mechanisms in these materials, and provide opportunities to further enhance performance.23–25
In this work, photoluminescence (PL) spectroscopy is used to probe the luminescence mechanism in LMOF-231, and DFT calculations are used to identify a secondary ligand with the appropriate electronic structure to improve the LMOF's photoluminescence quantum yield (PLQY). Based on the information from these spectroscopic and theoretical studies, a ligand substitution strategy is used to incorporate this secondary ligand into LMOF-231, creating LMOF-305, which sets a new record for LMOF based PC-WLED yellow phosphors with an internal quantum yield (IQY) of 88% under 455 nm excitation and an ideal emission peak of 550 nm, and polarized PL spectroscopy is used to validate the emission mechanism.
2. Experimental
2.1. Materials and synthesis
All materials were used as received from Sigma Aldrich. The chromophoric ligands H4tcbpe and H4tcbpe-F were synthesized using a previously reported method with some modifications (Scheme 1), as was LMOF-231.21,26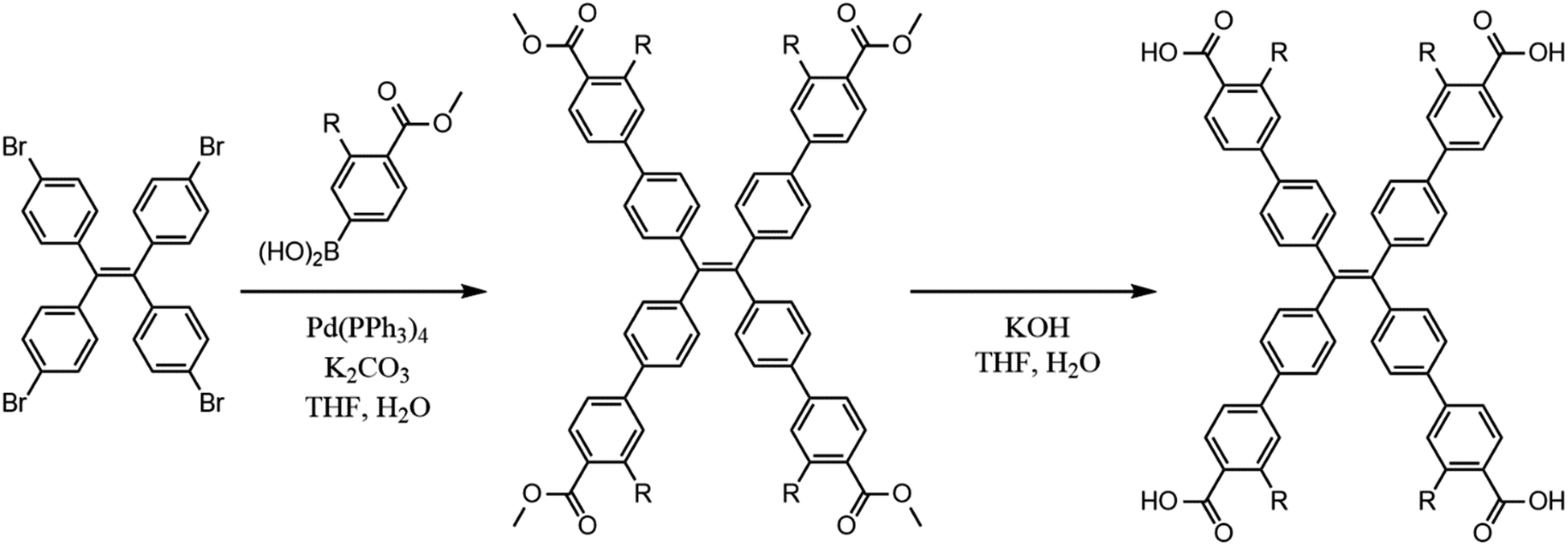 | ||
| Scheme 1 Synthesis of H4tcbpe (R = H) and H4tcbpe-F (R = F). | ||
LMOF-305 was synthesized solvothermally. H4tcbpe (0.0305 g, 0.0375 mmol), H4tcbpe-F (0.0332 g, 0.0375 mmol), and Zn(NO3)2·6H2O (0.149 g, 0.400 mmol) were dissolved in a glass vial with dimethylacetamide (DMA, 2 mL). The resulting solution was heated in a reaction oven at 120 °C for 24 hours, after which the product was collected via filtration as large, slightly yellow translucent crystals. Solvent exchange was performed by immersion in approximately 15 mL ethyl acetate for 24 hours, with the solvent being refreshed every two hours for the first eight hours, after which the sample was heated under vacuum at 50 °C overnight to give the outgassed product with approximately 58% yield with respect to H4tcbpe.
2.2 Single crystal structure determination
The selected crystal was mounted on MiTeGen® loops in Paratone oil on a Bruker D8 diffractometer equipped with a PHOTON100 CMOS detector and Oxford Cryosystems Cryostream 800 plus, on beamline 11.3.1 of the Advanced Light Source at LBNL. A sphere of data were collected at 100 K using Bruker APEX3 software27 in shutterless mode with ω rotations at fixed φ values at λ = 0.7749 Å, from a channel cut silicon [111] monochromator. The intensity data were integrated and corrections applied with SAINT v8.34a,28 absorption and other corrections were made using TWINABS 2012/1.29 Dispersion corrections appropriate for this wavelength were calculated using the Brennan method in XDIP with in WinGX.30 The structures were solved with a dual space method with SHELXT 2014/4 and refined using SHELXL 2014/7.31 Once the refinement had converged, SQUEEZE was used to mask the electron density in the pores.322.3 Physical characterizations
A Rigaku Ultima IV diffractometer was used to collect all powder X-ray diffraction (PXRD) data. Data collection was performed at room temperature using Cu Kα radiation (λ = 1.5406 Å), scanning across a 2θ range from 3° to 35° with a scan speed of 2° 2θ min−1 and a step size of 0.2°. A TA Instruments Q5000 was used to perform thermogravimetric analyses (TGA) of all samples. Samples were loaded into a Pt pan and heated under constant N2 flow (20 mL min−1) from 30 °C to 600 °C, with the temperature increasing at a constant rate of 10 °C min−1. SEM images were obtained using a Zeiss Sigma Field Emission SEM with Oxford INCA PentaFETx3 EDS system.2.4 Photoluminescence characterizations
Steady-state PL emission and excitation spectra were collected from dry powder samples using a Varian Cary Eclipse spectrophotometer at room temperature. Diffuse reflectance data were collected at room temperature using a Shimadzu UV-3600 spectrophotometer. Internal quantum yield was measured at room temperature for dry powder samples with a Hamamatsu C9220-03 spectrophotometer, using a 150 W Xenon monochromatic light source and integrating sphere. Polarized photoluminescence measurements were carried out in reflection mode of an inverted fluorescence microscope (Zeiss, AxioVert), using a 470 nm (±20 nm) excitation light and a 515 nm long-pass filter in the collection path. The excitation and emission passed thought the same microscope objective and a dichroic mirror was used to separate the excitation light from the emission light. Single crystals of LMOF-305 were positioned with their c-axis perpendicular to the excitation and collection directions. Polarizers were added to the excitation and collection paths to analyze the polarization of the emitted light from the crystal. Polarization bias in the equipment was quantified using an amorphous conjugated polymer sample to give an instrument correction factor. Subsequently, the polarized PL spectra from LMOF-305 crystals were corrected for polarization bias in the measurement using the instrument correction factor.2.5 Lifetime measurements
Temperature-dependent photoluminescence lifetime measurements were carried out in the solid state using powder samples that had been pressed into pellets on a cryostat (Cryo Industries of America, Inc., Model 6NSVT Variable Temperature Optical Top-Loading Nitrogen Cryostat) and temperature controller (Cryo-con 32B Temperature Controller). A pulsed laser excitation wavelength of 440 nm was employed with a laser repetition rate of 10 kHz or 20 kHz. The PL emission was collected from the sample by a convex lens and directed through a 550 ± 44 nm band-pass filter to an APD detector for luminescence decay measurements. The PL lifetime decay data were recorded using the time-correlated single photon counting method via a TimeHarp 260 Nano (PicoQuant) with down to 1 ns resolution. The PL lifetimes of the samples at different temperatures were extracted by fitting the PL decay curves with second-order exponential decay functions.2.6 Density functional theory (DFT) calculations
DFT calculations were performed on Gaussian 09, using the B3LYP3 hybrid functional and DGDZVP basis set.33–38 The geometries of all ligand molecules were optimized, with a frequency calculation performed after geometry optimization to confirm that the calculations resulted in a true minimum.3. Results and discussion
3.1 Structure and luminescence mechanism of LMOF-231
LMOF-231 or Zn2(tcbpe) is composed of a zinc-carboxylate chain forming an infinite secondary building unit running in the c direction, with each tcbpe4− ligand linking four of these chains linked together. The Zn2+ ions display a distorted tetrahedral geometry, with each ion bound to oxygen from four different ligand molecules, and the ligand molecules organized into close-packed columns running in the c direction (Fig. 1).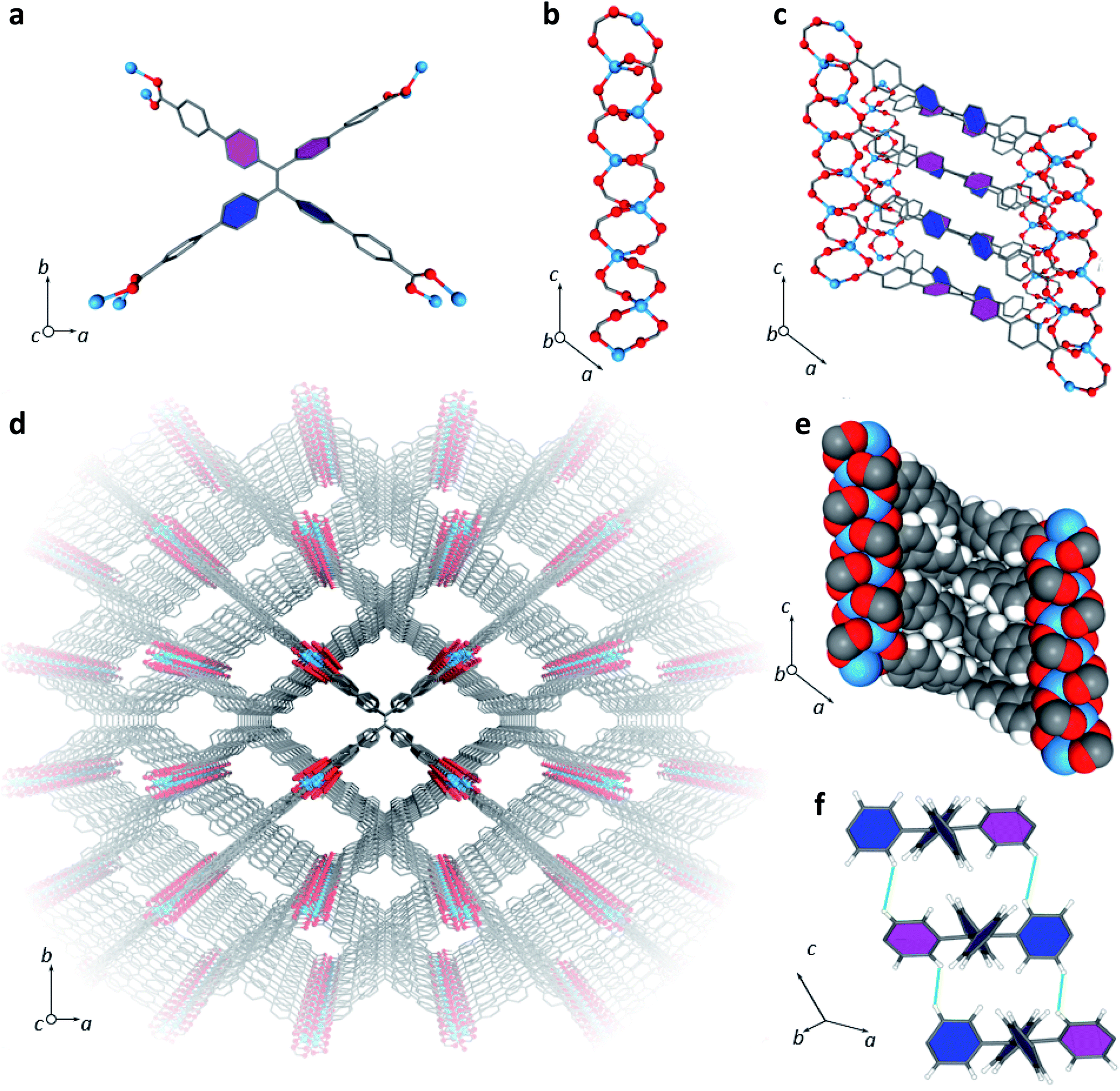 | ||
| Fig. 1 (a) A single tcbpe molecule coordinated to eight Zn2+ ions, with the pink-colored phenyl rings having a dihedral angle of 42.2° with respect to the central ethene, and the blue colored phenyl rings having dihedral angles of 58.7° with respect to the central ethene. (b) A segment of LMOF-231's infinite secondary building unit, which runs parallel to the c axis. (c) A section of LMOF-231 viewed along the b axis, showing a single column of tcbpe ligands linking four zinc-carboxylate chains parallel to the c axis. Pink-colored phenyl rings indicate an ethene-phenyl dihedral angle of 42.2°, while blue-colored phenyl rings indicate an ethene-phenyl dihedral angle of 58.7°. (d) The complete structure of LMOF-231 viewed along the c axis. (e) The structure of LMOF-231 viewed along the b axis using a space-filling model to illustrate the tight packing of the ligand molecules. (f) The central tetraphenylethene cores from three neighboring tcbpe ligands within a column, illustrating the edge-face-edge arrangement, with green lines showing close H–H interactions. Pink-colored phenyl rings indicate an ethene-phenyl dihedral angle of 42.2°, while blue-colored phenyl rings indicate an ethene-phenyl dihedral angle of 58.7°. Hydrogen have been omitted from Fig. 1a, c, and d for clarity (Zn = blue, O = red, C = grey, H = white). | ||
Within each ligand, the dihedral angles of the four phenyl rings connected to the ligand's central ethenyl moiety vary. Two phenyl rings bonded to one of the ethenyl carbons have dihedral angles of 42.2° relative to the central ethene, while the two phenyl rings bonded to the other ethenyl carbon have dihedral angles of 58.7° (Fig. 1). Each ligand is symmetrically related to its neighbors above and below it in the column through an inversion center; this results in the neighboring phenyl rings on different ligands being arranged in an edge-face-edge fashion, with close H–H interactions (atom–atom distances of approximately 2.4 Å) preventing significant rotation of the phenyl rings in the structure (Fig. 1). Additionally, DFT calculations indicated that the lowest energy configuration of a single ligand molecule has ethene-phenyl dihedral angles of 49.4°. The observed displacement from that value supports the identification of rigidifying H–H interactions.
The average layer spacing between ligands within these stacks, determined by simplifying the tcbpe ligand into a 2D rectangular surface with its corners defined by the four carboxylate carbons and measuring the distance separating these surfaces, is 5.4 Å. And since the ligands are arranged in a tilted fashion, the closest centroid–centroid distance between phenyl rings in neighboring ligands is 5.93 Å, which is distant enough to prevent significant pi–pi interactions that could lead to quenching.39
LMOF-231 emits yellow light at 550 nm under both blue and UV excitation, with an IQY of 96% and 76% under 420 and 455 nm excitation, respectively.40 Given the scale of Stokes shift, it is apparent that the absorbance of both 420 and 455 nm photons excites an electron from the ground state to a higher energy state than the state involved in emission, after which the electron rapidly relaxes to the emissive state. As the emission energy (Fig. S2†) is independent of the excitation wavelength, it is clear that electrons excited by both 420 nm and 455 nm relax to the same state before emitting (Fig. 2).
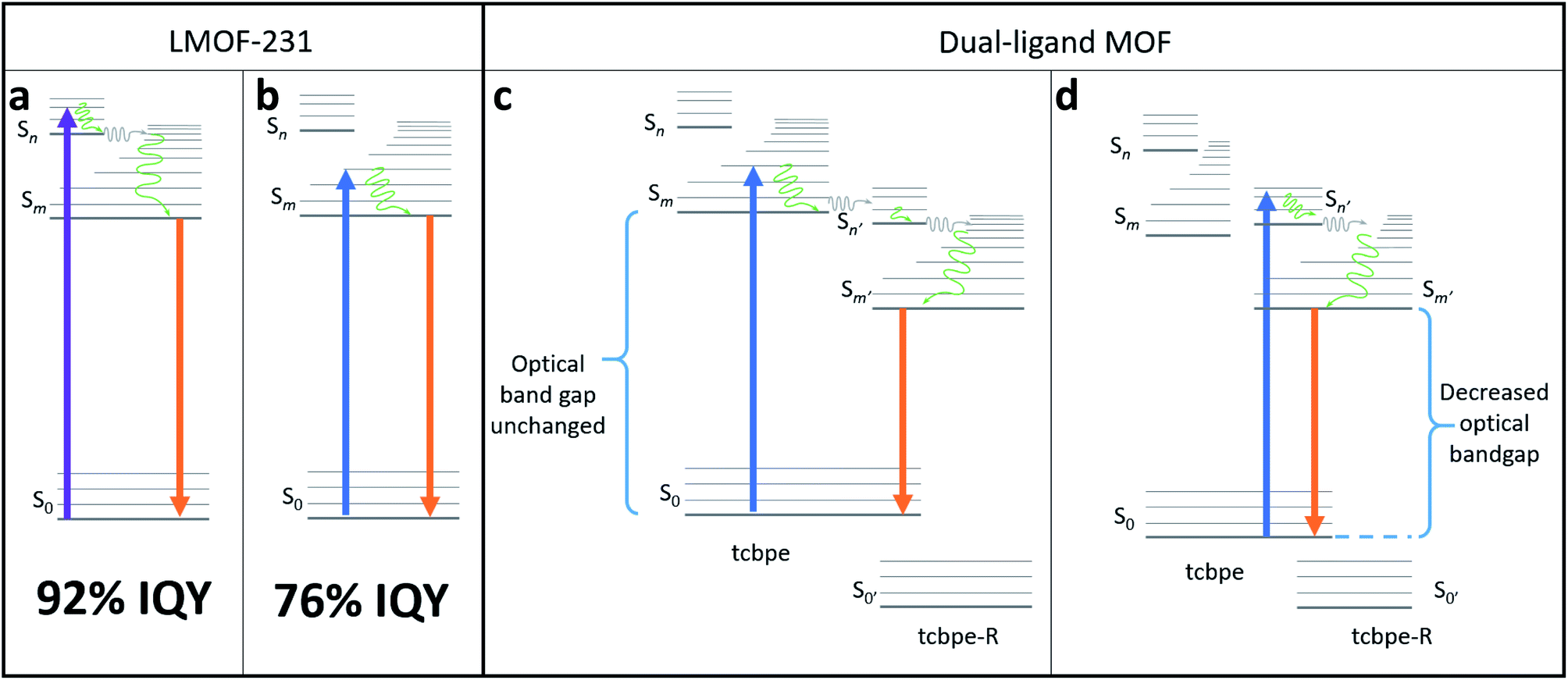 | ||
| Fig. 2 (a) Schematic illustrating a possible fluorescence mechanism for the more efficient “higher-energy pathway” in LMOF-231 following absorbance of a 420 nm photon. (b) Schematic illustrating a possible fluorescence mechanism for the less efficient “lower-energy pathway” in LMOF-231 following absorbance of a 455 nm photon. (c) Schematic demonstrating a possible fluorescence mechanism in a dual-ligand MOF composed of both tcbpe and a functionalized tcbpe with an offset HOMO–LUMO, in which absorbance on a 455 nm photon on one ligand is followed by electron transfer to a neighboring functionalized tcbpe ligand with lower-lying HOMO/LUMO energy levels. (d) Schematic demonstrating a possible fluorescence mechanism in a dual-ligand MOF composed of both tcbpe and a functionalized tcbpe with an offset HOMO–LUMO, in which absorbance on a 455 nm photon results in direct excitation from the non-functionalized ligand (tcbpe) to the functionalized ligand, injecting an excited electron directly in to the “higher-energy pathway”. | ||
The only clear difference between excitation at 420 nm and excitation at 455 nm is the energy level of the orbital that the excited electron resides immediately after absorbance of the photon, which is higher for the 420 nm photon. However, the fact that absorbance of the 420 nm photon resulted in emission with a higher quantum yield suggests that the “higher energy pathway” through which it decays is more efficient than the “lower energy pathway” available to the 455 nm photon (Fig. 2).
There are slight differences between the photoluminescent lifetimes of LMOF-231 under 380 nm (UV) and 440 nm (blue) excitation, supporting the existence of these two pathways (Table 1). The average amplitude weighted lifetime for LMOF-231 is approximately 4.14 ns under UV excitation and 3.81 ns under blue excitation—with the total decay being the sum of two processes (τ1 and τ2). However, under UV excitation, the faster τ1 process is more significant, while the slower τ2 dominates under blue excitation. Additionally, both τ1 and τ2 are approximately 1 ns slower under UV excitation than under blue excitation.
| Excitation | Average amplitude weighted τ | τ 1 (ns) | τ 2 (ns) |
|---|---|---|---|
| UV | 4.14 ns | 3.02 (56.3%) | 5.59 (43.7%) |
| Blue | 3.81 ns | 1.98 (35.6%) | 4.80 (64.4%) |
This suggests that the quantum yield of LMOF-231 under 455 nm excitation could potentially be improved by altering the material's electronic structure such that absorbance of a 455 nm photon activates the “higher energy pathway”, mimicking absorbance of a 420 nm photon. One possible way to accomplish this is through an adapted bandgap modulation approach,41 in which a functionalized tcbpe ligand with an offset HOMO–LUMO energy gap relative to the non-functionalized ligand is introduced into the LMOF as a secondary ligand to create a new dual-ligand MOF. This could achieve the desired change in one of two ways. If the ligand with higher-lying HOMO–LUMO energy levels is excited by a 455 nm photon, that excited electron could be transferred to the ligand with the lower-lying HOMO–LUMO energy levels at a higher energy state than was possible in the original absorbing ligand (Fig. 2c). Alternatively, absorbance of a photon may directly excite an electron between the primary and secondary ligands, with the offset HOMO–LUMO gaps of the two permitting the 455 nm photon to directly excite the electron into the “higher energy pathway” (Fig. 2d). As the hole will likely remain located in the higher-lying HOMO of the originally excited ligand, emission would need to occur between the two ligands. Experimentally, these two mechanisms can be distinguished by their optical bandgaps; for the first mechanism, the optical bandgap will be identical to native LMOF-231, while in the second mechanism, a decreased optical bandgap would be observed for the dual-ligand MOF. In either case, this interligand emission (Fig. 2c and d) would likely occur simultaneously with intraligand emission from both the primary and secondary ligands (Fig. 2b).
However, for bandgap modulation with a functionalized tcbpe as a secondary ligand to be effective, the luminescence process in the modulated LMOF must involve electron or energy transfer between the primary and secondary ligands. Given the close spacing between neighboring tcbpe ligands within LMOF-231 and the electron-rich aromatic core of the ligand, it was hypothesized that the luminescence process may involve interactions between neighboring ligands. This suggests that bandgap modulation would be an effective strategy, as long as the functionalized tcbpe ligand replace some of the non-functionalized tcbpe ligands without disturbing the overall framework structure.
3.2 Development of secondary ligand
Density Functional Theory (DFT) calculations were used as a screening method to identify the functionalized tcbpe-analogue with the appropriate electronic structure, and the version of tcbpe fluorinated at the ortho position relative to the carboxylate was found to meet the design requirements. Calculations indicated that this ligand, 1,1,2,2-tetrakis(4-(3-fluoro-4-carboxy-phenyl)phenyl)ethane or H4tcbpe-F, possessed HOMO and LUMO energy levels that are approximately 0.23 and 0.22 eV lower than those of H4tcbpe, respectively. This is ideal, as the offset energy levels in combination with a multi-ligand excitation process should allow the ligand to function as a bandgap modulator for excitation, while the nearly identical HOMO–LUMO energy gap should limit changes to the emission wavelength.Following synthesis, spectroscopic study indicated that the electronic properties of the fluorine-functionalized ligand were consistent with the relevant calculations. Diffuse reflectance data indicated that the two ligands had nearly identical optical bandgaps, and their excitation and emission spectra were nearly identical as well (Fig. 3).
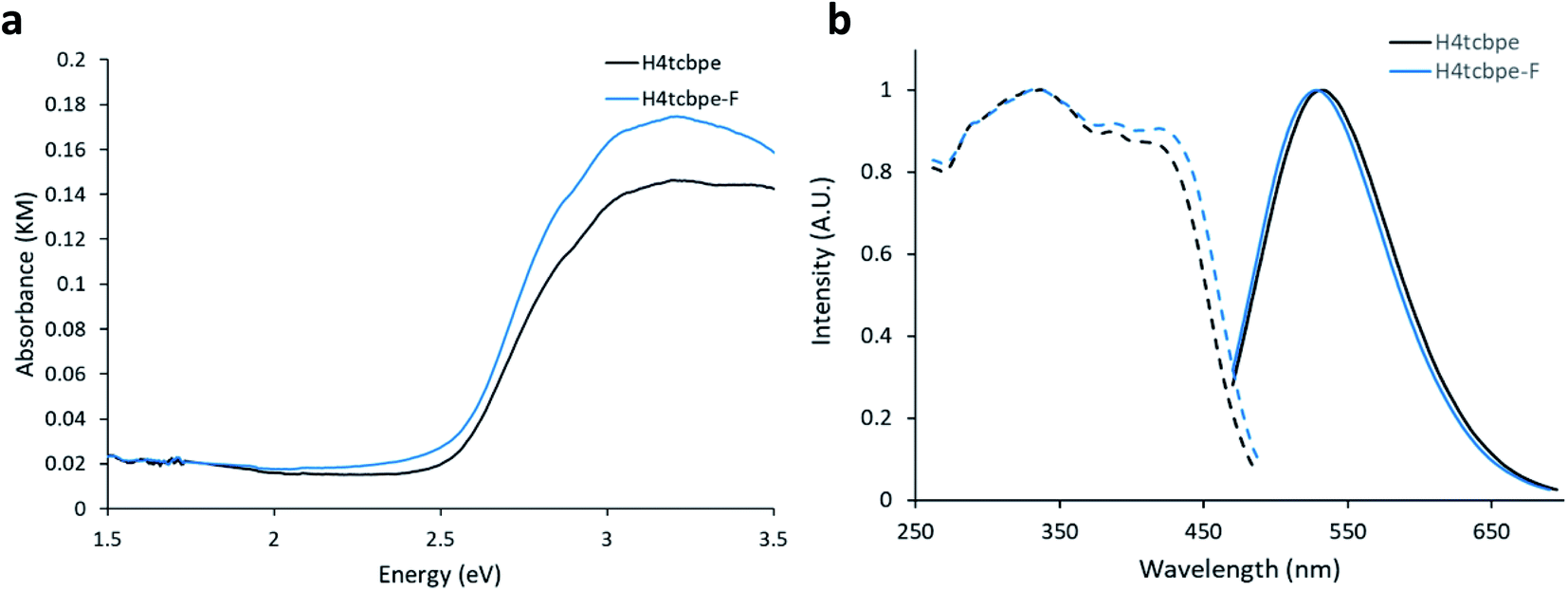 | ||
| Fig. 3 (a) Absorbance-analogue Kubelka–Munk function for H4tcbpe and H4tcbpe-F. (b) Excitation (dotted line) spectra for H4tcbpe and H4tcbpe-F at 550 nm emission, and emission spectra (solid line) for H4tcbpe and H4tcbpe-F under 455 nm excitation. | ||
3.3 Structure of LMOF-305
The multivariate LMOF-305 or Zn2(tcbpe)0.8(tcbpe-F)0.2 was synthesized solvothermally with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio for H4tcbpe and H4tcbpe-F. The resulting single crystals grew up to 2 mm long, in bloom-like bunches of rectangular crystals. Both PXRD and single crystal analysis confirmed that inclusion of the tcbpe-F ligand did not alter the crystal structure or phase of the LMOF. As shown in Table S1,† LMOF-305 crystallizes in the same space group as that of LMOF-231 with the same metal and ligand arrangement, except the distribution of ligand, where 20% of the tcbpe in LMOF-231 (Fig. 1) is replaced by tcbpe-F in LMOF-305. To determine stability under storage conditions, PXRD and QY data was measured on a sample of LMOF-305 that had been stored under air in ambient conditions for two months; the results are summarized in ESI Section S3.†
:1 molar ratio for H4tcbpe and H4tcbpe-F. The resulting single crystals grew up to 2 mm long, in bloom-like bunches of rectangular crystals. Both PXRD and single crystal analysis confirmed that inclusion of the tcbpe-F ligand did not alter the crystal structure or phase of the LMOF. As shown in Table S1,† LMOF-305 crystallizes in the same space group as that of LMOF-231 with the same metal and ligand arrangement, except the distribution of ligand, where 20% of the tcbpe in LMOF-231 (Fig. 1) is replaced by tcbpe-F in LMOF-305. To determine stability under storage conditions, PXRD and QY data was measured on a sample of LMOF-305 that had been stored under air in ambient conditions for two months; the results are summarized in ESI Section S3.†
Fragments of multiple crystals were analyzed using SEM-EDS to ensure that the distribution of tcbpe-F within the LMOF was homogenous (Fig. 4). The elemental analysis data confirmed that, although the synthetic ratio between H4tcbpe and H4tcbpe-F was 50:50, the resulting LMOF-305 crystal was composed of approximately 80% H4tcbpe and 20% H4tcbpe-F. This was consistent with the single crystal data, which yielded a 20% occupancy of F atoms at the relevant crystallographic site.
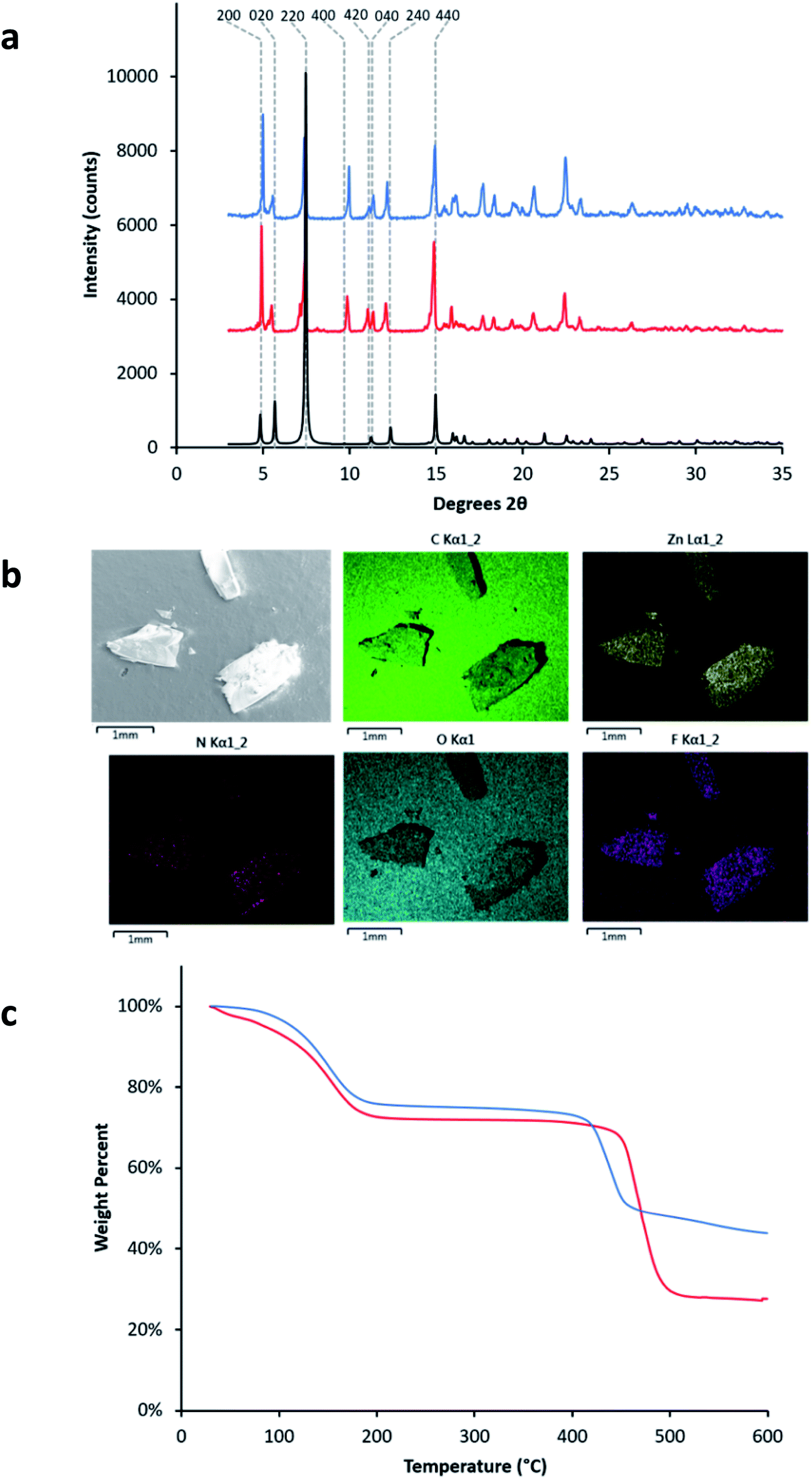 | ||
| Fig. 4 (a) PXRD patterns of LMOF-231 (red) and LMOF-305 (blue) overlaid with the simulated PXRD pattern of LMOF-231 (black). (b) SEM-EDS images of fragments from three different LMOF-305 crystals, mounted on graphite tape, showing the distribution of C, Zn, N, O, and F atoms within the samples. (c) Thermogravimetric (TG) curves for LMOF-231 (red) and LMOF-305 (blue). | ||
Despite the relatively low loading of the fluorinated ligand into the LMOF, the replacement of 20% of the tcbpe ligands with tcbpe-F leads to a structure in which approximately 50% of the remaining non-functionalized tcbpe ligands have one neighboring tcbpe-F ligand either above or below within the c-oriented ligand stacks. This assumes that two tcbpe-F ligands rarely neighbor each other, which is reasonable given the low functionalized ligand loading level and the fact that attempts to further increase the percentage of H4tcbpe-F within the structure were not successful, which is likely due to steric crowding around the zinc-carboxylate chain in the presence of multiple fluorinated ligands. This is consistent with the thermal stability of LMOF-305 as measured by TGA, which was slightly reduced relative to LMOF-231 and would be expected in the event that the inclusion of the fluorine increased steric strain near the zinc-carboxylate building unit (Fig. 4c). TG analysis also indicated that less solvent was present within the LMOF pore, which is consistent with the presence of fluorine slightly decreasing the available space (Fig. 4c).
3.4 Optical properties of LMOF-305
Following the successful inclusion of H4tcbpe-F into the Zn2(tcbpe) framework at 20% occupancy to give LMOF-305, the material's optical bandgap decreased by approximately 0.3 eV based on diffuse reflectance measurement, and the absorption intensity at 455 nm increased significantly (Fig. 5a). The excitation spectrum similarly redshifted to increase coverage at 455 nm, while changes to the emission spectrum were minor, as desired (Fig. 5b). Most importantly, the quantum yield of LMOF 305 under 455 nm excitation increased to 88%, a 12% increase over that of LMOF-231 (76%) under the same excitation energy.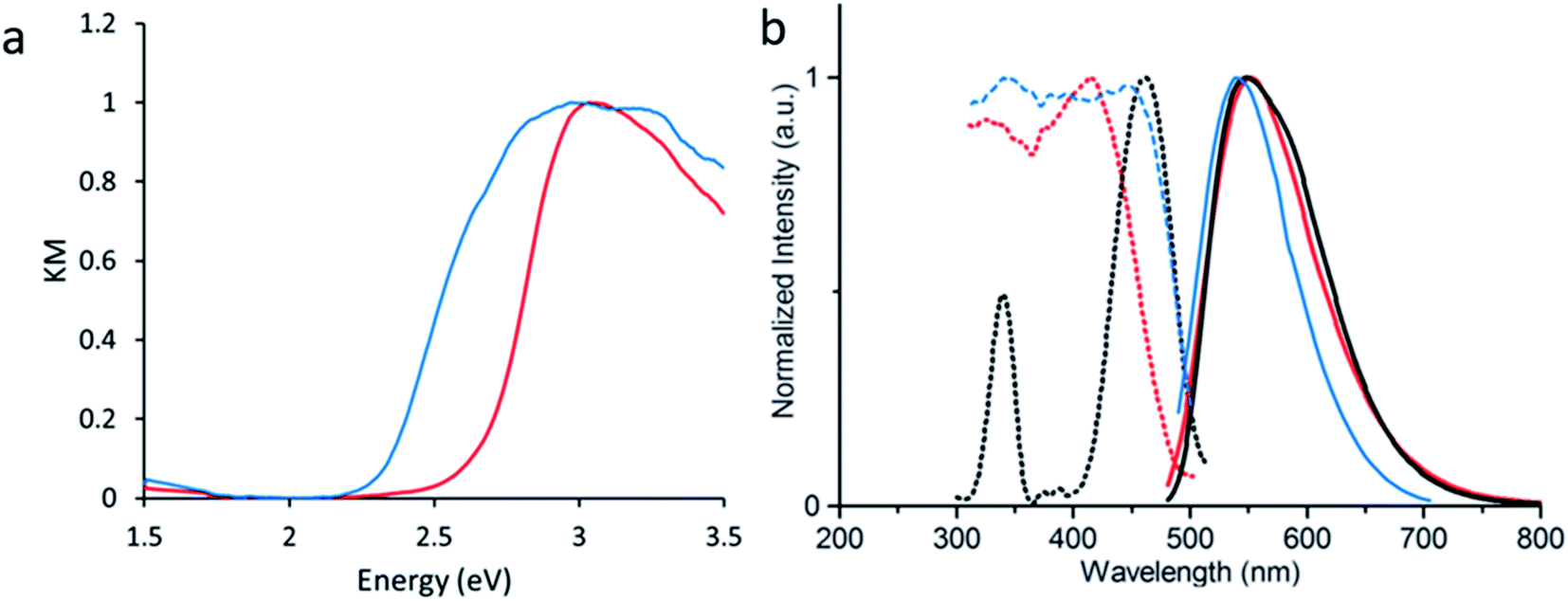 | ||
| Fig. 5 (a) UV-vis absorbance analogue Kubelka–Munk function derived from diffuse reflectance for LMOF-231 (red) and LMOF-305 (blue). (b) Excitation (dotted) and emission (solid) spectra for LMOF-231 (red), LMOF-305 (blue), and the commercial phosphor YAG:Ce (black). Excitation spectra were monitored at 550 nm emission, and emission spectra were collected under 455 nm excitation. | ||
Given that the emission is almost the same for the two LMOFs yet the Stokes shift is much smaller for the dual-ligand LMOF-305 (Fig. 5b), the density of electronic states at energies near the lowest tcbpe LUMO state must be enhanced either due to reduction in motion of the ligands, which is consistent with the expected steric influence of tcbpe-F on the framework SBU as discussed previously, or due to additional tcbpe-F states that are very close in energy to the tcbpe LUMO.
In order to demonstrate the effect of tcbpe-F on the PL of LMOF-305, three additional samples were prepared using tcbpe:tcbpe-F molar ratios of 85:15, 90:10, and 95:5, and their quantum yields were measured. Additionally, a physical mixture consisting of 80% H4tcbpe ligand and 20% H4tcbpe-F ligand—the ratio present in LMOF-305—was prepared and optically qualified. As the tcbpe-F content of the LMOF samples increased, the quantum yield under 455 nm excitation also increased, and the physical ligand mixture showed no improvement. These results, summarized in Table 2 below and in Fig. S3 and S4,† support the argument that an interaction between tcbpe and tcbpe-F is responsible for LMOF-305's elevated quantum yield, and that the specific arrangement imposed by the LMOF framework is required to enable that interaction.
:20 tcbpe:tcbpe-F, LMOF-305, and three LMOF samples prepared with varying tcbpe-F amounts
| Sample | Quantum yield (455 nm ex.) |
|---|---|
| 80:20 H4tcbpe:H4tcbpe-F |
52% |
| LMOF-305′ (5% H4tcbpe-F) | 57% |
| LMOF-305′′ (10% H4tcbpe-F) | 69% |
| LMOF-305′′′ (15% H4tcbpe-F) | 83% |
| LMOF-305 (20% H4tcbpe-F) | 88% |
To further probe the emission mechanism, temperature dependent photoluminescence lifetime data were collected for the bulk ligands H4tcbpe and H4tcbpe-F, as well as LMOF-231 and LMOF-305, from 77 K to 295 K under 440 nm excitation. Room temperature lifetime data for all four samples is presented below in Table 3, and the lifetime data at temperatures ranging from 77 K to 273 K, as well as the luminescence decay profiles, are given in ESI Section S5.†
| Sample | Average amplitude weighted τ | τ 1 (ns) | τ 2 (ns) |
|---|---|---|---|
| H4tcbpe | 3.44 ns | 2.11 (42.2%) | 4.49 (55.8%) |
| H4tcbpe-F | 3.57 ns | 2.34 (50.5%) | 4.83 (49.5%) |
| LMOF-231 | 3.80 ns | 1.98 (35.6%) | 4.80 (64.4%) |
| LMOF-305 | 4.05 ns | 3.05 (66.2%) | 6.00 (33.8%) |
Both LMOF samples possessed similar average amplitude weighted lifetimes of approximately 4 ns (3.81 ns for LMOF-231 and 3.98 ns for LMOF-305) at 273 K, with the total decay being the sum of two processes (τ1 and τ2). LMOF-305 showed longer lifetimes for τ1 and τ2, with τ1 (3.05 ns) being 54% longer than that of LMOF-231 (1.98 ns) and τ2 (6.00 ns) being 25% longer than that of LMOF-231 (4.80 ns). Despite this, the average amplitude-weighted lifetimes were very similar because the longer τ2 lifetime was dominant in LMOF-231, while the shorter τ1 lifetime was dominant in LMOF-305.
Importantly, the lifetime behavior displayed by LMOF-305 under 440 nm excitation is very similar to the previously-discussed lifetime behavior of LMOF-231 under 380 nm excitation. This is strong evidence that inclusion of the fluorinated ligand helped push emission towards the more efficient “higher-energy pathway”. It should also be noted that the lifetime of LMOF-305 is independent of excitation wavelength (Fig. S8 and Table S6†), which further indicates that the luminescence mechanism for the material is identical for both blue and UV photons, unlike LMOF-231.
A similar trend can also be observed when comparing the lifetime of H4tcbpe and H4tcbpe-F; both the τ1 and τ2 processes are slower at room temperature in H4tcbpe-F, but the relatively faster τ1 process plays a larger role in the fluorinated ligand. This suggests that the H4tcbpe-F ligand plays a relatively large role in the emission from LMOF-305, despite the fact that it is present at a much lower concentration than H4tcbpe, and is convincing evidence that excitation energy moves to the H4tcbpe-F ligand before emission. Furthermore, the fact that LMOF-305 has both a higher quantum yield than LMOF-231 and slower excited state decay processes indicates that the inclusion of H4tcbpe-F within LMOF-231 weakens nonradiative excited state recombination.
As discussed in Section 3.1, emission from LMOF-305 is expected to result from a combination of intermolecular processes (electron transfer between tcbpe and tcbpe-F), and intramolecular processes located in a single ligand molecule. Polarized photoluminescence measurements were carried out on LMOF-305 single crystals to investigate the relationship between these processes.
When excitation light was polarized parallel to the c-axis of the crystal, the PL emission intensity was found to be generally lower in intensity compared to the PL emission when the excitation light was polarized perpendicularly to the c-axis (Fig. 6a). This suggests that absorption is slightly stronger in the direction perpendicular to the c-axis (i.e., almost parallel to the a-axis). This may be because the conjugated ligands absorb more strongly when light is polarized parallel to the ligand chain which is oriented almost perpendicular to the c-axis. In most cases, when the excitation light polarization was parallel to the c-axis, the emission light was more strongly polarized in the perpendicular direction (Fig. 6b). Therefore, the dominant excitation and emission polarizations were orthogonal to one another, which indicates electronic interaction between ligands (intermolecular). However, when excitation light was polarized perpendicularly to the c-axis, the results were less straight-forward: in some cases the emission polarized along the c-axis was strongest (i.e., the emission polarization was orthogonal to the excitation polarization); in other cases the emission polarized perpendicular to the c-axis was stronger or was completely depolarized (Fig. S9†). These results suggest that the emission originates from combinations of both intermolecular (tcbpe to tcbpe-F) and intramolecular ligand excited states.
 | ||
| Fig. 6 (a) Non-polarized emission spectra from LMOF-305 under excitation polarized parallel with (black) and perpendicular to (red) the crystallographic c axis. (b) Polarized emission spectra from LMOF-305 oriented parallel with (green) and perpendicular to (blue) the crystallographic c axis, produced under polarized excitation that was oriented parallel to the crystallographic c axis, with a photograph of a LMOF-305 single crystal used in the polarized photoluminescence experiment inset in the upper right. (c) Diagram illustrating measurement geometry and crystal orientation with the excitation source polarized parallel with (top) and perpendicular to (bottom) the crystallographic c index, used to collect polarized emission spectra oriented parallel with (top) or perpendicular to (bottom) the crystallographic c axis. As the excitation and emission polarization filters are independent, emission spectra were also collected with the excitation and emission filters orthogonal to each other; in the interest of saving space, these measurement geometries are not shown. | ||
4. Conclusions
In order to develop LMOF-based phosphor materials capable of competing with the commercial yellow phosphor YAG:Ce for applications in PC-WLEDs, it is necessary to be able to obtain a yellow-emitting LMOF with quantum yield comparable to YAG:Ce under blue (455 nm) excitation. We have previously synthesized LMOF-231, based on the chromophoric ligand H4tcbpe. With a quantum yield of 76% (λex = 455 nm), it became the highest performing yellow phosphor among all LMOFs reported to date. However, further improvement was necessary to increase the material's competitiveness. To address this, spectroscopic measurements were used to assess the potential excitation mechanisms, and DFT calculations were performed to help design a ligand that could be doped into the LMOF-231 structure and function as a bandgap modulator. The ligand H4tcbpe-F was synthesized, and spectroscopic studies indicated that it possessed the qualities predicted by DFT calculations; namely, reduced HOMO and LUMO energies and a largely unchanged HOMO–LUMO energy gap. H4tcbpe-F was successfully doped into LMOF-231 at 20% occupancy, and the resulting LMOF-305 showed increased blue light absorption and a significantly higher quantum yield of 88.2% at 450 nm excitation, the highest value among all yellow-emitting LMOF phosphors reported to date. In addition, we conducted an in-depth spectroscopic study employing temperature dependent and polarized photoluminescence methods in order to investigate and to gain a fundamental understanding of the PL mechanism and the origin for strongly enhanced quantum yield of this LMOF. This work serves as an example of how understanding luminescence mechanisms can guide the rational design of materials with greatly improved performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
WPL, EV and JL acknowledge the financial support from the National Science Foundation (Grant No. DMR-1507210). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.References
- Energy Savings Forecast of Solid-State Lighting in General Illumination Applications, US Department of Energy, 2014 Search PubMed.
- Annual Energy Outlook 2019, US Energy Information Administration, 2019 Search PubMed.
- N. Horowitz, LED Lighting Could Save Developing Countries $40 Billion/yr, Natural Resources Defense Council, Inc., 2017 Search PubMed.
- G. Charalampides, K. Vatalis, V. Karayannis and A. Baklavaridis, Environmental Defects And Economic Impact On Global Market Of Rare Earth Metals, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 161, 012069 CrossRef.
- M. A. de Boer and K. Lammertsma, Scarcity of Rare Earth Elements, ChemSusChem, 2013, 6(11), 2045–2055 CrossRef CAS PubMed.
- H. Wang and J. Li, Microporous Metal–Organic Frameworks for Adsorptive Separation of C5–C6 Alkane Isomers, Acc. Chem. Res., 2019, 52(7), 1968–1978 CrossRef CAS PubMed.
- Z. Bao, G. Chang, H. Xing, R. Krishna, Q. Ren and B. Chen, Potential of microporous metal–organic frameworks for separation of hydrocarbon mixtures, Energy Environ. Sci., 2016, 9(12), 3612–3641 RSC.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Gas/vapour separation using ultra-microporous metal–organic frameworks: insights into the structure/separation relationship, Chem. Soc. Rev., 2017, 46(11), 3402–3430 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion, Nat. Rev. Mater., 2017, 2(8), 17045 CrossRef CAS.
- C. Wang, B. An and W. Lin, Metal–Organic Frameworks in Solid–Gas Phase Catalysis, ACS Catal., 2019, 9(1), 130–146 CrossRef CAS.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Metal–Organic Frameworks as Platforms for Functional Materials, Acc. Chem. Res., 2016, 49(3), 483–493 CrossRef CAS PubMed.
- C. He, D. Liu and W. Lin, Nanomedicine Applications of Hybrid Nanomaterials Built from Metal–Ligand Coordination Bonds: Nanoscale Metal–Organic Frameworks and Nanoscale Coordination Polymers, Chem. Rev., 2015, 115(19), 11079–11108 CrossRef CAS PubMed.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Luminescent sensors based on metal–organic frameworks, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- M. J. Hurlock, Y. Kan, T. Lécrivain, J. Lapka, K. L. Nash and Q. Zhang, Molecular Association-Induced Emission Shifts for E/Z Isomers and Selective Sensing of Nitroaromatic Explosives, Cryst. Growth Des., 2018, 18(10), 6197–6203 CrossRef CAS.
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Metal–organic framework-based materials: superior adsorbents for the capture of toxic and radioactive metal ions, Chem. Soc. Rev., 2018, 47(7), 2322–2356 RSC.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Metal–organic frameworks for the removal of toxic industrial chemicals and chemical warfare agents, Chem. Soc. Rev., 2017, 46(11), 3357–3385 RSC.
- W. P. Lustig and J. Li, Luminescent metal–organic frameworks and coordination polymers as alternative phosphors for energy efficient lighting devices, Coord. Chem. Rev., 2018, 373, 116–147 CrossRef CAS.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Metal–organic frameworks: functional luminescent and photonic materials for sensing applications, Chem. Soc. Rev., 2017, 46(11), 3242–3285 RSC.
- J. Cornelio, T.-Y. Zhou, A. Alkaş and S. G. Telfer, Systematic Tuning of the Luminescence Output of Multicomponent Metal–Organic Frameworks, J. Am. Chem. Soc., 2018, 140(45), 15470–15476 CrossRef CAS PubMed.
- W. P. Lustig, F. Wang, S. J. Teat, Z. Hu, Q. Gong and J. Li, Chromophore-Based Luminescent Metal–Organic Frameworks as Lighting Phosphors, Inorg. Chem., 2016, 55(15), 7250–7256 CrossRef CAS PubMed.
- Z. Hu, G. Huang, W. P. Lustig, F. Wang, H. Wang, S. J. Teat, D. Banerjee, D. Zhang and J. Li, Achieving exceptionally high luminescence quantum efficiency by immobilizing an AIE molecular chromophore into a metal–organic framework, Chem. Commun., 2015, 51(15), 3045–3048 RSC.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, Rigidifying Fluorescent Linkers by Metal–Organic Framework Formation for Fluorescence Blue Shift and Quantum Yield Enhancement, J. Am. Chem. Soc., 2014, 136(23), 8269–8276 CrossRef CAS PubMed.
- L. Hanna and J. V. Lockard, From IR to X-rays: gaining molecular level insights on metal–organic frameworks through spectroscopy, J. Phys.: Condens. Matter, 2019, 31(48), 483001 CrossRef CAS PubMed.
- E. A. Dolgopolova, A. M. Rice, M. D. Smith and N. B. Shustova, Photophysics, Dynamics, and Energy Transfer in Rigid Mimics of GFP-based Systems, Inorg. Chem., 2016, 55(15), 7257–7264 CrossRef CAS PubMed.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Photochemistry and photophysics of MOFs: steps towards MOF-based sensing enhancements, Chem. Soc. Rev., 2018, 47(13), 4710–4728 RSC.
- F. Wang, W. Liu, S. J. Teat, F. Xu, H. Wang, X. Wang, L. An and J. Li, Chromophore-immobilized luminescent metal–organic frameworks as potential lighting phosphors and chemical sensors, Chem. Commun., 2016, 52(67), 10249–10252 RSC.
- Bruker Apex3, Bruker Analytical X-ray, Systems Inc., Madison, WI, 2003 Search PubMed.
- S. Bruker, SAX Area-Detector Integration Program v7.60a, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2010 Search PubMed.
- R. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51(1), 33–38 CrossRef PubMed.
- L. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32(4), 837–838 CrossRef CAS.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64(1), 112–122 CrossRef CAS PubMed.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(Pt 1), 9–18 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- C. Sosa, J. Andzelm, B. C. Elkin, E. Wimmer, K. D. Dobbs and D. A. Dixon, A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds, J. Phys. Chem., 1992, 96(16), 6630–6636 CrossRef CAS.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation, Can. J. Chem., 1992, 70(2), 560–571 CrossRef CAS.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Reducing aggregation caused quenching effect through co-assembly of PAH chromophores and molecular barriers, Nat. Commun., 2019, 10(1), 169 CrossRef PubMed.
- Z. Hu, G. Huang, W. P. Lustig, F. Wang, H. Wang, S. J. Teat, D. Banerjee, D. Zhang and J. Li, Achieving exceptionally high luminescence quantum efficiency by immobilizing an AIE molecular chromophore into a metal–organic framework, Chem. Commun., 2015, 51(15), 3045–3048 RSC.
- Q. Gong, Z. Hu, B. J. Deibert, T. J. Emge, S. J. Teat, D. Banerjee, B. Mussman, N. D. Rudd and J. Li, Solution Processable MOF Yellow Phosphor with Exceptionally High Quantum Efficiency, J. Am. Chem. Soc., 2014, 136(48), 16724–16727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1946919. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05721h |
| This journal is © The Royal Society of Chemistry 2020 |