
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven spirocyclization of epoxides via dual titanocene and photoredox catalysis†
Shuangjie
Lin‡
,
Yuqing
Chen‡
,
Fusheng
Li
,
Caizhe
Shi
and
Lei
Shi
 *
*
Zhang Dayu School of Chemistry, State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, China. E-mail: shilei17@dlut.edu.cn
First published on 9th December 2019
Abstract
We describe the synergistic utilization of titanocene/photoredox dual catalysis driven by visible light for the radical opening/spirocyclization of easily accessible epoxyalkynes. This environmentally benign process uses the organic donor–acceptor fluorophore 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) as a photocatalyst and Hantzsch ester (HE) as an electron donor instead of stoichiometric metallic reductants. The photocatalytic conditions showed exceptionally high reactivity for the synthesis of privileged and synthetically challenging spirocycles featuring a spiro all-carbon quaternary stereocenter. Cyclic voltammetry (CV) studies suggest that Cp2TiIIICl is the catalytically active species.
Over the last few decades, radical-based transformations have been increasingly used in organic synthesis due to their salient features, such as ease of generation, mild reaction conditions, and broad functional group compatibility.1,2 As a mild single-electron-transfer (SET) reagent, titanocene monochloride (Cp2TiIIICl) is considered a formidable tool in contemporary radical chemistry due to its ability to promote various fundamental radical-based transformations.3–7 Cp2TiIIICl was first introduced by Nugent and RajanBabu as a very mild stoichiometric reagent for the reductive opening of epoxides.8–11 Later, the catalytic conditions developed by Gansäuer et al. (Scheme 1a)12 employing stoichiometric amounts of active metals in combination with 2,4,6-collidine·HCl further expanded its applications and led to the discovery of a number of novel transformations.13–16 The key to success was the formation of a stable complex A in reactions while decreasing the concentration of active Cp2TiIIICl.17,18 We were interested in the radical opening/cyclization reaction of epoxides which has attracted considerable attention from the synthetic community and has been used numerous times in the synthesis of natural products.19,20 Nevertheless, this reaction required stoichiometric metallic reductants and proceeded slowly particularly with sterically hindered substrates even with high catalyst loading.21 Therefore, the development of an eco-friendly and efficient catalytic system with an expanded substrate scope is highly desirable.
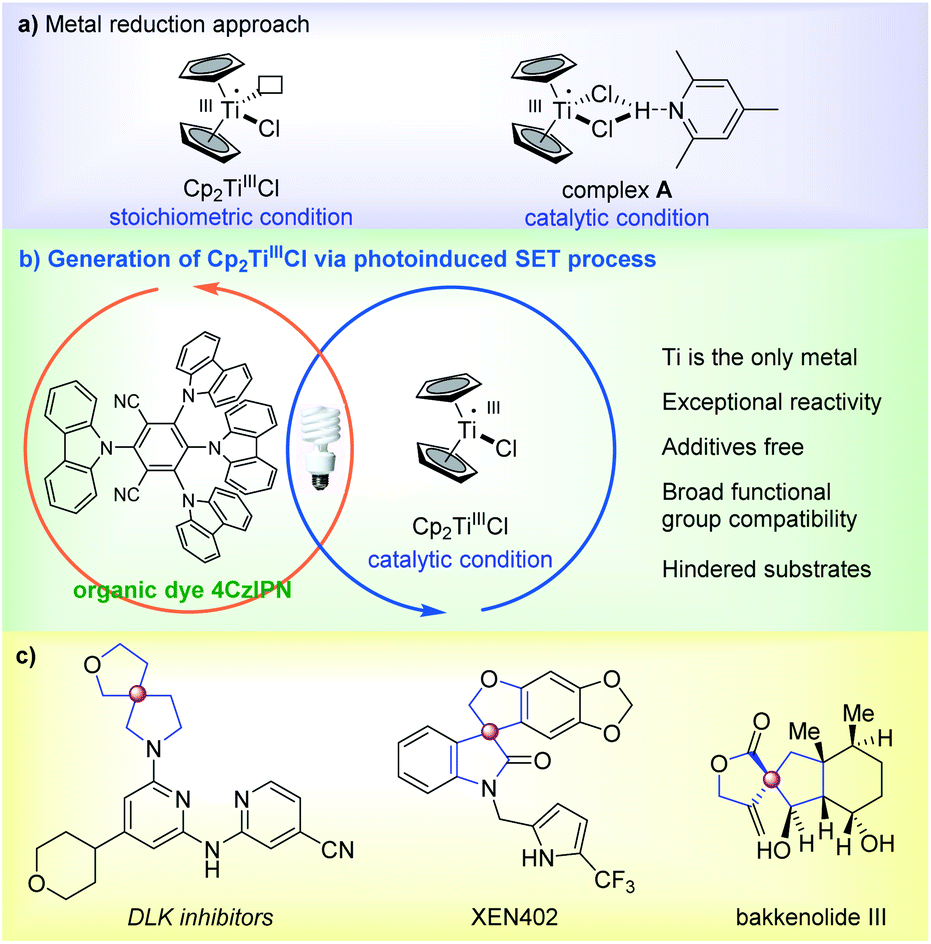 | ||
| Scheme 1 Cp2TiIIICl mediated radical opening/spirocyclization of epoxides; (a) generation of TiIIIvia a metal reduction approach; (b) dual titanocene/photoredox catalysis; (c) examples of drugs and natural products containing heterospirocycles. | ||
In recent years metallaphotoredox catalysis has been a new and rapidly growing research subject.22–29 Photoredox processes can directly modulate the oxidation state of metals by electron transfer (ET).30–33 Given that the generation of TiIII is a SET process, we envisioned that the reduction could be facilitated by a photoredox-controlled process while overcoming the aforementioned limitations. On the other hand, spirocycles bearing a chiral spiro all-carbon quaternary carbon are particularly attractive synthetic targets in pharmaceutical development (Scheme 1c).34–36 Such privileged rigid 3D structures offer the concomitant ability to project functionalities in all three-dimensional orientations and led to enhanced pharmacological activities of molecules. Thus significant attention has been paid to their synthesis.37,38 Against this backdrop, here we describe our efforts on the synthesis of various heterospirocycles with the aid of photoredox catalysis.
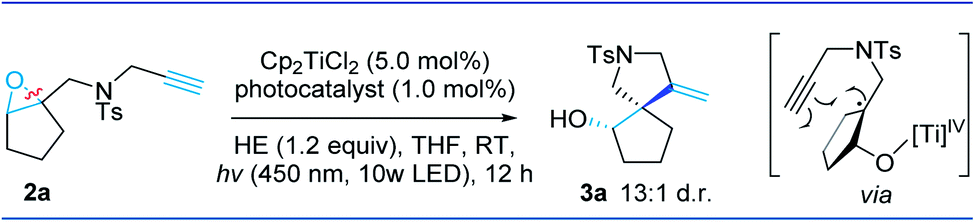
We chose epoxyalkyne 2a as a model substrate for optimization of reaction conditions. After a systematic variation of different reaction parameters, we were pleased to identify the optimal reaction conditions in which a mixture of Cp2TiCl2 (5.0 mol%), [Ir(dtbbpy)(ppy)2]PF6 (1a, 1.0 mol%, EIII/II1/2 = −1.51 V vs. SCE in MeCN), HE (1.2 equiv.) and 2a (1.0 equiv.) in THF at room temperature under the irradiation of a 10 W 450 nm light emitting diode (LED) lamp for 12 hours afforded the desired product 3a in an excellent yield of 96% (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.) upon isolation (entry 1). Using a commercial 23 W compact fluorescent lamp (CFL) instead of the 10 W 450 nm LED did not compromise the overall yield of the reaction (entry 2). Notably, when the loading of Cp2TiCl2 was decreased to as low as only 2.0 mol%, the reaction still led to full conversion and produced 3a in 95% yield (entry 3). Further screening of other photosensitizers revealed that the cheap and readily obtained organic dye 4CzIPN 1b is a competent alternative, which led to full conversion with 94% isolated yield (entry 4). Importantly, the reaction did not proceed in the absence of Cp2TiCl2, HE, the photocatalyst, or visible light (entries 5–8). Various solvents, including DMF, MeOH, DMSO, and MeCN, were screened, and they all resulted in poor conversion. The use of other organic electron donors, such as triethylamine, triethanolamine, and ascorbic acid, afforded the product in poor yield.
:1 d.r.) upon isolation (entry 1). Using a commercial 23 W compact fluorescent lamp (CFL) instead of the 10 W 450 nm LED did not compromise the overall yield of the reaction (entry 2). Notably, when the loading of Cp2TiCl2 was decreased to as low as only 2.0 mol%, the reaction still led to full conversion and produced 3a in 95% yield (entry 3). Further screening of other photosensitizers revealed that the cheap and readily obtained organic dye 4CzIPN 1b is a competent alternative, which led to full conversion with 94% isolated yield (entry 4). Importantly, the reaction did not proceed in the absence of Cp2TiCl2, HE, the photocatalyst, or visible light (entries 5–8). Various solvents, including DMF, MeOH, DMSO, and MeCN, were screened, and they all resulted in poor conversion. The use of other organic electron donors, such as triethylamine, triethanolamine, and ascorbic acid, afforded the product in poor yield.
With satisfactory reaction conditions established, we then explored the scope of the cyclization reaction using 4CzIPN as the photosensitizer. Positively, the cyclization reaction worked well and afforded the desired variably heterospirocyclic products in good to excellent yield (Tables 2 and 3). The reaction allows the rapid construction of various 5/5, 5/6, 5/7 and 5/8 spiro-ring fused systems (3a–3k) bearing tetrahydrofuran or pyrrolidine motifs via the 5-exo cyclization pathway. Interesting, the diastereoselectivity of the cyclization reaction is highly correlated with the ring size in the substrates. Heterospirocycles containing a 5/5 spiro-ring fused system (3a–3f) were obtained with surprisingly high diastereoselectivity. In some cases (3b, 3c, and 3e) only a single isomer was obtained. The product 3d with a sterically hindered t-butyloxy carbonyl (Boc) protecting group on the N atom was obtained with reduced diastereoselectivity (5:1 d.r.). The diastereoselectivities dropped in 5/6, 5/7 and 5/8 spiro-ring fused systems. Given that enantioenriched epoxides could be easily obtained (e.g. via sharpless asymmetric epoxidation), this strategy provides access to optically active spirocycles featuring an all-carbon quaternary stereocenter with the transfer of stereochemical information from epoxides (3c, 3e and 3f). Bis-heterospirocyclic scaffolds were frequently employed in pharmaceutical chemistry. For example, bis-heterospirocyclic 3d is the core structure of DLK inhibitors39 and XEN402 (ref. 40) (scheme 1c), which are used for treating neurodegeneration and congenital erythromelalgia respectively. Furthermore, 6-exo cyclization was also investigated under the standard conditions and smoothly produced a serious of drug-like 6-(trifluoromethyl)-3-pyridinesulfonyl piperidine derivatives including 6/5, 6/6 and 6/7 spiro-ring fused systems (5a–5k) in generally excellent yields. Moreover, cyclization reactions with epoxy-alkynes afforded products containing exocyclic-alkenes and free alcohols which were suitable for further functionalization. This approach provides access to a broad range of novel spirocyclic piperidine and pyrrolidine spirocycles which could be of interest to synthetic and medicinal chemists.
|
|
||
|---|---|---|
| Entry | Conditionsa | Yield (%)b |
| a Reaction conditions: 2a (100 mg, 0.1 M in THF). b Isolated yield. | ||
| 1 | [Ir(dtbbpy)(ppy)2]PF61a | 96 |
| 2 | 23 W CFL instead of 450 nm LED, 1a, 16 h | 95 |
| 3 | 2 mol% Ti, 1a, 24 h | 94 |
| 4 | 4CzIPN 1b, 16 h | 94 |
| 5 | No titanocene | 0 |
| 6 | No HE | 0 |
| 7 | No photocatalyst | 0 |
| 8 | No hν | 0 |
| 9 | DCE instead of THF, 1a | 93 |
| 10 | Et3N instead of HE, 1a | 9 |
| 11 | Na2CO3 (1.0 equiv.) as additive, 1a | 0 |
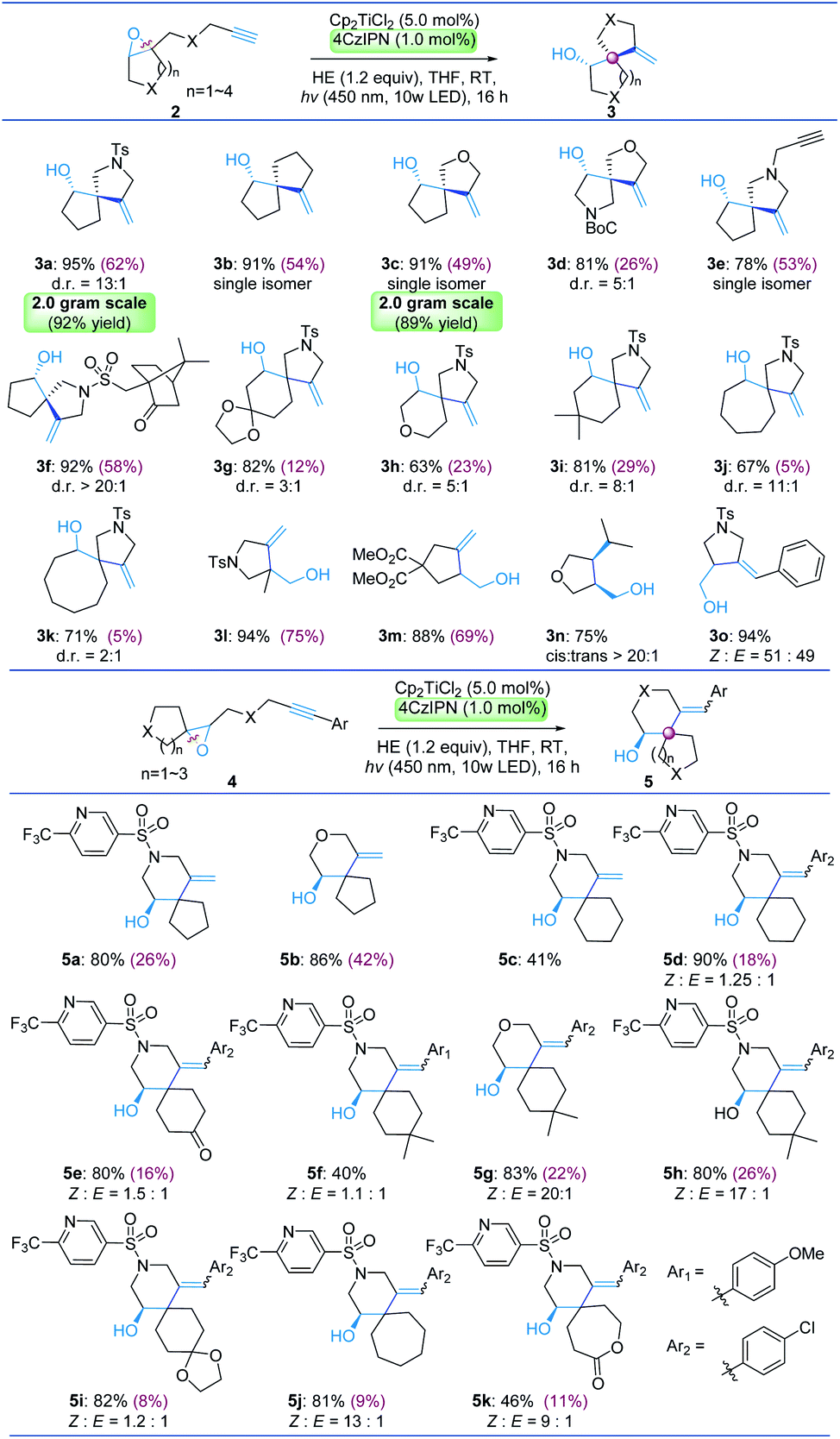

To examine the scalability of the reaction, gram-scale synthesis of 3a and 3c was performed under the standard conditions with 23 W CFL irradiation. Pleasingly, 92% (3a) and 89% (3c) isolated yields were obtained respectively without any deterioration. Furthermore, an additive-based investigation41 was performed and the results are summarized (Table 3). From this screening, we found that 11 out of 12 additives have no adverse impact on the yield of the reaction. The additives were recovered after the reaction, including benzoxazole 6a, quinazolinone 6b, collidine 6c, tetrahydroquinoline 6d, benzothiazole 6e, indole 6f, and benzofuran 6h. However, quinoxaline 6l strongly inhibited Ti catalysis and 3l was produced in only 23% yield. Notably, the reaction is compatible with various functional groups including phenols 6g, free alcohols and alkene 6j, iodobenzene 6i, ester 3m, ether 3n, dioxolane 5i, lactone 5k, and alkyne 3e. The generality of the dual Ti/4CzIPN catalysis system was further demonstrated by a highly related hydrogen transfer reaction of epoxides which could exclusively provide anti-Markovnikov alcohols (see the ESI S8†). The low-cost of 4CzIPN, broad compatibility with sensitive functional groups, and simple operation conditions are appealing for laboratory and industrial applications.
Importantly, the reactions were re-subjected to metal reduction catalytic conditions for comparison with the photocatalytic conditions and the yields are shown within parentheses (Table 2d). It clearly showed that the yields were generally lower. Particularly, we found that the yields dropped dramatically as the steric encumbrance of the substrates increased. For example, substrates containing dioxolane (2g, 4i) or a 7/8-membered-ring (2j, 2k, 4j) afforded the products (3g, 5i, 3j, 3k, 5j) in less than 10% yield with the recovery of the starting materials.
Catalytic species and mechanistic studies
To further understand the exceptional reactivity of the photocatalytic reaction system and the mechanism, a series of experiments were performed. First, the mixture of Cp2TiCl2, the photocatalyst (1a or 1b) and HE in THF was irradiated under a 23 W CFL or a 450 nm LED at room temperature. The color change from red (Cp2TiCl2) to green indicates that a TiIII species was formed (Fig. 1b). The results of analysis by ultraviolet-visible (UV-vis) spectroscopy further revealed that the reduction is fast. It was nearly complete within 20 minutes (Fig. 1a). On the other hand, no change of color was observed in the dark even at elevated temperatures, indicating that ground-state SET was not operative. The initial electron transfer during the SET process was investigated by fluorescence quenching experiments (Fig. 1c). The luminescence of 1a at its λmax (591 nm) in degassed THF was readily quenched by Cp2TiCl2, exhibiting Stern–Volmer kinetics with a rate of 3.8 × 109 L M−1 S−1. HE also quenched the luminescence of 1a with a rate of 4.5 × 107 L M−1 S−1. Therefore, both oxidative and reductive quenching of the excited state of 1a can occur.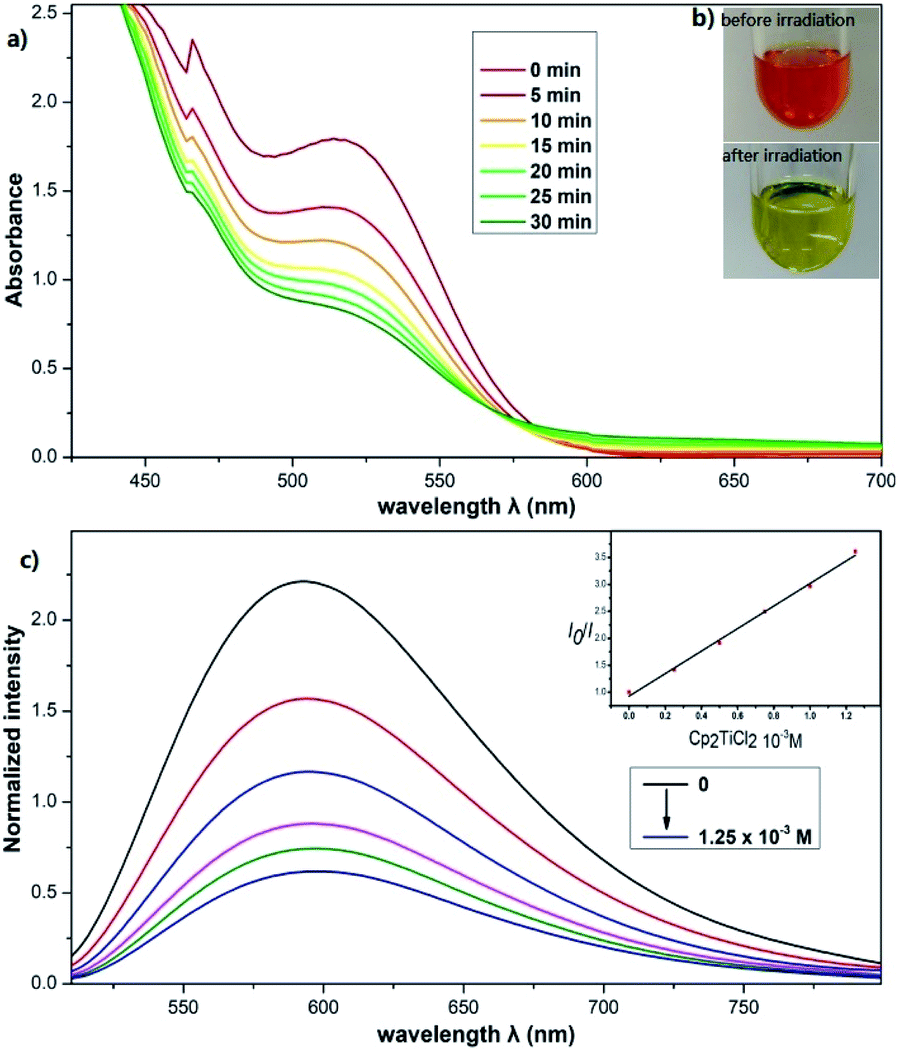 | ||
| Fig. 1 (a) UV-vis spectra showing the reduction of Cp2TiCl2 upon photoirradiation of a solution of Cp2TiCl2, [Ir(dtbbpy)(ppy)2]PF6 and HE in THF; the peak at 515 nm corresponds to Cp2TiCl2. (b) Photoirradiation (23 W CFL) of the mixture for 30 min. (c) [Ir(dtbbpy)(ppy)2]PF6 emission quenching by Cp2TiCl2. Experimental details are found in the ESI, S2.† | ||
Cyclic voltammetry (CV) studies were carried out and the results are shown in Fig. 2. In line with the previous CV studies of Zn/Cp2TiCl2 solutions in THF (green line), the broad oxidation wave appearing at −0.26 V (vs. SCE) is assigned to the (Cp2TiIIICl)2 dimer and Cp2TiIIICl monomer couple (designated 1/2).17,42 Interestingly, CV studies of TiIII species generated under photoreductive conditions (orange line) revealed the main oxidation wave appearing at −0.22 V (vs. SCE) which corresponds to the Cp2TiIIICl monomer. One concern was the intrinsic instability of Cp2TiCl2 under visible light which would decompose by losing a cyclopentadienyl ligand.43 Nevertheless, in the preliminary studies we found that the Cp2TiIIICl species exhibits special stability under photoreductive conditions (for detailed Experiments and discussion, please see the ESI S9†). Furthermore, the CV analysis of Zn/CpTiCl3 showed the main oxidation wave appearing at −0.63 V (vs. SCE) (see the ESI S4†). Therefore, the possibility of generation of the CpTiIIICl2 species via losing a cyclopentadienyl ligand under light could be ruled out.
 | ||
| Fig. 2 Cyclic voltammetry of 2 mM Cp2TiCl2/Zn (green line) and 2 mM Cp2TiCl2/[Ir(dtbbpy)(ppy)2]PF6/HE (orange line) recorded on a glassy carbon disk electrode with ν = 0.1 V s−1 in 0.2 M Bu4NPF6/THF. Experimental details are found in the ESI, S4.† The values are given in V vs. SCE and can be converted to V vs. Fc+/Fc by subtracting 0.52 V. | ||
Additionally, the reaction profiles were examined and are shown in Fig. 3. The photocatalytic reaction did not show an obvious incubation period. Moreover, the reactions proceeded pretty fast and completed (full conversion of substrates) within around 2 hours with 2a (red solid line) and even more sterically hindered 4i (blue solid line). In contrast, under the metal reduction catalytic conditions, the reactions proceeded slowly for the same substrates 2a (red dashed line) and 4i (blue dashed line). These results clearly showed the advantage of the current photocatalytic system. Kinetically, complex A (Scheme 1a) formed under metal reduction conditions is less reactive presumably due to the fact that it has no free coordination sites. In contrast, Cp2TiIIICl generated under photocatalytic conditions has a vacant site allowing the oxygen atom of the epoxide to perform a SET through an inner-sphere mechanism efficiently. Furthermore, the in situ-generated HP cation 7, which has a low pKa value compared with the collidine cation, can enhance the rate of protonation of the TiIV-alkoxy bond in the catalytic cycle. This is supported by the fact that the reaction is significantly inhibited by the addition of Na2CO3 or using triethylamine as a reductant (Table 1, entry 10 and 11).
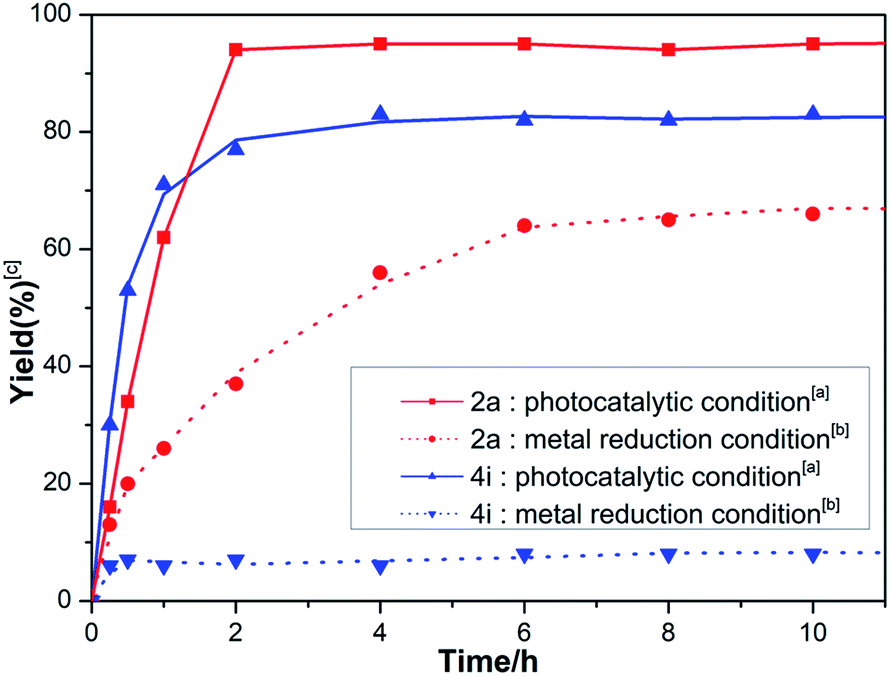 | ||
| Fig. 3 Reaction profiles for 2a and 4i (200 mg, 0.1 M in THF). [a] Photocatalytic conditions: 450 nm 10 W LED, 1a (1 mol%), Cp2TiCl2 (5 mol%), HE (1.2 eq). [b] Metal reduction conditions: CpTi2Cl2 (5 mol%), Zn (2.0 eq.), coll·HCl (2.5 eq.). [c] The yields of 3a or 5i were determined with 1H NMR. | ||
Based on these results, we tentatively propose the following catalytic cycle in Fig. 5. First, Cp2TiCl2 is reduced to Cp2TiIIICl by a photocatalyst (such as excited IrIII). The strong oxidant IrIV species is then reduced by HE to IrIII and produces HE+˙ concurrently. Cp2TiIIICl promoted the reductive opening of the epoxides. The resulting carbon radical 8 intramolecularly adds to the pendant alkyne to construct the spirocyclic structure. The newly formed vinyl radical 9 can abstract a C4–H hydrogen atom of the HE radical cation (HE+˙). This is because HE+˙ has a low homolytic bond-dissociation free energy for the C4–H bond (this energy requirement is 31.4 kcal mol−1 for acetonitrile).44 Deuterium labeling experiments also shows that THF may offer a H source for hydrogen atom transfer (Fig. 4). The in situ-produced Ti–oxygen bond is protonated by the HP cation and TiIV is released as well as the cyclization product. Finally, IrIII can regenerate Cp2TiIIICl for the next catalytic cycle.
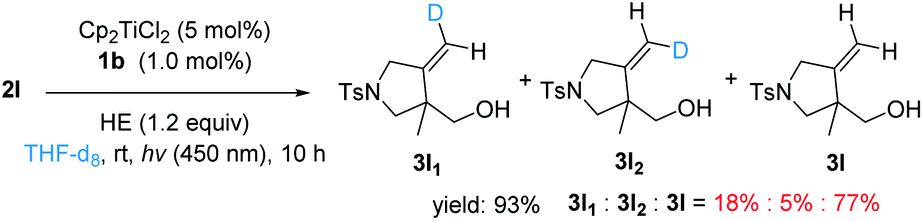 | ||
| Fig. 4 Cyclization of 2l in the presence of THF-d8. | ||
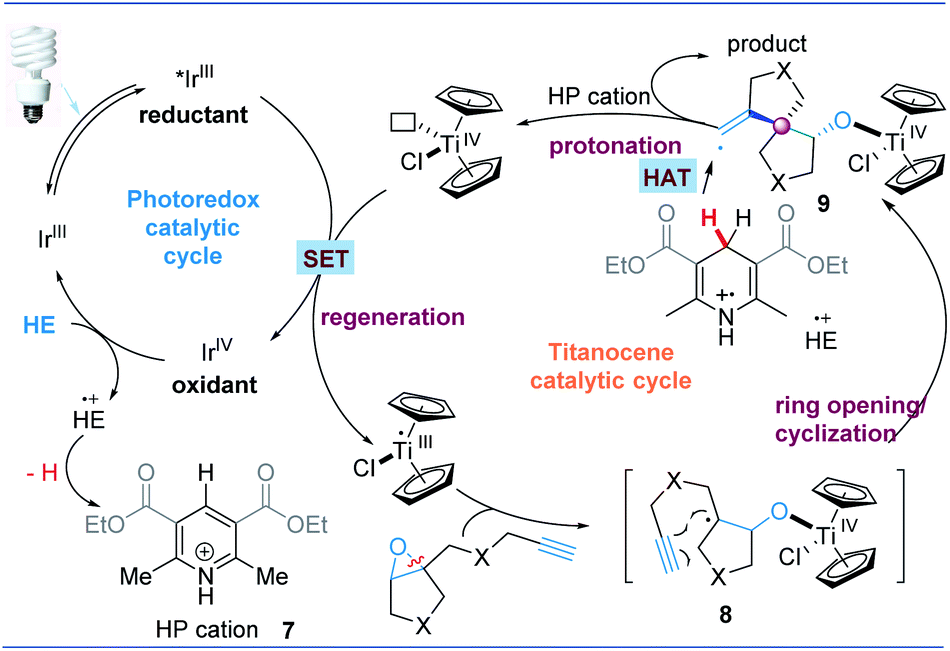 | ||
| Fig. 5 Proposed catalytic mechanism. | ||
Conclusions
In summary, we report a novel and greener dual catalyst system consisting of titanocene and the organic dye 4CzIPN for the radical spirocyclization of epoxides under photocatalytic conditions. This operationally simple, scalable, and efficient method provides a strategically distinct manner for the assembly of heterospirocycles featuring a spiro all-carbon quaternary stereocenter. Unlike the metal reduction catalytic conditions, mechanistic studies suggest that under photocatalytic conditions, TiIII is present as Cp2TiCl which is readily accessible to epoxides. As expected, the photocatalytic conditions showed exceptional high reactivity for the assembly of heterospirocycles. In this respect, the photocatalytic approach complements the metal reduction approach. Development of new transformations based on photocatalytic reactions is currently under way in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Fundamental Research Funds for the Central Universities (No. DUT19LK31). We thank WATTCAS CHEM-TECH Co., Ltd. for kindly providing the photoreactor.Notes and references
- M. P. Plesniak, H. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1(10), 77 CrossRef.
- A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd., 2012 Search PubMed.
- A. Rosales, I. Rodríguez-García, J. Muñoz-Bascón, E. Roldan-Molina, N. M. Padial, L. P. Morales, M. García-Ocaña and J. E. Oltra, Eur. J. Org. Chem., 2015, 21, 4567–4591 CrossRef.
- Y. Zhang, E. Vogelsang, Z. Qu, S. Grimme and A. Gansäuer, Angew. Chem., Int. Ed., 2017, 56(41), 12654–12657 CrossRef CAS.
- K. Ye, T. MCallum and S. Lin, J. Am. Chem. Soc., 2019, 141(24), 9548–9554 CrossRef CAS.
- L. H. Leijendekker, J. Weweler, T. M. Leuther and J. Streuff, Angew. Chem., Int. Ed., 2017, 56(22), 6103–6106 CrossRef CAS PubMed.
- J. Muñoz-Bascón, I. Sancho-Sanz, E. Álvarez-Manzaneda, A. Rosales and J. E. Oltra, Chem.–Eur. J., 2012, 18(45), 14479–14486 CrossRef.
- W. A. Nugent and T. V. RajanBabu, J. Am. Chem. Soc., 1988, 110, 8561–8562 CrossRef CAS.
- T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116(3), 986–997 CrossRef CAS.
- M. L. H. Green and C. R. Lucas, J. Chem. Soc., Dalton Trans., 1972, 1000–1003 RSC.
- J. M. Birmingham, A. K. Fischer and G. Wilkinson, Naturwissenschaften, 1955, 42, 96 CrossRef CAS.
- A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120(49), 12849–12859 CrossRef.
- J. Justicia, J. L. Oller-López, A. G. Campaña, J. E. Oltra, J. M. Cuerva, E. Buñuel and D. J. Cárdenas, J. Am. Chem. Soc., 2005, 127(42), 14911–14921 CrossRef CAS.
- J. Friedrich, M. Dolg, A. Gansäuer, D. Geich-Gimbel and T. Lauterbach, J. Am. Chem. Soc., 2005, 127(19), 7071 CrossRef CAS.
- J. Friedrich, K. Walczak, M. Dolg, F. Piestert, T. Lauterbach, D. Worgull and A. Gansäuer, J. Am. Chem. Soc., 2008, 130(5), 1788–1796 CrossRef CAS PubMed.
- A. Gansäuer, D. Worgull, K. Knebel, I. Huth and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48(47), 8882–8885 CrossRef PubMed.
- A. Gansäuer, C. Kube, K. Daasbjerg, R. Sure, S. Grimme, G. D. Fianu, D. V. Sadasivam and R. A. Flowers, J. Am. Chem. Soc., 2014, 136(4), 1663–1671 CrossRef PubMed.
- J. Gordon, S. Hildebrandt, K. R. Dewese, S. Klare, A. Gansäuer, T. V. RajanBabu and W. A. Nugent, Organometallics, 2018, 37(24), 4801–4809 CrossRef CAS PubMed.
- N. M. Padial, E. Roldan-Molina, A. Rosales, M. Álvarez-Corral, I. Rodríguez-García, M. Muñoz-Dorado and J. E. Oltra, Studies in Natural Products Chemistry, 2018, vol. 55, pp. 31–71 Search PubMed.
- S. P. Morcillo, D. Miguel, A. G. Campana, L. Alvarez De Cienfuegos, J. Justicia and J. M. Cuerva, Org. Chem. Front., 2014, 45(25), 15–33 RSC.
- L. Shi, K. Meyer and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49(48), 9250–9253 CrossRef CAS.
- J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1(7), 52 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113(7), 5322–5363 CrossRef CAS.
- B. L. Tóth, O. Tischler and Z. Novák, Tetrahedron Lett., 2016, 57(41), 4505–4513 CrossRef.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49(8), 1566–1577 CrossRef CAS.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49(7), 1429–1439 CrossRef CAS.
- D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49(10), 2295–2306 CrossRef CAS.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116(17), 10035–10074 CrossRef CAS.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., 2019, 131(19), 6212–6224 CrossRef.
- K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139(28), 9467–9470 CrossRef CAS.
- Q. Meng, S. Wang and B. König, Angew. Chem., Int. Ed., 2017, 56(43), 13426–13430 CrossRef CAS.
- Z. Zhang, R. B. Richrath and A. Gansäuer, ACS Catal., 2019, 9(4), 3208–3212 CrossRef CAS.
- D. C. Fabry and M. Rueping, Merging Visible Light, Acc. Chem. Res., 2016, 49(9), 1969–1979 CrossRef CAS.
- Y. Zheng and C. M. Tice, Expert Opin. Drug Discovery, 2016, 11(9), 831–834 CrossRef.
- Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24(16), 3673–3682 CrossRef CAS.
- L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13(39), 9907–9933 RSC.
- N. J. Flodén, A. Trowbridge, D. Willcox, S. M. Walton, Y. Kim and M. J. Gaunt, J. Am. Chem. Soc., 2019, 141(21), 8426–8430 CrossRef.
- N. A. Weires, Y. Slutskyy and L. E. Overman, Angew. Chem., Int. Ed., 2019, 58(25), 8561–8565 CrossRef CAS.
- B. A. Chalyk, M. V. Butko, O. O. Yanshyna, K. S. Gavrilenko, T. V. Druzhenko and P. K. Mykhailiuk, Chem.–Eur. J., 2017, 23(66), 16782–16786 CrossRef CAS.
- S. K. Bagal, A. D. Brown, P. J. Cox, K. Omoto, R. M. Owen, D. C. Pryde, B. Sidders, S. E. Skerratt, E. B. Stevens, R. I. Storer and N. A. Swain, J. Med. Chem., 2013, 56(3), 593–624 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5(7), 597–601 CrossRef CAS.
- R. J. Enemærke, J. Larsen, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2004, 126(25), 7853–7864 CrossRef.
- M. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff and J. Fouassier, Macromolecules, 2011, 45(1), 356–361 CrossRef.
- X. Zhu, H. Li, Q. Li, T. Ai, J. Lu, Y. Yang and J. Cheng, Chem.–Eur. J., 2003, 9(4), 871–880 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05601g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |